Metabolic Engineering Fundamentals: From Core Concepts to Biomedical Applications
This comprehensive guide provides researchers, scientists, and drug development professionals with essential knowledge in metabolic engineering.
Metabolic Engineering Fundamentals: From Core Concepts to Biomedical Applications
Abstract
This comprehensive guide provides researchers, scientists, and drug development professionals with essential knowledge in metabolic engineering. Covering foundational principles, cutting-edge methodologies, optimization strategies, and validation techniques, it bridges basic science with clinical applications. The content explores how engineered metabolic pathways enable sustainable production of therapeutic compounds, drug precursors, and biomarkers, incorporating the latest advances in AI, computational modeling, and heterologous expression systems for biomedical innovation.
Core Principles and Historical Evolution of Metabolic Engineering
Metabolic engineering is a discipline dedicated to the optimization of native metabolic pathways and regulatory networks, or the assembly of heterologous metabolic pathways, for the production of targeted molecules using molecular, genetic, and combinatorial approaches [1]. The primary goal is to generate efficient microbial cell factories that produce cost-effective molecules at an industrial scale from renewable feedstocks [2] [1]. Since the term was coined in the late 1980s-early 1990s, the field has expanded substantially, moving beyond manipulations of single enzymes to encompass the holistic design and optimization of entire metabolic networks [2]. This evolution has been driven by advances in adjacent fields, including DNA sequencing, genetic tool development, and sophisticated analytical and modeling techniques [2] [3].
The applications of metabolic engineering are vast, spanning the production of active pharmaceutical ingredients (APIs), specialty chemicals, biofuels, and bulk chemicals [2]. A key advantage over traditional synthetic organic chemistry is the ability to produce complex natural products that are otherwise difficult or impossible to synthesize chemically [2]. The field is guided by the central metrics of titer, yield, and rate (TYR), which have become the benchmarks for evaluating the cost-competitiveness of an engineered cell factory [1].
Core Principles and Methodologies
The practice of metabolic engineering requires the consideration of multiple, interconnected factors. Successful projects typically involve [1]:
- Selection of a Host Organism: Choosing a safe, robust host with genetic and physiological advantages for the target product (e.g., E. coli, S. cerevisiae, or the oleaginous yeast Yarrowia lipolytica).
- Pathway Understanding: Comprehensive knowledge of the metabolic pathways, co-factor balances (e.g., NADPH, ATP), and regulatory networks involved.
- Genetic Tool Availability: Access to effective enzymes, genetic elements, and efficient transformation technologies.
- Analytical and Modeling Frameworks: The use of stoichiometric, thermodynamic, and kinetic analyses to identify and overcome pathway bottlenecks.
The process can be conceptualized as an iterative Design-Build-Test-Learn (DBTL) cycle [1]. In this framework, computational models are used to design a cell factory, which is then constructed and tested experimentally. The resulting data is analyzed to refine the model and inform the next cycle of engineering, progressively optimizing the system [1] [3].
Key Engineering Strategies at the Network Level
Modern metabolic engineering has shifted from targeting a handful of genes to implementing complex designs requiring the modification of dozens of genes across diverse metabolic functions [1]. A systematic computational study evaluating 12,000 biosynthetic scenarios revealed that over 70% of product pathway yields can be improved by introducing appropriate heterologous reactions, and identified 13 universal engineering strategies [4]. The five most effective strategies, applicable to over 100 products, are summarized in the table below.
Table 1: High-Impact Metabolic Engineering Strategies for Breaking Stoichiometric Yield Limits
| Strategy Category | Specific Mechanism | Example Action | Key Impact |
|---|---|---|---|
| Carbon-Conserving | Non-oxidative glycolysis (NOG) | Replaces the classic Embden-Meyerhof-Parnas glycolysis pathway [4] | Increases yield of acetyl-CoA-derived products (e.g., farnesene, PHB) [4] |
| Carbon-Conserving | Reductive TCA cycle | Assimilates COâ‚‚ and fixes it into central metabolism [4] | Enhances carbon efficiency for various products |
| Energy-Conserving | ATP-efficient pathways | Utilizes NADH-generating glyceraldehyde-3-phosphate dehydrogenase [4] | Improves ATP yield and overall metabolic efficiency |
| Energy-Conserving | Bypassing ATP-inefficient steps | Replaces phosphoenolpyruvate carboxykinase with an ATP-insensitive enzyme [4] | Conserves ATP, increasing energy available for biosynthesis |
| Cofactor Balancing | Transhydrogenase cycles | Shuttles reducing equivalents between NADH and NADPH pools [4] | Balances cofactor availability, relieving thermodynamic constraints |
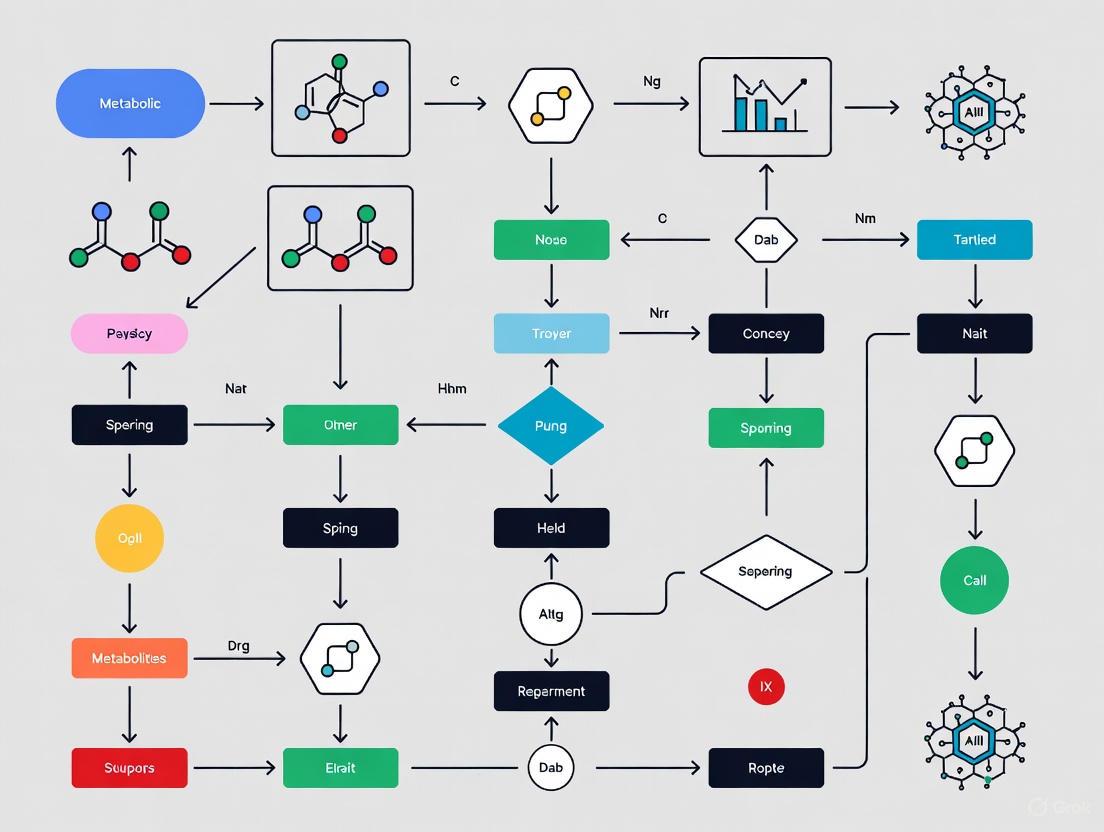
The following workflow diagram illustrates how these strategies are integrated into a modern, model-driven metabolic engineering pipeline.
The Scientist's Toolkit: Computational and Analytical Frameworks
The complexity of modern metabolic engineering necessitates a suite of sophisticated computational tools to model, predict, and analyze cellular metabolism. These tools are integral to the "Design" and "Learn" phases of the DBTL cycle.
Metabolic Modeling and Flux Analysis
Genome-Scale Metabolic Models (GEMs) are comprehensive representations of an organism's metabolism, integrating all metabolic reactions annotated from its genome [4]. GEMs are typically used with Flux Balance Analysis (FBA), a constraint-based method that predicts steady-state metabolic fluxes to optimize a biological objective, such as biomass growth or product formation [4] [3]. For more advanced analysis, 13C-based Metabolic Flux Analysis (13C-MFA) is considered the gold standard for experimentally estimating intracellular metabolic fluxes. It involves culting microbes on 13C-labeled carbon substrates, measuring the resulting isotope patterns in metabolites, and using computational optimization to identify the flux distribution that best fits the experimental data [5].
Table 2: Key Computational Tools and Platforms for Metabolic Engineering
| Tool Name | Primary Function | Key Features | Application in Metabolic Engineering |
|---|---|---|---|
| GEMs + FBA [4] [3] | Predicting metabolic flux distributions | Genome-scale network reconstruction; constraint-based optimization | In silico prediction of gene knockout targets, nutrient requirements, and theoretical yield limits. |
| QHEPath [4] | Quantitative heterologous pathway design | Algorithm to identify heterologous reactions that break host's yield limit | Systematically design pathways to surpass stoichiometric yield limits of native host metabolism. |
| Fluxer [6] | Flux network visualization | Web application for FBA and visualization of GEMs as interactive graphs | Visualize major metabolic pathways and identify key routes between metabolites of interest. |
| mfapy [5] | 13C-MFA data analysis | Open-source Python package for non-linear optimization of flux distributions | Estimate intracellular fluxes from isotope labeling data; supports custom model development and experimental design. |
| CSMN [4] | Cross-species metabolic network modeling | Integrated model combining reactions from multiple organisms and databases | Serves as a universal biochemical reaction database for heterologous pathway design in non-native hosts. |
| Blestriarene A | Blestriarene A, MF:C30H26O6, MW:482.5 g/mol | Chemical Reagent | Bench Chemicals |
| Dracaenoside F | Dracaenoside F|Supplier | Dracaenoside F is a steroidal saponin for research use. Isolated from Dracaena sp. For Research Use Only. Not for human or veterinary use. | Bench Chemicals |
Visualizing Metabolic Networks and Regulation
Effective visualization is critical for interpreting the vast amounts of data generated from GEMs and flux analyses. Tools like Fluxer enable the automated visualization of complete GEMs, displaying flux distributions as spanning trees, dendrograms, or complete graphs to help researchers identify the most important pathways contributing to a product of interest [6]. Beyond fluxes, understanding regulation is key. The concept of Regulatory Strength (RS) provides a quantitative measure of how strongly a metabolite (effector) up- or down-regulates a reaction step compared to its non-regulated state [7] [8]. This allows for the visualization of inhibitory or activating interactions within a network, which is crucial for explaining why metabolic fluxes are at certain levels even when substrate and product concentrations suggest otherwise [7].
The diagram below illustrates how these computational and data layers integrate to form a comprehensive understanding of a metabolic network, from gene to function.
Experimental Protocols and Applications
Protocol for Computational Strain Design Using GEMs
This protocol outlines the use of genome-scale models to predict gene knockout targets for maximizing product yield [4] [3].
- Model Selection and Preparation: Obtain a curated GEM for your host organism (e.g., from the BiGG Models database). Ensure the model includes a reaction for the synthesis of your target product. If not, add the necessary heterologous reactions.
- Define Constraints and Objective: Set constraints to reflect the experimental conditions, such as substrate uptake rate (e.g., glucose uptake = 10 mmol/gDW/h). Define the objective function to be maximized, typically the flux through the product synthesis reaction.
- In silico Gene Deletion Analysis: Use a algorithm such as OptKnock. Perform simulations that systematically "knock out" one or more gene-associated reactions in the model while maximizing the product formation objective.
- Output and Interpretation: The algorithm will output a set of gene deletion candidates that couple product formation to growth. These predictions represent potential metabolic engineering targets.
- Validation and Iteration: Construct the proposed strain in the laboratory and characterize its performance in bioreactors. Use omics data (transcriptomics, metabolomics) from the engineered strain to refine the model and identify the next cycle of targets.
Case Studies in Industrial Production
Metabolic engineering has successfully led to the commercial production of a diverse range of molecules. Key examples include:
- 1,3-Propanediol and 1,4-Butanediol: These bulk chemicals, used as polymer precursors, are produced in engineered E. coli by DuPont and Genomatica, respectively. The engineering involved transferring genes from Klebsiella pneumoniae and S. cerevisiae into E. coli to create a novel pathway from glucose [2] [1].
- Artemisinic Acid: A precursor to the antimalarial drug artemisinin is produced in engineered S. cerevisiae by Amyris. This project involved transferring the complex biosynthetic pathway from the plant Artemisia annua into yeast, and is a prime example of how metabolic engineering can rival synthetic chemistry for producing complex APIs [2] [1].
- Advanced Biofuels: Engineered microbes now produce hydrocarbons compatible with existing infrastructure. For example, linear alkanes and alkenes for diesel and jet fuel are produced via the fatty acid biosynthetic pathway, while branched hydrocarbons are produced via the isoprenoid pathway [2]. These fuels often diffuse out of the cells and phase-separate, simplifying downstream purification [2].
Metabolic engineering has matured from a discipline focused on single-enzyme manipulations to a sophisticated field of network-level design. The integration of systems biology, computational modeling, and advanced genetics through the DBTL cycle has enabled the rational development of microbial cell factories for a sustainable bio-economy. The future of the field lies in enhancing the predictability of models by further integrating regulatory and kinetic information, improving the scale and precision of genome editing, and developing more dynamic control systems to autonomously manage metabolic resources. As these tools advance, the scope of products accessible through biological production will continue to expand, solidifying metabolic engineering as a cornerstone of modern industrial biotechnology.
Metabolic engineering leverages cellular pathways to produce valuable chemicals, fuels, and therapeutics. Its foundation rests on seminal Nobel Prize-winning discoveries that have elucidated how cells convert energy, recycle components, sense their environment, and catalyze reactions. This guide synthesizes these foundational discoveries into a coherent framework for researchers and drug development professionals, connecting historical insights to modern engineering principles. By understanding the mechanistic basis of metabolism—from the central Krebs cycle to engineered enzymes—we can better design microbial cell factories and therapeutic interventions. The following sections detail the key discoveries, their experimental proofs, and the practical tools they have inspired.
Nobel Prize-Winning Discoveries in Metabolism
Several Nobel Prizes have been awarded for discoveries that form the bedrock of our understanding of cellular metabolism. The table below summarizes the most critical ones for metabolic engineering.
Table 1: Foundational Nobel Prizes in Metabolic Research
| Year | Laureate(s) | Key Discovery | Significance for Metabolic Engineering |
|---|---|---|---|
| 1953 | Hans Krebs [9] | The Citric Acid Cycle (Krebs Cycle) | Defined the central pathway for the oxidation of acetyl-CoA to produce energy and precursor metabolites. |
| 1953 | Fritz Lipmann [9] | Coenzyme A and its importance for intermediary metabolism | Identified the essential cofactor (CoA) that activates metabolic intermediates, such as acetyl-CoA, for entry into the Krebs cycle. |
| 2016 | Yoshinori Ohsumi [10] | Mechanisms of Autophagy | Elucidated the pathway for cellular recycling, allowing cells to degrade and reuse cytoplasmic components, a key consideration in cellular efficiency. |
| 2019 | W. G. Kaelin Jr., P. J. Ratcliffe, G. L. Semenza [11] | Oxygen-sensing mechanism of cells via the HIF-1α pathway | Revealed how cells sense and adapt to oxygen availability, a critical factor in large-scale bioreactor fermentations and tumor metabolism. |
| 2018 | Frances H. Arnold [12] [13] | Directed evolution of enzymes | Pioneered a method to engineer highly efficient and novel enzymes for industrial catalysis, including the synthesis of pharmaceuticals and biofuels. |
Detailed Analysis of Key Discoveries
The Krebs Cycle and Coenzyme A: The Energetic Core
The 1953 Nobel Prize awarded to Hans Krebs and Fritz Lipmann established the core of energetic metabolism [9]. Krebs identified the cyclic metabolic pathway that converts the energy in carbohydrates, fats, and proteins into usable chemical energy. Lipmann discovered coenzyme A (CoA), the critical molecule that "activates" metabolic fragments, most notably two-carbon acetyl groups, for entry into this cycle.
- Molecular Mechanism: The Krebs cycle occurs in the mitochondrial matrix. Acetyl-CoA, the activated form of acetate, condenses with oxaloacetate (a four-carbon compound) to form citrate. Through a series of eight enzymatic steps, citrate is progressively decarboxylated and oxidized, regenerating oxaloacetate. These reactions produce ATP, the high-energy electron carriers NADH and FADH2, and CO2. NADH and FADH2 then feed electrons into the electron transport chain to drive the production of most of the cell's ATP.
- Experimental Foundation: Krebs's work involved studying the oxygen consumption of pigeon breast muscle suspensions. By adding various suspected metabolic intermediates and measuring their effect on the rate of oxygen consumption, he was able to deduce the cyclic nature of the pathway. Lipmann's discovery of Coenzyme A was the result of persistent biochemical fractionation and experimentation, isolating the heat-stable cofactor necessary for the acetylation of sulfonamides.
- Metabolic Engineering Context: The Krebs cycle is a primary source of precursor metabolites for biosynthesis. To engineer a cell for high-yield production of a target compound, one must often divert carbon flux away from the Krebs cycle. This requires a deep understanding of its regulation and the availability of key intermediates like α-ketoglutarate (for glutamate synthesis) and oxaloacetate (for amino acid and lysine synthesis).
Autophagy: Cellular Recycling and Quality Control
Yoshinori Ohsumi's 2016 Nobel Prize-winning work defined the molecular mechanisms of autophagy, a fundamental process for degrading and recycling cellular components [10].
- Molecular Mechanism: Autophagy (meaning "self-eating") involves the sequestration of cytoplasmic cargo (damaged organelles, protein aggregates) within a double-membraned vesicle called an autophagosome. The autophagosome then fuses with the lysosome (or vacuole in yeast) to form an autolysosome, where the encapsulated contents are degraded by hydrolases and the resulting macromolecules are recycled back into the cytoplasm.
- Experimental Foundation: Ohsumi's groundbreaking experiment used baker's yeast (Saccharomyces cerevisiae) as a model. He engineered mutant yeast strains lacking vacuolar degradation enzymes and then starved the cells to induce autophagy. Under the microscope, he observed the accumulation of non-degraded autophagosomes within the vacuole, proving the existence of autophagy in yeast. This visual assay provided a powerful tool for his subsequent screening of yeast mutants, which led to the identification of the first essential autophagy genes (ATG genes) [10].
- Metabolic Engineering Context: In bioproduction, autophagy can be a double-edged sword. It can be engineered to recycle nutrients during stress, sustaining cell viability and productivity in long-term fermentations. Conversely, it can also degrade engineered enzymes or pathway intermediates. Understanding and controlling autophagic flux is therefore crucial for optimizing microbial cell factory performance.
Oxygen Sensing: The HIF Pathway and Metabolic Adaptation
The 2019 Nobel Prize to William G. Kaelin Jr., Sir Peter J. Ratcliffe, and Gregg L. Semenza was for their discovery of how cells sense and adapt to oxygen availability [11].
- Molecular Mechanism: The central player is the transcription factor HIF (Hypoxia-Inducible Factor). Under normal oxygen levels (normoxia), HIF-α subunits are continuously synthesized but rapidly degraded. This degradation is triggered by oxygen-dependent prolyl hydroxylases (PHDs), which mark HIF-α for recognition by the VHL E3 ubiquitin ligase complex, leading to its proteasomal degradation. Under low oxygen (hypoxia), PHD activity is inhibited, allowing HIF-α to stabilize, enter the nucleus, dimerize with HIF-β, and activate the transcription of hundreds of genes involved in angiogenesis, erythropoiesis, and metabolic adaptation [11].
- Metabolic Adaptation: A key metabolic shift induced by HIF is the switch from oxidative phosphorylation to glycolysis. HIF activates genes for glucose transporters and glycolytic enzymes while suppressing mitochondrial function. This allows cells to generate ATP without consuming oxygen, a critical adaptation in solid tumors and ischemic tissues.
- Experimental Foundation: Key experiments included Semenza's identification of HIF binding to the EPO gene enhancer, Ratcliffe's demonstration of the ubiquitous nature of oxygen sensing, and Kaelin's discovery that the VHL tumor suppressor protein was necessary for HIF degradation. The final piece was the simultaneous discovery by both Ratcliffe and Kaelin that oxygen-dependent prolyl hydroxylation is the signal for VHL binding [11].
- Metabolic Engineering Context: Oxygen availability is a major scale-up challenge in industrial bioreactors. Understanding the HIF pathway provides insights into how production cells (including mammalian cell cultures) respond to hypoxic pockets in large fermenters. Engineering strategies may involve modulating HIF pathway components to enhance cell viability and product yield under sub-optimal oxygen conditions.
Diagram: The HIF Oxygen-Sensing Pathway
Directed Evolution of Enzymes: Engineering Novel Catalysts
Frances H. Arnold was awarded the 2018 Nobel Prize in Chemistry for pioneering the directed evolution of enzymes, a method that allows engineers to create optimized biocatalysts for specific industrial processes [12] [13].
- Core Principle: Directed evolution mimics natural selection in a laboratory setting. It involves iterative rounds of (1) introducing genetic diversity into a protein-coding gene and (2) screening or selecting the resulting variant proteins for the desired enhanced or novel function.
- Experimental Workflow:
- Gene Diversification: The gene of interest is mutated using methods like error-prone PCR to create a large library of variants.
- Expression: The gene library is expressed in a host system (e.g., bacteria).
- Screening/Selection: The population is subjected to a high-throughput screen or selection that identifies variants with improved performance (e.g., activity in organic solvents, higher thermostability, or novel catalytic function).
- Gene Amplification: The genes from the best-performing variants are isolated and used as the template for the next round of evolution.
- Metabolic Engineering Context: Directed evolution is a powerful tool for creating custom enzymes that are not found in nature. This allows metabolic engineers to design new biosynthetic pathways or optimize existing ones. Examples cited for Arnold's work include evolving enzymes for the environmentally friendly production of pharmaceuticals and the biosynthesis of renewable biofuels like isobutanol [13].
Diagram: The Directed Evolution Workflow
The Scientist's Toolkit: Essential Reagents and Protocols
Key Research Reagent Solutions
Modern metabolic research relies on a suite of reagents and tools to probe pathway function.
Table 2: Essential Reagents for Metabolic Pathway Analysis
| Reagent / Tool | Function / Application | Example in Context |
|---|---|---|
| 2-Deoxy-D-Glucose | Glycolysis Inhibitor | Competitively inhibits hexokinase, allowing measurement of glycolytic dependency in ATP production assays [14]. |
| Oligomycin A | ATP Synthase Inhibitor | Inhibits mitochondrial oxidative phosphorylation, allowing quantification of its contribution to total ATP production [14]. |
| Metformin | AMPK Activator / Complex I Inhibitor | Induces metabolic stress and is used to study cellular adaptation to energy deprivation, relevant in cancer and diabetes research [14]. |
| Luminescent ATP Assay | Quantify Cellular ATP Levels | Provides a high-throughput, direct readout of cellular energy status after metabolic perturbation [14]. |
| Metabolic Pathway Databases (KEGG, MetaCyc) | Reference for Pathway Reconstruction | Provide curated maps of metabolic reactions and pathways across different organisms, essential for pathway comparison and design [15]. |
| 16-Deoxysaikogenin F | 16-Deoxysaikogenin F, MF:C30H48O3, MW:456.7 g/mol | Chemical Reagent |
| Alpinumisoflavone acetate | Alpinumisoflavone acetate, MF:C22H18O6, MW:378.4 g/mol | Chemical Reagent |
A Representative Experimental Protocol: Metabolic Dependency Analysis
A 2024 protocol provides a modern method for analyzing the relative contribution of different metabolic pathways to ATP production, a key question in metabolic engineering and cancer biology [14]. This method is high-throughput and directly measures ATP, the functional energy output.
Protocol: Analyzing Energy Metabolic Pathway Dependency [14]
- Cell Seeding: Harvest and count cells (e.g., HepG2 liver carcinoma cell line). Seed cells at a uniform density in a 96-well plate.
- Perturbation/Treatment: Treat cells with the compound of interest (e.g., Metformin) to perturb metabolism. Include appropriate control groups.
- Systematic Metabolic Inhibition: In a separate plate, treat cells with a panel of specific metabolic inhibitors to block specific pathways:
- Glycolysis Inhibition: Use 2-Deoxy-D-Glucose.
- Oxidative Phosphorylation Inhibition: Use Oligomycin A.
- Other inhibitors can target fatty acid oxidation or amino acid metabolism.
- Viability and ATP Assay: After an incubation period, assay each well for both cell viability (e.g., using an XTT-based assay) and ATP content (using a luminescent ATP detection assay kit).
- Data Calculation and Analysis:
- Normalize the luminescent ATP readings to the cell viability data to obtain a viability-corrected ATP level.
- Calculate the dependency on a specific pathway using the following type of calculation:
- Glycolytic Capacity = (ATP level from untreated cells - ATP level after Oligomycin treatment) / (ATP level from untreated cells)
- The results provide a quantitative profile of how a cell relies on different pathways for its energy needs.
The journey from foundational Nobel Prize discoveries to modern metabolic engineering is a powerful demonstration of how basic biological research enables technological innovation. The discovery of core pathways like the Krebs cycle and autophagy, the elucidation of sensory systems like the HIF oxygen-sensing pathway, and the development of powerful engineering tools like directed evolution provide a comprehensive toolkit for today's researchers. By integrating these historical foundations with contemporary high-throughput protocols and computational analyses, scientists can continue to push the boundaries of what is possible in the production of renewable chemicals, advanced biofuels, and next-generation therapeutics.
Central carbon metabolism (CCM) constitutes the fundamental biochemical network responsible for the conversion of carbon-containing molecules into energy, reducing power, and precursor metabolites essential for cell growth, proliferation, and survival [16]. This network acts as the core "processing hub" within the cell, tightly linking numerous catabolic and anabolic processes [17]. For researchers in metabolic engineering and drug development, a rigorous understanding of CCM is indispensable. It provides the foundational knowledge required to rationally redesign microbial hosts for the sustainable production of valuable chemicals, biofuels, and active pharmaceutical ingredients (APIs) [2]. The core of CCM primarily comprises three interconnected pathways: Glycolysis (the Embden-Meyerhof-Parnas pathway, or EMP pathway), the Tricarboxylic Acid (TCA) Cycle, and the Pentose Phosphate Pathway (PPP) [17] [16]. These pathways collectively transform simple sugars into a diverse set of metabolic intermediates that serve as building blocks for biosynthesis.
Table 1: Core Components of Central Carbon Metabolism
| Pathway Name | Primary Function | Key Inputs | Key Outputs | Cellular Location |
|---|---|---|---|---|
| Glycolysis (EMP) | Glucose breakdown to pyruvate, net ATP/NADH production | Glucose, ATP, NAD+ | Pyruvate, ATP, NADH | Cytoplasm |
| TCA Cycle | Complete oxidation of acetyl-CoA, high-yield NADH/FADH2 generation | Acetyl-CoA, NAD+, FAD, GDP/ADP | ATP/GTP, NADH, FADH2, CO2 | Mitochondrial Matrix |
| Pentose Phosphate Pathway (PPP) | Generation of NADPH and pentose sugars | Glucose-6-phosphate, NADP+ | Ribose-5-phosphate, NADPH, CO2 | Cytoplasm |
The Embden-Meyerhof-Parnas (EMP) Pathway: Glycolysis
Glycolysis is a universal metabolic pathway involving the ten-step conversion of a single glucose molecule into two pyruvate molecules within the cytoplasm [17]. This process is divided into two distinct phases: an energy-investment phase and an energy-payoff phase.
Experimental Analysis of Glycolytic Flux
A key methodology for quantifying flux through glycolysis is Metabolic Flux Analysis (MFA). This technique relies on feeding cells substrates labeled with stable isotopes (e.g., ^13C-glucose) and tracking the incorporation of these labels into downstream metabolites using mass spectrometry [16] [18]. The resulting isotopic distribution data allows for the quantitative determination of intracellular metabolic reaction rates. For dynamic profiling, Fluxomics approaches combine this isotopic labeling with mathematical models, such as flux balance analysis (FBA), to estimate the flow of metabolites through the network under different genetic or environmental perturbations [16].
Table 2: Glycolysis (EMP Pathway) Reaction Sequence and ATP Balance
| Step | Reactants | Products | Enzyme | ATP/NADH Balance |
|---|---|---|---|---|
| 1 | Glucose | Glucose-6-phosphate | Hexokinase | -1 ATP |
| 2 | Glucose-6-phosphate | Fructose-6-phosphate | Phosphohexose isomerase | - |
| 3 | Fructose-6-phosphate | Fructose-1,6-bisphosphate | Phosphofructokinase-1 | -1 ATP |
| 4 | Fructose-1,6-bisphosphate | Glyceraldehyde-3-phosphate (G3P) & Dihydroxyacetone phosphate | Aldolase | - |
| 5 | Dihydroxyacetone phosphate | Glyceraldehyde-3-phosphate (G3P) | Triose phosphate isomerase | - |
| 6 | Glyceraldehyde-3-phosphate + NAD+ | 1,3-Bisphosphoglycerate + NADH | Glyceraldehyde-3-phosphate dehydrogenase | +2 NADH* |
| 7 | 1,3-Bisphosphoglycerate + ADP | 3-Phosphoglycerate + ATP | Phosphoglycerate kinase | +2 ATP |
| 8 | 3-Phosphoglycerate | 2-Phosphoglycerate | Phosphoglycerate mutase | - |
| 9 | 2-Phosphoglycerate | Phosphoenolpyruvate | Enolase | - |
| 10 | Phosphoenolpyruvate + ADP | Pyruvate + ATP | Pyruvate kinase | +2 ATP |
| Net Yield per Glucose | 2 ATP, 2 NADH, 2 Pyruvate |
Note: Values account for the doubling of all molecules from one glucose to two G3P molecules.
The Tricarboxylic Acid (TCA) Cycle
The TCA cycle, also known as the Krebs or citric acid cycle, is the central aerobic hub for oxidizing acetyl-CoA derived from carbohydrates, fats, and proteins [19]. Located in the mitochondrial matrix, it completes the energy-yielding oxidation of carbon fuels and provides key precursors for biosynthesis [17].
Protocol: Investigating Cycle Activity with Isotope Tracing
The operation of the TCA cycle can be studied using ^13C-glutamine or ^13C-glucose tracing followed by analysis via Liquid Chromatography-Mass Spectrometry (LC-MS). Cells are cultured in media containing the labeled substrate. Metabolites are then extracted and analyzed to determine the ^13C enrichment pattern in TCA cycle intermediates (e.g., citrate, α-ketoglutarate, succinate). The mass isotopomer distributions reveal the relative flux through various segments of the cycle and ancillary pathways, such as reductive carboxylation, which is often upregulated in cancer cells [16].
Table 3: TCA Cycle Reactions and Energy Carriers Generated
| Step | Reaction | Enzyme | Energy Carriers Produced | Type of Reaction |
|---|---|---|---|---|
| 0 | Oxaloacetate + Acetyl-CoA → Citrate | Citrate synthase | - | Aldol condensation |
| 1 | Citrate cis-Aconitate Isocitrate | Aconitase | - | Dehydration/Hydration |
| 2 | Isocitrate + NAD+ → α-Ketoglutarate + CO2 | Isocitrate dehydrogenase | 1 NADH | Oxidative decarboxylation |
| 3 | α-Ketoglutarate + NAD+ + CoA → Succinyl-CoA + CO2 | α-Ketoglutarate dehydrogenase | 1 NADH | Oxidative decarboxylation |
| 4 | Succinyl-CoA + GDP/Pi → Succinate + GTP/ATP | Succinyl-CoA synthetase | 1 GTP/ATP | Substrate-level phosphorylation |
| 5 | Succinate + Ubiquinone (Q) → Fumarate | Succinate dehydrogenase | 1 FADH2 (as QH2) | Oxidation |
| 6 | Fumarate + H2O → L-Malate | Fumarase | - | Hydration |
| 7 | L-Malate + NAD+ → Oxaloacetate + NADH | Malate dehydrogenase | 1 NADH | Oxidation |
| Total per Acetyl-CoA | 3 NADH, 1 FADH2, 1 GTP/ATP |
The Pentose Phosphate Pathway (PPP)
The PPP operates in the cytoplasm parallel to glycolysis and serves two critical biosynthetic roles: generating ribose-5-phosphate for nucleotide synthesis and producing NADPH for reductive biosynthesis and oxidative stress defense [17] [16]. The pathway consists of an oxidative and a non-oxidative phase.
Methodology: Quantifying NADPH Production via the PPP
The flux through the oxidative branch of the PPP can be specifically measured by monitoring the release of ^14CO2 from glucose labeled at the C1 position (1-^14C-glucose). As the first step of the PPP is the decarboxylation of glucose-6-phosphate, the amount of CO2 released from C1 is directly proportional to PPP activity. This can be compared to CO2 release from other labeled positions (e.g., 6-^14C-glucose) to differentiate PPP flux from glycolytic flux [16].
Table 4: Pentose Phosphate Pathway Phases and Outputs
| Phase | Key Reactions | Key Enzymes | Primary Outputs |
|---|---|---|---|
| Oxidative | Glucose-6-phosphate → 6-Phosphoglucono-δ-lactone | Glucose-6-phosphate dehydrogenase | 2 NADPH (per G6P entering oxidative phase) |
| 6-Phosphoglucono-δ-lactone → 6-Phosphogluconate | Lactonase | ||
| 6-Phosphogluconate → Ribulose-5-phosphate + CO2 | 6-Phosphogluconate dehydrogenase | 1 Ribulose-5-phosphate | |
| Non-Oxidative | Ribulose-5-phosphate Ribose-5-phosphate Xylulose-5-phosphate | Pentose phosphate isomerase & epimerase | Various sugar phosphates (C3, C4, C5, C6, C7) |
| Transketolase & Transaldolase Reactions | Transketolase, Transaldolase | Fructose-6-phosphate, Glyceraldehyde-3-phosphate |
Regulation of Central Metabolic Pathways
The flux through central carbon metabolism is precisely controlled via multiple regulatory mechanisms to maintain metabolic homeostasis and respond to cellular energy demands and nutrient availability [17] [16].
Allosteric Regulation: Key enzymes are modulated by effectors that signal the cell's energy status. For instance, phosphofructokinase-1 (PFK-1), the rate-limiting enzyme of glycolysis, is allosterically inhibited by high levels of ATP and activated by AMP, signaling low energy [17] [16]. Similarly, citrate synthase, the first enzyme of the TCA cycle, is inhibited by ATP and succinyl-CoA [17].
Covalent Modification: Enzyme activity is rapidly and reversibly modified through processes like phosphorylation. Glycogen synthase is inhibited by phosphorylation, redirecting carbon flow away from storage and into glycolysis when energy is needed [17].
Feedback Inhibition: The end-products of pathways inhibit earlier steps. Accumulation of ATP feeds back to inhibit PFK-1, preventing excessive glycolysis when energy is abundant [17] [16].
Substrate Availability: High glucose concentrations activate hexokinase, driving glucose into the metabolic network [17].
Metabolic Engineering Applications and Experimental Workflow
Metabolic engineering applies a Design-Build-Test-Learn (DBTL) cycle to construct efficient microbial cell factories for producing molecules ranging from biofuels to pharmaceuticals [1] [2]. Central carbon metabolism is a primary target for these engineering efforts.
A Metabolic Engineering DBTL Workflow
A standard DBTL cycle for engineering CCM involves [1]:
- Design: Using genomic and modeling tools (e.g., Flux Balance Analysis on a Genome-Scale Model, or GSM) to identify gene knockout, knockdown, or overexpression targets to redirect flux toward a desired product.
- Build: Employing molecular biology techniques (e.g., CRISPR-Cas9, promoter engineering, gene assembly) to implement the genetic modifications in a host organism (e.g., E. coli, S. cerevisiae, or the oleaginous yeast Yarrowia lipolytica).
- Test: Culturing the engineered strain and applying analytical techniques like LC-MS, NMR, and stable isotope tracing (e.g., with ^13C-glucose) to measure metabolite levels, product titer, yield, productivity (TYR metrics), and actual metabolic fluxes (MFA).
- Learn: Analyzing the T&L data to identify unforeseen bottlenecks (e.g., regulatory issues, toxic intermediate accumulation, cofactor imbalances) and inform the next round of design.
Success Stories in Metabolic Engineering
- Artemisinic Acid: The precursor to the antimalarial drug artemisinin is now produced in engineered S. cerevisiae by integrating plant-derived genes into the host's native isoprenoid pathway, which draws on acetyl-CoA from central metabolism [2].
- 1,3-Propanediol and 1,4-Butanediol: These bulk chemicals are commercially produced in engineered E. coli by introducing heterologous pathways that divert central metabolic intermediates like dihydroxyacetone phosphate (glycolysis) and succinyl-CoA (TCA cycle) [1] [2].
- Advanced Biofuels: Hydrocarbons with properties similar to diesel and jet fuel have been produced by engineering the fatty acid (from acetyl-CoA) and isoprenoid biosynthetic pathways in various microbial hosts [2].
Table 5: Essential Research Reagents and Tools for Metabolic Pathway Analysis
| Tool/Reagent | Function/Application | Example Use Case |
|---|---|---|
| ^13C-labeled Substrates (e.g., ^13C-Glucose, ^13C-Glutamine) | Stable isotope tracers for Metabolic Flux Analysis (MFA) | Quantifying carbon flow and pathway fluxes in live cells [16]. |
| LC-MS / GC-MS Systems | Analytical instruments for identifying and quantifying metabolites | Measuring concentrations and ^13C isotopic enrichment in metabolic intermediates [16] [20]. |
| Genome-Scale Metabolic Models (GEMs) | Computational models of an organism's entire metabolism | Predicting growth phenotypes, gene essentiality, and optimal genetic modifications in silico [21] [18]. |
| CRISPR-Cas9 Tools | For precise genome editing (knockouts, knock-ins, point mutations) | Engineering microbial hosts to delete competing pathways or insert heterologous genes [1]. |
| KEGG / MetaCyc Databases | Curated databases of metabolic pathways and enzymes | Retrieving reference pathways and enzyme information for pathway design [22]. |
| Metabolic Network Tools (e.g., MetaDAG, GEMsembler) | Software for reconstructing, visualizing, and comparing metabolic networks | Analyzing and comparing metabolic networks across different organisms or conditions [22] [21]. |
This guide provides an in-depth examination of the primary molecular carriers of metabolic energy and reducing power: adenosine triphosphate (ATP) and nicotinamide adenine dinucleotide (NADH). Framed within the context of metabolic engineering, this resource is designed for researchers, scientists, and drug development professionals seeking a foundational understanding of these crucial cofactors. Cellular metabolism encompasses thousands of reactions necessary for growth and proliferation, requiring both Gibbs free energy and molecular building blocks [23]. ATP and NADH sit at the core of this network, serving as universal currencies for energy transfer and redox reactions. Their fundamental principles are foundational for efforts in metabolic engineering, which modifies and optimizes biochemical pathways in microorganisms to produce valuable compounds [24]. A thorough grasp of their function is essential for designing new biochemical pathways or redesigning existing ones for applications in biofuel, pharmaceutical, and chemical production.
Adenosine Triphosphate (ATP): The Primary Energy Currency
Structure and Chemical Properties
Adenosine triphosphate (ATP) is a nucleoside triphosphate consisting of three primary components: a nitrogenous base (adenine), the sugar ribose, and a triphosphate group [25]. The triphosphate unit comprises three phosphoryl groups labeled alpha (α), beta (β), and gamma (γ) [25]. In neutral aqueous solutions at physiological pH, ATP exists primarily as the ion ATPâ´â» [25]. A key property is its ability to bind divalent metal cations, particularly magnesium (Mg²âº), with high affinity. The resulting ATP-Mg²⺠complex is the predominant cellular form and is crucial for most enzymatic interactions involving ATP [25].
Energetics of ATP Hydrolysis
The hydrolysis of ATP is a highly exergonic reaction that provides the driving force for countless cellular processes.
- Hydrolysis to ADP: ATP + H₂O → ADP + Pi with ΔG°' = -30.5 kJ/mol (-7.3 kcal/mol) [25]
- Hydrolysis to AMP: ATP + H₂O → AMP + PPi with ΔG°' = -45.6 kJ/mol (-10.9 kcal/mol) [25]
Under actual intracellular conditions, where the ATP/ADP ratio is maintained far from equilibrium, the free energy change (ΔG) for ATP hydrolysis is even more favorable, reaching approximately -57 kJ/mol (-12 kcal/mol) [25] [26]. This significant release of free energy qualifies the phosphoanhydride bonds in ATP as "high-energy bonds," not because of special chemical properties, but because their hydrolysis releases a large amount of usable energy under cellular conditions [26].
ATP-Dependent Coupling of Metabolic Reactions
ATP functions as a universal energy currency by coupling its exergonic hydrolysis to endergonic biochemical reactions, making them thermodynamically favorable. A classic example is the first reaction in glycolysis, the conversion of glucose to glucose-6-phosphate [26]. This reaction is energetically unfavorable on its own (ΔG°′= +3.3 kcal/mol) but becomes favorable when coupled to ATP hydrolysis (ΔG°′= -7.3 kcal/mol), yielding a net ΔG°′ of -4.0 kcal/mol for the coupled reaction [26]. This coupling mechanism is universal across living cells, allowing ATP to drive essential processes including muscle contraction, nerve impulse propagation, active transport, and biosynthesis of macromolecules [25] [26].
Table 1: Standard Free Energy of ATP Hydrolysis Under Different Conditions
| Reaction | Conditions | ΔG°' (kJ/mol) | ΔG°' (kcal/mol) |
|---|---|---|---|
| ATP → ADP + Pi | Standard Biochemical Conditions | -30.5 | -7.3 |
| ATP → AMP + PPi | Standard Biochemical Conditions | -45.6 | -10.9 |
| ATP → ADP + Pi | Typical Cellular Conditions | ~ -57 | ~ -12 |
NADH: The Key Electron Carrier in Redox Reactions
Structure and Redox Function
Nicotinamide adenine dinucleotide (NADH) exists in two interconvertible forms: the oxidized form (NADâº) and the reduced form (NADH). Its primary role is to serve as a reversible carrier of reducing equivalents (electrons and protons) in metabolic pathways. The nicotinamide ring is the active site where a hydride ion (Hâ») is transferred during redox reactions. This reversible reduction of NAD⺠to NADH is central to metabolic energy extraction.
Generation and Utilization in Catabolic Pathways
NADH is predominantly generated during the oxidative phases of catabolism. Key generating reactions include:
- Glycolysis: Glyceraldehyde-3-phosphate is oxidized to 1,3-bisphosphoglycerate, coupled to the reduction of NAD⺠to NADH [26].
- Citric Acid Cycle: Multiple steps within the mitochondrial matrix involve oxidation of substrates like isocitrate, α-ketoglutarate, and malate, producing NADH [27].
The reducing power of NADH is primarily utilized in the mitochondrial electron transport chain. NADH donates its electrons to Complex I (NADH dehydrogenase), initiating the process of oxidative phosphorylation [27]. This electron transfer is crucial for establishing the proton gradient that drives ATP synthesis.
Energy Equivalency of NADH
The oxidation of NADH is a highly exergonic reaction. The transfer of a pair of electrons from NADH to oxygen has a standard free energy change (ΔG°′) of -52.5 kcal/mol [27]. This substantial release of energy is harnessed gradually as electrons pass through the electron transport chain. This process ultimately leads to the synthesis of approximately 2.5 to 3 molecules of ATP per NADH molecule oxidized, depending on the organism and specific cellular conditions [27]. This quantitative relationship is fundamental for calculating metabolic yields.
Integrated Metabolic Pathways and Energy Yield
The complete oxidation of glucose demonstrates the integrated roles of ATP and NADH in energy metabolism. The process involves three major stages: glycolysis, the citric acid cycle, and oxidative phosphorylation.
Glycolysis: Substrate-Level Phosphorylation
In the cytosol, glycolysis breaks down one glucose molecule into two pyruvate molecules, yielding a net gain of 2 ATP molecules and 2 NADH molecules [26]. This ATP is produced via substrate-level phosphorylation, where a high-energy phosphate is directly transferred to ADP from a metabolic intermediate like 1,3-bisphosphoglycerate or phosphoenolpyruvate [26] [27].
Oxidative Phosphorylation: Chemiosmotic Coupling
The majority of ATP from glucose oxidation is generated through oxidative phosphorylation in the mitochondria. This process involves two coupled phases:
- Electron Transport Chain: Electrons from NADH (and FADHâ‚‚) are transferred through a series of protein complexes (I-IV) in the inner mitochondrial membrane. This exergonic electron flow is coupled to the active pumping of protons from the matrix to the intermembrane space, creating an electrochemical gradient [27].
- ATP Synthesis: The potential energy stored in the proton gradient is harvested by ATP synthase (Complex V). The energetically favorable flow of protons back into the matrix through ATP synthase drives the mechanical rotation of its subunits, catalyzing the synthesis of ATP from ADP and Pi—a process known as chemiosmotic coupling [27].
The complete oxidation of one glucose molecule via glycolysis, the citric acid cycle, and oxidative phosphorylation yields approximately 30-32 molecules of ATP [27]. This high yield underscores the efficiency of cellular respiration in harnessing the energy stored in nutrient molecules through the coordinated action of ATP, NADH, and other cofactors.
Diagram 1: Central Energy Metabolism Pathway
Quantitative Analysis of Energy Equivalents
Understanding the quantitative yield of ATP from different substrates and cofactors is critical for metabolic flux analysis and pathway engineering. The following table summarizes key energy equivalents in central metabolism.
Table 2: Energy Equivalents and ATP Yield in Glucose Metabolism
| Molecule / Pathway | ATP Yield (Molecules per Glucose) | Primary Metabolic Process |
|---|---|---|
| Glycolysis (Net) | 2 | Substrate-Level Phosphorylation |
| NADH from Glycolysis | 3-5* | Oxidative Phosphorylation |
| NADH from Citric Acid Cycle | 15 | Oxidative Phosphorylation |
| FADHâ‚‚ from Citric Acid Cycle | 3 | Oxidative Phosphorylation |
| Citric Acid Cycle (GTP) | 2 | Substrate-Level Phosphorylation |
| Total from Complete Oxidation | 30-32 | Combined Processes |
Note: The yield depends on the shuttle system used to transfer electrons from cytosolic NADH into the mitochondria.
Table 3: Standard Free Energy Changes of Key Metabolic Reactions
| Reaction | ΔG°' (kJ/mol) | ΔG°' (kcal/mol) | Location/Pathway |
|---|---|---|---|
| Glucose + 6 O₂ → 6 CO₂ + 6 H₂O | -2870 | -686 | Overall Cellular Respiration |
| ATP → ADP + Pi | -30.5 | -7.3 | Universal |
| PEP → Pyruvate | -61.9 | -14.8 | Glycolysis |
| 1,3-BPG → 3-PG | -49.3 | -11.8 | Glycolysis |
| NADH + ½ O₂ → NAD⺠+ H₂O | -219.7 | -52.5 | Electron Transport Chain |
Metabolic Engineering Applications and Protocols
Foundational Principles in Metabolic Engineering
Metabolic engineering is "the modification and optimization of metabolic pathways, mainly in microorganisms, by altering genes, nutrient uptake, or metabolic flow to allow the production of novel compounds" [24]. The field relies on a systematic approach, often involving an analytical phase (pathway analysis) followed by a synthesis phase (pathway modification) [24]. A core principle is the redirection of metabolic flux toward a desired product, which requires a deep understanding of the energy and redox balances governed by ATP and NADH.
Essential Research Reagents and Host Organisms
Successful metabolic engineering relies on a standardized toolkit of reagents and host organisms.
Table 4: Key Research Reagent Solutions in Metabolic Engineering
| Reagent / Organism | Function / Characteristic | Application Example |
|---|---|---|
| Escherichia coli | Model bacterium; rapid growth; well-established genetic tools [24] | Production of interferon, insulin, growth hormone [24] |
| Saccharomyces cerevisiae | Baker's yeast; non-pathogenic; established fermentation technology [24] | Biosynthesis of isoprenoids; lactic acid production [24] |
| CRISPR-Cas Systems | Precision genome editing for gene knockout, knockdown, or insertion [24] | Deleting competing pathways or inserting heterologous genes |
| Plasmid Vectors | Carriers for introducing foreign genetic material into a host organism [24] | Expressing enzymes from a non-host organism to extend a pathway |
| Analytical Tools (e.g., LC-MS, GC-MS) | Tracking metabolites and quantifying pathway fluxes [24] [28] | Identifying metabolic bottlenecks during strain development |
Protocol: Computational Modeling of Metabolic Pathways
Computational models are indispensable for predicting the behavior of engineered metabolic systems before laboratory implementation.
Objective: To create a quantitative model of a target biochemical pathway for in silico analysis and optimization. Methodology:
- System Definition: Define the set of reactants (species and enzymes) and the stoichiometry of all reactions in the pathway [28].
- Kinetic Law Selection: Assign appropriate kinetic laws (e.g., Mass-Action, Michaelis-Menten) to each reaction. Mass-action kinetics are often suitable for initial modeling, where the reaction rate is proportional to reactant concentrations [28].
- Model Representation: Represent the network, for instance, using a Petri net formalism. Basic component patterns can be defined, such as a binding reaction (P1 + P2 → P3) and an unbinding reaction (P3 → P1* + P2*) [28].
- Parameterization: Populate the model with kinetic rate constants, which can be obtained from literature, databases, or experimental fitting [29].
- Simulation and Analysis: Use a system of ordinary differential equations (ODEs) to simulate the system's dynamics over time. Analyze flux distributions and identify control points [28] [23].
- Uncertainty Quantification and Experimental Design: Apply Bayesian optimal experimental design (BOED) to identify which experimental measurements would most effectively reduce uncertainty in model predictions, thereby guiding efficient laboratory work [29].
Diagram 2: Metabolic Engineering Workflow
Protocol: Quantitative Analysis of ATP and NADH in Cultured Cells
Measuring intracellular ATP and NADH/NAD⺠ratios is crucial for assessing the metabolic state of engineered strains.
Objective: To quantify the energy and redox states of microbial production hosts under different fermentation conditions. Materials:
- Cell Culture: Engineered E. coli or S. cerevisiae strain.
- ATP Assay Kit: Luciferin-luciferase based bioluminescence assay.
- NAD/NADH Assay Kit: Enzymatic cycling assay.
- Quenching Solution: Cold methanol or perchloric acid for rapid metabolite arrest.
- Cell Disruption System: Bead beater or sonicator.
- Luminometer or Plate Reader: For detecting assay signals.
Procedure:
- Culture and Sampling: Grow the engineered host in a controlled bioreactor. At defined time points (e.g., during exponential growth and production phase), rapidly extract a known volume of culture.
- Metabolite Quenching: Immediately quench the sample in cold quenching solution (-40°C methanol) to instantaneously halt all metabolic activity and preserve in vivo metabolite levels.
- Metabolite Extraction: Pellet the quenched cells and resuspend in an appropriate extraction buffer to release intracellular ATP and NAD(H). Use separate extraction protocols for NAD⺠and NADH to preserve the in vivo ratio (e.g., acidic extraction for NADâº, basic for NADH).
- Assay Performance: Follow the specific protocols of the commercial assay kits.
- For ATP: Mix the sample with luciferase reagent and measure the resulting bioluminescence, which is proportional to ATP concentration.
- For NAD/NADH: Use enzymatic reactions that reduce a tetrazolium salt to a colored formazan product, the formation of which is proportional to the concentration of NAD(H).
- Data Analysis: Calculate concentrations by comparing sample readings to standard curves. Normalize values to cell density (OD₆₀₀) or protein content. The NADH/NAD⺠ratio is a key indicator of the cellular redox state.
ATP and NADH are fundamental cofactors that power and regulate cellular metabolism. ATP serves as the universal energy currency, with its hydrolysis driving endergonic processes, while NADH acts as a central carrier of reducing power, feeding electrons into the energy-yielding pathway of oxidative phosphorylation. Their quantitative yields and interactions form the basis for calculating metabolic efficiency. In metabolic engineering, manipulating the pathways that generate and consume these cofactors is a primary strategy for optimizing the production of biofuels, pharmaceuticals, and renewable chemicals. A deep, quantitative understanding of ATP and NADH is therefore not merely an academic exercise but a prerequisite for the rational design of efficient microbial cell factories.
The selection of an appropriate host organism is a foundational decision in metabolic engineering and biopharmaceutical development. These microbial cell factories are engineered to produce complex molecules, from life-saving therapeutic proteins to high-value industrial compounds. The field primarily relies on two well-established microbial workhorses: the prokaryotic bacterium Escherichia coli and various eukaryotic yeast species. Each platform offers a distinct set of advantages and limitations based on its unique cellular machinery, post-translational capabilities, and cultivation requirements. Understanding the core characteristics of these hosts is essential for designing efficient metabolic pathways, optimizing bioprocesses, and successfully bringing new products from the laboratory to the market. This guide provides an in-depth technical overview of E. coli and yeast platforms, compares their capabilities through structured data, outlines key experimental methodologies for their engineering, and explores emerging trends shaping the future of microbial biotechnology.
Comparative Analysis of Major Production Platforms
Escherichia coli Platforms
Escherichia coli is one of the most widely used and well-understood prokaryotic hosts for recombinant protein production and metabolic engineering. Its rapid growth, high achievable cell densities, and extensive genetic toolbox make it a default choice for many applications [30]. The genetics of E. coli are the most comprehensively understood in the microbial world, facilitating straightforward genetic manipulation [30]. A key advantage of this platform is the rapid strain development cycle; a microbial strain for heterologous protein production can be developed in as little as four weeks, and short fermentation batch cycles (around one week) make it highly attractive for fast-paced development and production [30].
However, E. coli, being a prokaryote, lacks the cellular machinery for performing eukaryotic post-translational modifications, such as glycosylation, which is essential for the activity and stability of many therapeutic proteins [30]. It is also prone to forming inclusion bodies—aggregates of misfolded protein—which can complicate downstream processing, although this can sometimes be an advantage for initial product concentration and isolation. Recent research continues to expand E. coli's capabilities, as demonstrated by the engineering of the E. coli W strain for enhanced flavonoid glycosylation. This strain shows superior tolerance to toxic substrates and, when optimized through Adaptive Laboratory Evolution (ALE) and metabolic engineering, can efficiently utilize sucrose to produce high-value compounds like chrysin-7-O-glucoside at bench-scale titers reaching 1844 mg/L [31].
Yeast Platforms
Yeasts, as eukaryotic organisms, bridge the gap between simple bacterial systems and complex mammalian cell cultures. The most common yeast species used in production include Saccharomyces cerevisiae, Komagataella phaffii (formerly Pichia pastoris), and Hansenula polymorpha [32]. Yeasts offer several practical advantages, including ease of genetic engineering, rapid growth, high biomass yield, and the absence of endotoxins [32]. A significant advantage over E. coli is their ability to perform certain post-translational modifications and secrete correctly folded proteins into the culture supernatant, simplifying downstream purification [30].
S. cerevisiae has a long history of safe use in food and pharmaceutical production, with commercialized vaccines for hepatitis B and human papillomavirus (HPV) [32]. K. phaffii has gained prominence due to its strong, inducible promoters like AOX1, which enable very high levels of protein expression—sometimes constituting up to 30% of total cell protein [32]. It can achieve high cell densities and high product titers of secreted proteins (>3 g/L) [30]. Furthermore, the development of "customized" glycosylation pathways in yeasts like P. pastoris is a significant advancement, allowing for the humanization of protein glycosylation patterns, which is critical for many therapeutic biologics [30].
Quantitative Platform Comparison
Table 1: Key Characteristics of E. coli and Yeast Expression Platforms
| Feature | E. coli | Yeast (e.g., K. phaffii) |
|---|---|---|
| Cell Type | Prokaryote | Eukaryote |
| Growth Rate | Very High (doubling time ~20 min) | High (doubling time ~1-2 hrs) |
| Post-Translational Modification | Limited (no glycosylation) | Capable (glycosylation possible) |
| Protein Secretion | Generally limited; often forms inclusion bodies | Efficient secretion possible with appropriate signals |
| Typical Product Titer | Varies; high for some proteins [31] | >3 g/L for secreted proteins [30] |
| Cost & Scalability | Low-cost media, highly scalable | Low-cost media, highly scalable |
| Regulatory Status | Well-established for many products [30] | GRAS status; approved for human vaccines [32] [30] |
| Genetic Tools | Extensive and highly advanced [30] | Advanced, but clonal variation can require more screening [30] |
Table 2: Commercial and Industrial Market Context (2025)
| Platform | Market Context | Key Applications |
|---|---|---|
| E. coli Strains | Market size estimated at USD 2.26 Bn in 2025 [33] | Recombinant protein production, biopharmaceutical development, industrial processes [33] |
| Yeast | Market size estimated at USD 4.19 Bn in 2025 [34] | Baker's yeast (38.7%), therapeutic proteins, vaccines, bioethanol [32] [34] |
Detailed Experimental Methodologies
Strain Engineering and Selection
A critical first step in metabolic engineering is the introduction of heterologous DNA into the host organism. For E. coli, this is typically achieved via plasmid-based expression cassettes (e.g., ColE1, p15A), which allow for rapid gene expression and high copy numbers [30]. In contrast, for yeasts like K. phaffii, expression constructs are usually integrated directly into the host chromosome via homologous recombination. While this creates mitotically stable strains, it can also lead to significant clonal variation in productivity, necessitating the screening of hundreds or even thousands of transformants to identify high-producing clones [30]. This process is greatly enhanced by automated high-throughput screening methods.
Promoter selection is another vital component. E. coli systems often use inducible promoters like the T7 lac promoter or constitutive promoters of varying strengths. In K. phaffii, the methanol-inducible AOX1 promoter is one of the strongest and most widely used, though constitutive promoters such as GAP are also common [32]. The genetic engineering toolkit has been expanded with advanced techniques like CRISPR-Cas9, which allows for precise and efficient genome editing in both E. coli and yeast, accelerating the construction of complex production strains [33].
Metabolic Engineering and Bioprocess Optimization
Overcoming metabolic limitations is key to achieving high yields. A prime example is the engineering of E. coli W for flavonoid glycosylation [31]. The success of this platform relied on several interconnected strategies:
- Adaptive Laboratory Evolution (ALE): The native E. coli W strain was subjected to ALE to enhance its ability to utilize sucrose as a carbon source, improving its growth and robustness [31].
- Metabolic Rerouting: Key genes in central carbon metabolism (xylA, zwf, pgi) were knocked out to redirect carbon flux away from biomass and toward the synthesis of uridine diphosphate glucose (UDPG), the essential precursor for glycosylation [31].
- Pathway Overexpression: Genes encoding enzymes for the glycosylation pathway, including a sucrose phosphorylase (for breaking down sucrose) and a glycosyltransferase (YjiC from Bacillus licheniformis), were overexpressed to drive the conversion of chrysin to chrysin-7-O-glucoside (C7O) [31].
The diagram below illustrates the overall workflow for developing such a platform.
Diagram 1: Strain and Bioprocess Development Workflow. This chart outlines the key stages in developing a high-performance production strain, from initial host selection to final scaled-up production, highlighting the iterative nature of metabolic engineering.
Following strain construction, bioprocess optimization is critical for scaling up production. This involves moving from shake flasks to controlled bioreactors. Key parameters to optimize include pH, dissolved oxygen, temperature, and nutrient feeding strategies. For the engineered E. coli W platform, implementing a fed-batch process in a 3 L bioreactor was essential to achieve the reported high titer of 1844 mg/L C7O, as it allowed for careful control of substrate and toxin levels [31]. Similarly, fed-batch bioprocesses are used in yeast cultivations, such as for a recombinant vaccine against Entamoeba histolytica in K. phaffii, where optimization led to a 12-fold increase in production compared to shake flasks [32].
Essential Research Reagents and Tools
The experimental workflows described rely on a suite of specialized reagents and tools. The following table details key components of a metabolic engineer's toolkit.
Table 3: The Scientist's Toolkit: Key Research Reagents and Materials
| Item | Function | Example Use Case |
|---|---|---|
| Expression Vectors | Plasmids for gene cloning and expression. | Shuttle vectors for E. coli (e.g., ColE1 origin) or integrative plasmids for yeast (e.g., for K. phaffii) [32] [30]. |
| Inducible Promoters | DNA sequences that control gene expression in response to a signal. | AOX1 promoter in K. phaffii (induced by methanol) [32]; T7/lac promoter in E. coli (induced by IPTG). |
| Engineering Tools (CRISPR-Cas9) | Molecular scissors for precise genome editing. | Knocking out genes like pgi or zwf in E. coli to reroute metabolic flux [31] [33]. |
| Specialized Media Components | Nutrients and inducers for selective growth and protein production. | Using sucrose as a carbon source for engineered E. coli W [31] or methanol for induction in K. phaffii [32]. |
| Chromatography Resins | Matrices for purifying target proteins from cell lysates or culture supernatant. | Protein A chromatography for antibody purification; ion exchange and affinity chromatography for general protein purification [32]. |
Emerging Trends and Future Directions
The field of microbial production is continuously evolving, driven by technological advancements and market demands. Several key trends are shaping its future:
- Advanced Genetic Engineering: The application of CRISPR-Cas9 and synthetic biology tools is becoming standard, allowing for more precise and efficient strain modification. This facilitates the creation of E. coli and yeast strains with optimized genomes for higher protein yields and better post-translational modifications [33].
- Platform Specialization and Robustness: There is a growing focus on engineering non-model but inherently robust strains, such as E. coli W, which demonstrates enhanced tolerance to toxic compounds like flavonoids compared to the standard K-12 strain [31]. This highlights a move towards tailoring the host organism to the specific stresses of the production process.
- Glyco-engineering in Yeast: A significant innovation trend is the humanization of yeast glycosylation pathways. Companies are engineering yeast strains like P. pastoris to produce proteins with human-like N-glycans, making yeast a more viable and cost-effective alternative to mammalian cells for complex therapeutic proteins [30].
- AI and Automation Integration: The use of artificial intelligence (AI) and machine learning is rising to manage the complexity of metabolic networks. AI can analyze large datasets to predict optimal gene edits and fermentation conditions. Furthermore, automated high-throughput screening is becoming indispensable for rapidly identifying top-performing yeast clones amidst significant clonal variation [35] [30].
- Sustainability Drivers: The push for environmentally friendly solutions is increasing interest in using microbial hosts for sustainable production of biofuels, bioplastics, and the conversion of waste streams, reinforcing the role of metabolic engineering in the circular bioeconomy [33].
E. coli and yeast platforms form the cornerstone of modern industrial biotechnology. The choice between them hinges on the specific requirements of the target molecule, particularly the need for post-translational modifications, tolerance to process conditions, and overall production economics. E. coli remains the champion for rapid, high-yield production of proteins that do not require eukaryotic processing, while yeast offers a powerful eukaryotic alternative with superior secretion and evolving glycosylation capabilities. For beginners in metabolic engineering, mastering the genetic tools, metabolic strategies, and bioprocess principles associated with these two dominant platforms provides a strong foundation for contributing to the future of biomanufacturing. The ongoing integration of advanced gene editing, automation, and AI-driven design promises to further enhance the productivity and scope of these versatile microbial cell factories.
The field of metabolic engineering has undergone a fundamental transformation, evolving from a discipline focused on single-gene manipulations to one that embraces the complexity of entire biological systems. This evolution from genetic engineering to systems biology represents a paradigm shift in how researchers approach the design and optimization of biological systems for industrial and therapeutic applications. Where early metabolic engineering relied on sequential trial-and-error modifications, the modern approach leverages computational modeling, multi-omics data integration, and machine learning to develop predictive understanding of cellular behavior. This comprehensive review examines the technological advances driving this evolution, detailing the experimental methodologies and computational frameworks that now enable researchers to bridge the gap between genetic modifications and system-level phenotypes.
The significance of this transition extends across multiple industries, from sustainable energy to pharmaceutical development. In biofuel production, for instance, the integration of systems biology has enabled the engineering of microbial chassis with significantly enhanced capabilities. Advanced biofuels derived from non-food lignocellulosic feedstock demonstrate how systems-level approaches can address both economic and sustainability challenges that limited earlier generations of biofuel technology [36]. Similarly, in pharmaceutical development, the ability to map intricate interaction networks between different layers of biological molecules has created new opportunities for investigating complex disease etiology and identifying therapeutic targets [37].
The Generational Evolution of Bioengineering Approaches
The progression from simple genetic manipulations to sophisticated systems-level engineering can be observed through the development of biofuel technologies, which serve as an exemplary case study of this evolution. Each generation represents not only technical advancement but also a fundamental shift in engineering philosophy.
Table 1: Generational Evolution of Bioengineering Approaches in Biofuel Production
| Generation | Feedstock | Engineering Approach | Key Technologies | Limitations |
|---|---|---|---|---|
| First | Food crops (corn, sugarcane) | Conventional fermentation | Transesterification, distillation | Food vs. fuel competition, high land use |
| Second | Non-food lignocellulosic biomass | Microbial strain engineering | Enzymatic hydrolysis, fermentation | Biomass recalcitrance, process complexity |
| Third | Microalgae | Photosynthetic efficiency optimization | Photobioreactors, hydrothermal liquefaction | Scale-up challenges, production costs |
| Fourth | Engineered microorganisms & synthetic systems | Synthetic biology, systems-level design | CRISPR-Cas, pathway engineering, AI-driven optimization | Regulatory hurdles, technical complexity |
First-generation biofuels primarily relied on conventional fermentation and distillation of food crops like corn and sugarcane, employing basic genetic engineering techniques to improve yield but facing significant limitations regarding food competition and land use [36]. Second-generation approaches transitioned to non-food lignocellulosic biomass, requiring more sophisticated microbial engineering to efficiently convert resistant plant materials into fermentable sugars. This generation saw the development of specialized enzymes such as cellulases, hemicellulases, and ligninases to break down recalcitrant biomass, alongside engineering of microbial hosts like S. cerevisiae for improved xylose utilization [36].
The third generation marked a shift toward photosynthetic microorganisms, particularly microalgae, with engineering efforts focused on enhancing photosynthetic efficiency and lipid accumulation. This approach resolved the food-versus-fuel dilemma but introduced new challenges in scaling and economic viability [36]. Contemporary fourth-generation biofuel production fully embraces systems biology, integrating synthetic biology tools with computational modeling to create engineered microbial systems capable of producing advanced drop-in fuels. These systems employ CRISPR-Cas for precise genome editing, de novo pathway engineering for compounds like butanol and isoprenoids, and AI-driven optimization to overcome previous yield limitations [36]. Notable achievements include 91% biodiesel conversion efficiency from microbial lipids and a threefold increase in butanol yield in engineered Clostridium species [36].
Fundamental Technological Transitions
From Single-Gene Editing to Programmable Genome Engineering
The development of increasingly sophisticated DNA manipulation tools has been instrumental in enabling the transition to systems biology. Early genetic engineering depended on homologous recombination and basic recombinase systems (Cre-lox, Flp-FRT) that required pre-engineered recognition sequences and offered limited programmability [38]. While valuable for specific applications, these technologies were poorly suited for systems-level engineering due to their low throughput and inability to perform complex multiplexed edits.
The advent of CRISPR-based systems has dramatically expanded engineering capabilities. Initial CRISPR-Cas9 systems enabled targeted double-strand breaks, allowing more precise gene edits but still relying on endogenous DNA repair mechanisms that often produced heterogeneous outcomes [38]. The development of homology-directed repair (HDR) strategies improved editing precision but remained constrained by cell cycle dependence and competition with error-prone non-homologous end joining pathways [38].
Recent advances have overcome these limitations through several innovative approaches:
CRISPR-Assisted Transposase (CAST) Systems: These technologies combine CRISPR targeting with transposase-mediated DNA insertion, enabling precise integration of large DNA fragments (up to 30 kb) without double-strand breaks [38]. Type I-F CAST systems achieve this through a Cascade complex (Cas6, Cas7, Cas8) for target recognition and a heteromeric transposase (TnsA, TnsB, TnsC) for DNA integration approximately 50 bp downstream of the target site [38]. Type V-K systems utilize the single-effector Cas12k with integration occurring 60-66 bp downstream of the PAM site [38].
Prime Editing: This more recent innovation uses catalytically impaired Cas enzymes fused to reverse transcriptase, enabling precise point mutations and small insertions without double-strand breaks [38].
Vibrio natriegens Toolkit (Vnat Collection): This comprehensive, modular genetic toolkit exemplifies modern engineering approaches, featuring optimized Golden Gate assembly with improved junction sequences that achieve up to 300-fold increased assembly efficiency, novel operon connectors for multi-gene pathway construction, and refined NT-CRISPR methods that eliminate intermediate purification steps [39].
Diagram 1: Evolution of genetic engineering technologies from traditional recombinase-based methods to modern DSB-free systems.
The Rise of Multi-Scale Modeling and Omics Integration
Where early metabolic engineering focused on individual pathways, systems biology approaches now integrate multiple layers of biological information through sophisticated computational frameworks. This integration occurs across two primary domains: Systems Biology (SB) and Process Systems Engineering (PSE), which are increasingly converging into the unified discipline of Biotechnology Systems Engineering (BSE) [40].
Systems Biology provides mathematical and computational methods for understanding biological phenomena across different omics levels, utilizing several key modeling approaches:
Constraint-Based Modeling: This approach treats metabolic fluxes as decision variables in biologically inspired optimization problems, addressing system underdetermination by considering biologically relevant objective functions (e.g., maximizing growth) subject to mass-balance and physiological constraints [40]. When solved under pseudo-steady-state assumptions, it provides metabolic flux distribution snapshots for given temporal states.
Kinetic Modeling: Unlike constraint-based approaches, kinetic modeling explicitly describes fluxes as time-dependent functions governed by enzyme kinetics and metabolite concentrations, capturing accumulation of both metabolic intermediates and extracellular species [40]. Though more biologically insightful, these models present numerical challenges for optimization and parameterization.
Process Systems Engineering focuses on mathematical modeling and computer-aided methods for design, optimization, and control at macroscopic scales, emphasizing bioreactor-level variables like feed rates, oxygen availability, temperature, and pH [40]. Control strategies range from conventional proportional-integral-derivative (PID) control to advanced model predictive control (MPC) and reinforcement learning (RL) [40].
The emerging Biotechnology Systems Engineering framework integrates these approaches, creating multi-scale models that link intracellular metabolism with bioreactor dynamics and overall biomanufacturing facility performance [40]. This integration enables adaptive learning, continuous model updating, and self-adaptive optimization through digital twins that combine mechanistic modeling with machine learning [40].
Table 2: Multi-Omics Data Types and Their Applications in Metabolic Engineering
| Omics Layer | Analytical Focus | Engineering Applications | Data Sources |
|---|---|---|---|
| Genomics | DNA sequence and structure | Identification of metabolic potential, CRISPR target selection | Whole-genome sequencing, SNP arrays |
| Transcriptomics | RNA expression levels | Analysis of regulatory mechanisms, promoter engineering | RNA-seq, microarrays |
| Proteomics | Protein abundance and modifications | Enzyme expression optimization, metabolic flux analysis | Mass spectrometry, protein arrays |
| Fluxomics | Metabolic reaction rates | Pathway flux quantification, bottleneck identification | 13C tracing, metabolic flux analysis |
| Metabolomics | Metabolite concentrations | Pathway dynamics, intermediate accumulation | GC/MS, LC/MS, NMR |
Experimental Frameworks and Methodologies
The Design-Build-Test-Learn Cycle for Metabolic Pathway Engineering
The DBTL cycle represents a systematic framework for metabolic engineering that embodies the integration of genetic engineering with systems biology principles. This iterative approach provides structure to the engineering process while incorporating computational tools at each stage [41].
Design Phase: Computational tools pathway design, enzyme selection, and pathway discovery using scientific programming environments like Scientific Python [41]. This phase leverages both evidence-based networks (constructed from experimentally validated interactions in databases) and statistically inferred networks (derived from multi-omics data correlation analyses) [37]. For plant natural products, this may involve identifying key enzymes and transcription factors in biosynthetic pathways for CRISPR targeting [42].
Build Phase: Modern molecular toolkit implementation using standardized assembly systems such as Golden Gate modular cloning [39]. This phase employs specialized genetic toolkits like the Vnat Collection for Vibrio natriegens, which includes characterized inducible promoters for precise orthogonal regulation and optimized junction sequences that dramatically increase assembly efficiency [39]. For eukaryotic systems, this may involve delivery methods like viral vectors or electroporation for CRISPR components and donor DNA.
Test Phase: Comprehensive characterization using multi-omics profiling and analytical chemistry methods. This includes transcriptomic analysis to verify intended regulatory changes, proteomic validation of enzyme expression, metabolomic profiling of pathway intermediates and products, and fluxomic analysis to quantify metabolic reaction rates [40] [37]. Advanced biosensors can be incorporated for real-time monitoring of metabolic states [40].
Learn Phase: Data analysis and model refinement using machine learning approaches. This phase leverages the experimental data to improve predictive models, identify unanticipated interactions, and generate new hypotheses for the next DBTL cycle [43]. Techniques include network propagation to prioritize targets, functional module identification, and comparative network analysis to understand intervention effects [37].
Diagram 2: The Design-Build-Test-Learn (DBTL) cycle for systematic metabolic pathway engineering.
CRISPR-Cas Experimental Protocol for Plant Metabolic Engineering
The application of CRISPR systems in plant metabolic engineering exemplifies the integration of precise genetic tools with systems-level understanding. The following protocol outlines key steps for enhancing production of plant natural products (PNPs):
Target Identification: Select key enzymes or transcription factors in PNP biosynthetic pathways through multi-omics data integration and evidence-based network analysis [42] [37]. Priority should be given to nodes with high betweenness centrality in metabolic networks, as these represent potential bottlenecks with system-wide influence.
Guide RNA Design: Design specific gRNAs with minimal off-target potential using specialized software. For multiplexed editing, select gRNAs targeting multiple pathway genes simultaneously to address complex metabolic regulation.
Vector Construction: Assemble CRISPR construct using plant-optimized expression systems, incorporating appropriate promoters (e.g., Ubiqutin for monocots, 35S for dicots) and selectable markers. For large DNA integration, consider CAST systems adapted for plant applications [38].
Plant Transformation: Deliver constructs using Agrobacterium-mediated transformation, biolistics, or protoplast transfection based on plant species and experimental requirements.
Screening and Validation: Identify successful edits through PCR genotyping and sequencing. For metabolic engineering applications, screen multiple independent lines to account for position effects and somaclonal variation.
Metabolic Phenotyping: Conduct comprehensive metabolomic analysis of edited lines compared to wild-type controls. Employ LC-MS/MS or GC-MS to quantify target compounds and pathway intermediates.
Systems-Level Analysis: Integrate transcriptomic, proteomic, and metabolomic data to construct updated network models that capture the system-wide effects of genetic interventions and identify potential compensatory mechanisms [37].
Essential Research Reagents and Tools
The modern metabolic engineer's toolkit comprises integrated computational and molecular resources that enable systems-level approaches to biological design.
Table 3: Essential Research Reagent Solutions for Modern Metabolic Engineering
| Category | Specific Tools | Function | Applications |
|---|---|---|---|
| Genetic Toolkits | Vnat Collection [39] | Modular genetic parts for Vibrio natriegens | Fast-growing bacterial chassis engineering |
| Genome Editing Systems | CRISPR-Cas9, Prime Editing, CAST systems [38] [42] | Targeted gene knock-out, knock-in, and base editing | Pathway engineering, gene regulation |
| Assembly Systems | Golden Gate, Gibson Assembly | Modular DNA construction | Pathway assembly, vector engineering |
| Modeling Software | Constraint-based modeling tools, Kinetic modeling platforms | Metabolic network simulation and prediction | Strain design, pathway prediction |
| Machine Learning Libraries | TensorFlow, PyTorch, Scikit-learn [43] | Data-driven model development | Omics data analysis, predictive modeling |
| Biosensors | Transcription factor-based, FRET-based | Metabolite sensing and real-time monitoring | High-throughput screening, dynamic control |
| Multi-omics Platforms | RNA-seq, LC-MS/MS, GC-MS | Comprehensive molecular profiling | Systems-level analysis, DBTL cycles |
Future Perspectives: AI and Industry 4.0 in Biotechnology
The continued evolution of metabolic engineering toward systems biology approaches is increasingly intersecting with artificial intelligence and Industry 4.0 technologies, creating new paradigms for biological engineering.
AI-Driven Biological Design: Machine learning is transforming multiple aspects of metabolic engineering, from pathway construction and optimization to genetic editing optimization and production scale-up [43]. ML algorithms can predict enzyme performance, identify optimal genetic modifications, and guide experimental design, significantly reducing the empirical screening burden. The integration of mechanistic models with machine learning creates hybrid approaches that leverage both first principles and data-driven insights [43].
Digital Twins and Real-Time Optimization: The concept of digital twins – virtual replicas of biological systems that update continuously with real-time data – represents the cutting edge of systems biology applications [40]. These tools enable adaptive model updating and self-adaptive optimization through integration with bioreactor monitoring systems. By combining mechanistic models with machine learning, digital twins enhance predictive capabilities and support real-time decision making [40].
Multi-Scale Control Systems: Future bioprocesses will implement integrated control strategies that synergistically combine external bioreactor control with in-cell controllers encoded by engineered genetic circuits [40]. This approach enables dynamic management of metabolic trade-offs (e.g., growth vs. production) in response to changing process conditions, optimizing overall system performance.
Network-Based Therapeutic Discovery: In pharmaceutical applications, network biology approaches that bridge quantitative genetics and multi-omics will play an increasingly important role in identifying diagnostic biomarkers and therapeutic targets for complex diseases [37]. Strategies such as network propagation, functional module analysis, and comparative dynamic networks provide powerful methods for translating genetic associations into biological insights and clinical applications [37].
The ongoing formalization of Biotechnology Systems Engineering as a unified discipline will be crucial for advancing these capabilities, requiring interdisciplinary education that bridges traditional boundaries between biology, engineering, and computer science [40]. This integration will enable the next generation of bioengineers to fully leverage systems-level approaches for sustainable biomanufacturing, therapeutic development, and fundamental biological discovery.
Practical Strategies and Biomedical Applications in Metabolic Engineering
Metabolic engineering aims to rewire the metabolism of microorganisms to transform them into efficient cell factories for producing valuable chemicals, fuels, and pharmaceuticals. The foundation of this discipline rests on precise genetic tools that enable researchers to manipulate metabolic pathways with unprecedented accuracy and efficiency. Among the most powerful techniques in the modern metabolic engineer's arsenal are CRISPR interference (CRISPRi), gene knockouts, and heterologous expression. These tools collectively enable the downregulation of competing pathways, the complete elimination of gene function, and the introduction of entirely new metabolic capabilities into host organisms. The advent of CRISPR-based technologies, in particular, has revolutionized the field by providing simple, efficient, and highly programmable methods for genetic manipulation that overcome the limitations of earlier technologies like zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) [44] [45]. This technical guide explores the principles, applications, and methodologies of these core genetic tools within the context of metabolic engineering, providing both theoretical foundations and practical experimental protocols.
Technology Fundamentals and Comparative Analysis
CRISPR Interference (CRISPRi)
CRISPR interference represents a refined application of the CRISPR-Cas system that enables precise gene downregulation without permanent DNA alteration. The system utilizes a catalytically deactivated Cas9 (dCas9) protein, generated through point mutations (H840A and D10A) that abolish its endonuclease activity while retaining its DNA-binding capability [46] [45]. When guided by a sequence-specific single-guide RNA (sgRNA), dCas9 binds to target DNA sequences and creates a physical block that inhibits transcription initiation or elongation by RNA polymerase, effectively repressing gene expression [46]. The versatility of CRISPRi can be enhanced by fusing dCas9 to transcriptional repressor domains such as MXII, KRAB, or CS, which further strengthen repression efficiency through chromatin modification [46].
A significant advancement in CRISPRi methodology involves the integration of computational prediction tools to identify optimal gene targets for downregulation. For instance, the FluxRETAP (Flux-Reaction Target Prioritization) algorithm has demonstrated remarkable success in predicting gene knockdown targets that substantially increase isoprenol titers in Pseudomonas putida KT2440, outperforming conventional non-computational, pathway-guided selection approaches [47]. This combination of computational prediction and precise genetic intervention represents a powerful strategy for redirecting metabolic flux toward desired products.
Gene Knockouts
Gene knockouts involve the complete and permanent disruption of target genes, preventing the production of functional gene products. Unlike CRISPRi, which provides tunable but reversible suppression, knockout strategies result in irreversible gene elimination. Traditional methods for generating knockouts include homologous recombination, PEG/CaClâ‚‚-mediated transformation, and Agrobacterium-mediated transformation [48]. However, CRISPR-based knockout systems using catalytically active Cas9 have dramatically improved the efficiency and specificity of this process [46].
The CRISPR-Cas9 system induces double-strand breaks (DSBs) at specific genomic locations guided by sgRNA. Cellular repair of these breaks occurs primarily through two pathways: non-homologous end joining (NHEJ) and homologous recombination (HR) [44]. NHEJ is an error-prone process that often results in small insertions or deletions (indels) at the break site, leading to frameshift mutations and premature stop codons that effectively knockout gene function [44]. In contrast, HR enables precise gene replacement or insertion using donor DNA templates but occurs less frequently in most microbial systems [44].
Heterologous Expression
Heterologous expression refers to the introduction of genetic material from a foreign source into a host organism to confer new metabolic capabilities. This technique enables the production of novel compounds that the host would not naturally synthesize, expanding the bioproduction landscape considerably [48]. Successful heterologous expression requires several key components: (1) identification and isolation of the target gene or biosynthetic gene cluster (BGC) from the donor organism; (2) selection of appropriate expression vectors with suitable promoters, ribosomal binding sites, and terminators; (3) choice of compatible host organisms such as Escherichia coli, Bacillus subtilis, Saccharomyces cerevisiae, or specialized fungal hosts like Aspergillus nidulans and Aspergillus oryzae [48].
A critical consideration in heterologous expression is the potential for metabolic burden on the host organism, which can reduce growth rates and overall productivity. Chromosomal integration of heterologous pathways is often preferred over plasmid-based expression due to greater genetic stability and reduced metabolic burden, though copy number control becomes more challenging [45].
Table 1: Comparison of Key Genetic Tools in Metabolic Engineering
| Feature | CRISPRi | Gene Knockouts | Heterologous Expression |
|---|---|---|---|
| Genetic Outcome | Reversible gene downregulation | Permanent gene disruption | Introduction of foreign genetic material |
| Mechanism | dCas9-mediated transcription blockade | Cas9-induced DSB with NHEJ repair | Integration or plasmid-based expression of foreign DNA |
| Applicability | Tunable suppression of essential and non-essential genes | Disruption of non-essential genes | Pathway engineering for novel compound production |
| Advantages | Precise, programmable, tunable, suitable for essential genes | Complete elimination of gene function, stable phenotype | Expands host metabolic capabilities, enables novel compound production |
| Limitations | Requires sustained dCas9/sgRNA expression, potential incomplete suppression | Unsuitable for essential genes, potential off-target effects | Metabolic burden, codon usage compatibility, protein folding issues |
Experimental Protocols and Workflows
CRISPRi Workflow for Metabolic Flux Redirection
The following protocol outlines the implementation of CRISPRi for metabolic engineering applications, based on successful approaches used in Pseudomonas putida for enhanced production of sustainable aviation fuel precursors [47]:
Target Identification: Utilize computational tools such as FluxRETAP to identify potential gene targets whose knockdown may enhance flux toward the desired metabolic product [47].
sgRNA Design and Array Construction: Design sgRNAs with complementary sequences (typically 20 nt) to the target genes. For multiplexed repression, construct gRNA arrays using assembly methods such as VAMMPIRE (Versatile Assembly Method for MultiPlexing CRISPRi-mediated downREgulation), which enables accurate assembly of constructs containing up to five sgRNA arrays with reduced context dependency and uniform, position-independent gene downregulation [47].
Vector Construction: Clone the dCas9 gene (optionally fused to repressor domains for enhanced efficiency) and sgRNA array into appropriate expression vectors. Consider inducible promoters for tight control of dCas9 expression.
Transformation and Screening: Introduce the constructed vector into the host organism using transformation methods appropriate for the specific host (e.g., electroporation for bacteria). Screen for successful transformants using selective markers and verify dCas9 expression and sgRNA incorporation.
Evaluation of Knockdown Efficiency: Quantify gene repression using qRT-PCR to measure transcript levels and assess metabolic flux changes through targeted metabolomics or product titer measurements.
Diagram 1: CRISPRi Experimental Workflow for Metabolic Engineering
Gene Knockout Protocol Using CRISPR-Cas9
This protocol describes the generation of gene knockouts in microbial hosts using the CRISPR-Cas9 system, with specific considerations for various bacterial and fungal systems [48] [46]:
Target Selection: Identify the gene to be knocked out, prioritizing non-essential genes based on existing genomic knowledge or essentiality predictions.
sgRNA Design: Design sgRNAs with 20-nt spacer sequences complementary to the 5' region of the target gene, ensuring the presence of a PAM sequence (NGG for SpCas9) adjacent to the target site.
Repair Donor Design (for homology-directed repair): For precise deletions or when incorporating selection markers, design a donor DNA template with homology arms (typically 500-1000 bp) flanking the target site.
Vector Construction: Clone the Cas9 gene and sgRNA into an appropriate expression vector. For hosts with inefficient NHEJ, include a repair donor template for homology-directed repair.
Transformation: Introduce the CRISPR construct into the host organism using species-appropriate methods (electroporation for bacteria, PEG-mediated transformation for fungi).
Screening and Verification: Screen for successful knockouts using antibiotic selection (if markers are incorporated) or phenotypic screening. Verify knockout at the DNA level through PCR and sequencing.
Curing of CRISPR Plasmids: In strains where continuous Cas9 expression is undesirable, employ plasmid curing techniques to remove the CRISPR machinery after successful knockout.
Heterologous Expression Workflow
The successful implementation of heterologous expression for natural product biosynthesis involves the following key steps, particularly relevant for fungal secondary metabolites [48]:
Gene Cluster Identification: Mine the genome of the donor organism to identify biosynthetic gene clusters (BGCs) of interest using tools such as antiSMASH, BLAST, or ARTS [48].
Host Selection: Choose an appropriate heterologous host based on compatibility with the biosynthetic pathway. Common prokaryotic hosts include E. coli and B. subtilis, while eukaryotic hosts include S. cerevisiae, A. nidulans, and A. oryzae [48].
Vector Design and Assembly: Design expression vectors containing strong, host-compatible promoters, appropriate ribosomal binding sites (for prokaryotes), and selection markers. For large BGCs, consider bacterial artificial chromosomes (BACs) or similar systems.
Pathway Engineering: Optimize codon usage to match the host organism's preferences. Consider modular assembly for large pathways to enable troubleshooting and optimization.
Transformation and Screening: Introduce the constructed vector into the host organism and screen for successful transformants. Use analytical methods (HPLC, LC-MS) to detect the production of target compounds.
Pathway Optimization: Once functional expression is confirmed, optimize production through promoter engineering, ribosome binding site modification, or co-expression of accessory proteins.
Diagram 2: Heterologous Expression Workflow for Natural Product Biosynthesis
Advanced Applications and Combinatorial Approaches
The true power of modern genetic tools emerges when they are deployed in combinatorial strategies that simultaneously manipulate multiple genetic targets. The CRISPR-AID (CRISPR Activation, Interference, and Deletion) system exemplifies this approach, utilizing three orthogonal CRISPR proteins to enable transcriptional activation (CRISPRa), transcriptional interference (CRISPRi), and gene deletion (CRISPRd) concurrently in Saccharomyces cerevisiae [49]. This tri-functional system enables comprehensive rewiring of metabolic networks by simultaneously upregulating bottleneck enzymes, downregulating competing pathways, and eliminating non-essential genes that divert flux away from the desired product [49].
In practice, CRISPR-AID has demonstrated remarkable success in optimizing complex phenotypes. For β-carotene production in yeast, the application of CRISPR-AID resulted in a 3-fold increase in production in a single engineering step [49]. Similarly, when applied to optimize the display of an endoglucanase on the yeast surface, combinatorial testing of multiple metabolic engineering targets using CRISPR-AID achieved a 2.5-fold improvement in enzyme display [49]. These results highlight the advantage of testing genetic modifications in combinations rather than sequentially, as they can reveal synergistic interactions that would be missed in conventional approaches.
Table 2: Performance Metrics of Genetic Tools in Metabolic Engineering Applications
| Application | Host Organism | Tool Used | Outcome | Key Metric |
|---|---|---|---|---|
| Isoprenol Production | Pseudomonas putida | CRISPRi with FluxRETAP | Enhanced precursor production | 1.5 g/L titer achieved by knocking down PP_4118 [47] |
| β-Carotene Production | Saccharomyces cerevisiae | CRISPR-AID (Combinatorial) | Increased product yield | 3-fold improvement in production [49] |
| Endoglucanase Display | Saccharomyces cerevisiae | CRISPR-AID (Combinatorial) | Enhanced enzyme display | 2.5-fold improvement [49] |
| Fungal Secondary Metabolites | Various Fungi | Heterologous Expression | Access to novel compounds | >90% of BGCs are silent and uncharacterized [48] |
Research Reagent Solutions
Table 3: Essential Research Reagents for Genetic Tool Implementation
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| CRISPR Proteins | SpCas9, dCas9, SaCas9, St1Cas9, LbCpf1 | DNA cleavage or binding | Orthogonal proteins enable multiplexing; dCas9 for CRISPRi [46] [49] |
| Transcriptional Regulators | VP64, p65AD, Rta (for activation); MXI1, KRAB (for repression) | Enhance CRISPRa/CRISPRi efficiency | Optimal activation/repression domains vary by CRISPR protein [46] [49] |
| Assembly Systems | VAMMPIRE, Golden Gate Assembly | Multiplex gRNA array construction | Enables coordinated regulation of multiple genes [47] |
| Bioinformatics Tools | FluxRETAP, antiSMASH, BLAST, ARTS | Target identification and prioritization | Computational prediction enhances success rate [47] [48] |
| Host Organisms | E. coli, B. subtilis, S. cerevisiae, P. putida, A. nidulans | Production chassis | Selection depends on pathway requirements and genetic accessibility [48] [46] |
| Delivery Methods | Electroporation, PEG-mediated transformation, Agrobacterium-mediated transformation | Introduction of genetic material | Method depends on host organism and vector system [48] [50] |
Diagram 3: CRISPR System Components and Their Functional Relationships
The integration of CRISPRi, gene knockouts, and heterologous expression represents a powerful toolkit for addressing the complex challenges of metabolic engineering. While each technique offers distinct advantages for specific applications, their combinatorial implementation enables comprehensive rewiring of cellular metabolism that transcends the capabilities of individual approaches. The continued development of these technologies—particularly in areas such as orthogonal CRISPR systems, computational prediction tools, and advanced delivery methods—promises to further accelerate the design-build-test-learn cycle in metabolic engineering. As these tools become increasingly sophisticated and accessible, they will undoubtedly play a central role in the development of next-generation microbial cell factories for sustainable production of fuels, chemicals, and pharmaceuticals.
{#content#}
This whitepaper provides an in-depth technical guide on metabolic pathway engineering for the bioproduction of biliverdin and mandelic acid, two high-value compounds with significant pharmaceutical applications. Aimed at researchers and scientists entering the field of metabolic engineering, this document details the rational design of microbial cell factories, leveraging case studies that demonstrate the integration of computational design, enzyme engineering, and process optimization. We present structured data, experimental protocols, and visual workflows to illustrate the core principles of diverting cellular metabolism toward target compounds. By framing these concepts within the context of sustainable drug development, this guide serves as a foundational resource for pioneering green manufacturing routes in the pharmaceutical industry.
Pathway engineering is a cornerstone of industrial biotechnology, involving the modification and optimization of metabolic pathways in microorganisms to produce valuable chemicals from renewable resources. This approach stands in contrast to traditional chemical synthesis, which often relies on petrochemical feedstocks, involves harsh reaction conditions, and generates substantial waste. The paradigm shift towards microbial cell factories offers a sustainable alternative, enabling the production of complex molecules with high stereoselectivity under mild, bio-catalytic conditions [51] [52].
The core philosophy involves a "design-build-test-learn" cycle. Rational design starts with the selection of a host organism (e.g., E. coli, Corynebacterium glutamicum) and the identification or creation of a biosynthetic pathway to the target molecule. The build phase involves genetic modifications to implement this pathway, often requiring the introduction of heterologous genes and the deletion of competing pathways. The engineered strain is then tested in bioreactors, and performance data is analyzed to learn and inform the next cycle of design optimizations [53] [54]. This iterative process, accelerated by advances in synthetic biology and automation, is reshaping the production landscape for pharmaceuticals, materials, and fine chemicals [54].
Case Study 1: Biliverdin Production inCorynebacterium glutamicum
Biliverdin is a prospective recyclable antioxidant and a crucial precursor for chromophores used in optogenetics and medical research. Its traditional production via chemical oxidation of bilirubin from mammalian bile is fraught with challenges, including isomer separation, impurities, and environmental concerns. A bio-based production using a microbial cell factory presents a sustainable and efficient alternative [53].
Pathway Design and Engineering Strategies
The engineering of C. glutamicum for biliverdin production focused on the heme biosynthesis pathway, as heme is the direct precursor of biliverdin. A key innovation was the suggestion and utilization of a novel, thermodynamically favorable coporphyrin-dependent pathway (mediated by hemQ) over the more commonly noted protoporphyrin-dependent pathway (mediated by hemN) [53].
A systematic push and pull strategy guided by in vitro thermodynamic analysis was employed:
- Push Module: Thermodynamic analysis identified hemA (glutamyl-tRNA reductase) and hemL (glutamate-1-semialdehyde aminotransferase) as potential bottleneck enzymes in the pathway. Overexpression of a mutated, stabilized hemA (hemAM) from Salmonella typhimurium and hemL from E. coli was used to push carbon flow towards heme synthesis, resulting in a 5.48-fold increase in heme production [53].
- Pull Module: The native heme oxygenase gene hmuO was overexpressed to pull the carbon flux from heme to the final product, biliverdin [53].
Experimental Protocol & Key Results
The experimental workflow for building the biliverdin production strain involved the following key steps [53]:
- Host Strain Selection: C. glutamicum ATCC 13826, a glutamate-overproducing strain, was chosen for its natural abundance of glutamate, a key precursor in the C5 pathway for heme synthesis.
- In vitro Thermodynamic Analysis: The Gibbs free energy (ΔG'°) of each reaction in the heme biosynthesis pathway was calculated to identify thermodynamically unfavorable, rate-limiting steps.
- Modular Genetic Construction: Genes for the push (hemAM, hemL) and pull (hmuO) modules were assembled on plasmids and introduced into the host strain. The hemQ-mediated pathway was reinforced.
- Fed-Batch Fermentation: The optimized strain, C. glutamicum BV004, was cultivated in a 5 L bioreactor with minimal medium using glucose as the sole carbon source.
Through modular optimization and fed-batch fermentation, the engineered strain achieved a final biliverdin titer of 68.74 ± 4.97 mg/L, the highest reported titer at the time of the study [53].
Biliverdin Production Data
Table 1: Key performance metrics for biliverdin production in C. glutamicum [53].
| Metric | Value / Specification |
|---|---|
| Host Organism | Corynebacterium glutamicum ATCC 13826 |
| Carbon Source | Glucose |
| Engineering Strategy | Push-and-pull, Novel coproporphyrin-dependent pathway |
| Key Genetic Modifications | Overexpression of hemAM, hemL, hemQ, hmuO |
| Shake Flask Titer | 11.38 ± 0.47 mg/L |
| Bioreactor Titer (5 L) | 68.74 ± 4.97 mg/L |
| Cultivation Scale | 5 L Bioreactor |
Case Study 2: Mandelic Acid Production inEscherichia coli
Mandelic acid (MA) is a valuable α-hydroxy acid widely used as a chiral synthon in the synthesis of antibiotics, cosmetics, and fine chemicals. While existing chemical synthesis methods are well-established, they raise concerns regarding toxicity and environmental pollution. Metabolic engineering offers a direct, fermentative route to optically pure MA from renewable glucose [55] [52].
Pathway Design and Engineering Strategies
The de novo biosynthesis of MA in E. coli was achieved by engineering the central metabolic and L-phenylalanine pathways. The key heterologous enzyme introduced was a hydroxymandelate synthase (HMAS), which catalyzes the conversion of the intermediate phenylpyruvate to S-mandelic acid [55] [52].
Critical engineering strategies included:
- Precursor Supply Enhancement: The shikimate pathway was enhanced to increase the supply of the precursors erythrose-4-phosphate (E4P) and phosphoenolpyruvate (PEP), which are essential for phenylpyruvate synthesis [55].
- Competing Pathway Knockout: Genes encoding for aromatic amino acid aminotransferases (tyrB, aspC) and branch-point enzymes for L-tyrosine and L-tryptophan synthesis (tyrA, trpE) were deleted to minimize carbon flux diversion [52].
- CRISPR Interference (CRISPRi): To further fine-tune metabolic flux, CRISPRi was employed to repress competing pathways, redirecting flux toward MA production [55].
- High-Cell-Density Cultivation (HCDC): The process was scaled in a 5 L bioreactor under HCDC conditions to demonstrate industrial potential [55].
Experimental Protocol & Key Results
A representative protocol for engineering E. coli for MA production involves [55] [52]:
- Enzyme Screening: Screen and identify an efficient HMAS homolog (e.g., from Actinosynnema mirum).
- Strain Construction: Clone HMAS and overexpress key enzymes of the shikimate pathway (e.g., deregulated DAHP synthase aroFfbr). Use CRISPRi to knock down competing genes.
- Fermentation in Bioreactor: Cultivate the engineered strain in a 5 L bioreactor with ZYM-5052 or similar medium, using fed-batch strategies for high-cell-density cultivation.
This systematic approach enabled the achievement of an MA titer of 9.58 g/L, the highest reported for microbial production, demonstrating a robust and scalable process [55].
Mandelic Acid Production Data
Table 2: Key performance metrics for mandelic acid production in E. coli [55] [52] [54].
| Metric | Value / Specification |
|---|---|
| Host Organism | Escherichia coli |
| Carbon Source | Glucose |
| Engineering Strategy | Shikimate pathway enhancement, Competing pathway deletion, CRISPRi |
| Key Genetic Modifications | Expression of heterologous HMAS, Deletion of tyrB, aspC, etc. |
| Reported Titer (Shake Flask) | 0.74 g/L (S-MA) [52] |
| Reported Titer (Bioreactor) | 9.58 g/L [55] |
| Cultivation Scale | 5 L Bioreactor |
The Scientist's Toolkit: Essential Research Reagents
Successful pathway engineering relies on a suite of molecular biology and bioprocess tools. The table below lists essential reagents and their functions based on the cited case studies.
Table 3: Key research reagents and materials used in metabolic pathway engineering.
| Reagent / Material | Function in Research | Example from Case Studies |
|---|---|---|
| Hydroxymandelate Synthase (HMAS) | Key heterologous enzyme that catalyzes the formation of mandelic acid from phenylpyruvate. | Identified from Actinosynnema mirum for efficient MA synthesis in E. coli [55]. |
| Heme Oxygenase (hmuO) | Terminal enzyme that converts heme to biliverdin, "pulling" flux in the pathway. | Native hmuO from C. glutamicum was overexpressed [53]. |
| Plasmids (e.g., pEKEx2, pSU series) | Vectors for the expression of heterologous and regulatory genes. | pEKEx2 used for expressing hemAL in C. glutamicum; pSUFAAQ used for aroFfbr, pheAfbr, and hmaS in E. coli [53] [52]. |
| CRISPR-dCas9 System | For targeted repression of gene expression (CRISPRi) to downregulate competing pathways. | Used in E. coli to repress genes that divert flux away from the MA pathway [55]. |
| Glucose (Minimal Medium) | A defined, renewable carbon source for microbial cultivation, ensuring process sustainability and reproducibility. | Used as the sole carbon source in fed-batch fermentations for both biliverdin and MA production [53] [55]. |
| Inducers (e.g., IPTG, Arabinose) | Chemicals used to precisely control the timing and level of expression of pathway genes. | IPTG used for CRISPRi induction; Arabinose used for protein expression in E. coli [55]. |
| Yunnandaphninine G | Yunnandaphninine G, MF:C30H47NO3, MW:469.7 g/mol | Chemical Reagent |
| 3-Epiglochidiol | 3-Epiglochidiol, MF:C30H50O2, MW:442.7 g/mol | Chemical Reagent |
Comparative Analysis & Future Outlook
The case studies on biliverdin and mandelic acid production, while targeting different molecules and hosts, share a common methodological framework grounded in systems metabolic engineering. Both exemplify the critical importance of host selection—C. glutamicum for its native heme pathway and GRAS status, and E. coli for its well-characterized genetics and rapid growth. Furthermore, both studies move beyond simple gene overexpression, employing sophisticated strategies like thermodynamic analysis to identify push modules and CRISPRi for precise flux control.
Future advancements in the field are poised to build upon these foundations. The integration of artificial intelligence and machine learning with protein design and omics data will accelerate the discovery and optimization of enzymes and pathways [51] [56]. The concept of biofoundries—automated platforms for genetic design and strain construction—is already demonstrating its power, enabling the rapid prototyping of strains for dozens of target compounds, including mandelic acid, in remarkably short timeframes [54]. Finally, the consideration of biomechanics, such as the mechanical environment in bioreactors and molecular dynamics simulations of enzyme-substrate interactions, provides a transformative perspective for further enhancing the efficiency and stereoselectivity of biosynthesis systems [51].
Visualizing the Engineered Pathways
The following diagrams illustrate the core metabolic pathways and engineering strategies described in this whitepaper.
Biliverdin Biosynthesis Pathway
Mandelic Acid Biosynthesis Pathway
{#/content#}
Cofactor Engineering and Regeneration Strategies for Enhanced Flux
Cofactor engineering is a cornerstone of modern metabolic engineering, focused on optimizing the availability, balance, and regeneration of essential enzyme cofactors to drive metabolic flux toward desired products. Cofactors are non-protein molecules, such as nicotinamide adenine dinucleotide phosphate (NADPH), adenosine triphosphate (ATP), and acetyl coenzyme A (acetyl-CoA), that are indispensable for enzymatic activity and cellular metabolism [57]. They act as carriers of energy, electrons, or functional groups in approximately 1,610 enzymatic reactions, including those catalyzed by transferases, oxidoreductases, lyases, ligases, isomerases, and hydrolases [57]. In the context of microbial cell factories, pathway reconstitution for high-efficiency chemical production often leads to unbalanced intracellular redox states and energy deficits, limiting titers, yields, and productivities [58]. Cofactor engineering addresses these limitations by systematically redesigning central metabolism to enhance redox homeostasis and energy regeneration, thereby increasing the flux through engineered pathways.
The physiological functions of cofactors are multifaceted. NADPH/NADP+ serves as a primary electron donor in anabolic reactions, with more than 100 enzymatic reactions linked to NADPH [57]. ATP/ADP functions as the universal energy currency, powering biosynthetic reactions and cellular maintenance [57]. Acetyl-CoA connects various metabolic reactions, providing carbon sources and energy, and serving as a precursor for isoprenoids, fatty acids, terpenoids, and polyketides [57]. Efficient cofactor regeneration is crucial because these molecules are consumed in stoichiometric amounts during biocatalytic processes, and their de novo synthesis is costly for the cell [59]. By managing their concentrations and forms through cofactor engineering, metabolic engineers can direct metabolic flux to target metabolites, maintain redox balance, and achieve higher production of valuable chemicals [57].
Core Cofactor Regeneration Strategies
NAD(P)H Regeneration Systems
Regeneration of reduced nicotinamide cofactors is vital for driving NADPH-dependent or NADH-dependent enzymatic reactions. Several enzymatic systems have been developed for efficient in vivo and in vitro cofactor regeneration.
Phosphite Dehydrogenase (PtxD) System: The PtxD system oxidizes phosphite to phosphate, concurrently reducing NADP+ to NADPH. This system is particularly valuable because the reaction is practically irreversible, providing a strong thermodynamic driving force. A recent application demonstrated its use in regulating lactate-based copolymer biosynthesis in E. coli. By implementing a PtxD-based NADH regeneration module, researchers decoupled cofactor supply from central carbon metabolism, which increased the lactate fraction in poly(3-hydroxybutyrate-co-lactate) [P(3HB-co-LA)] to 39.0 mol% on xylose without disrupting bacterial growth. The genomic integration of ptxD proved superior to plasmid-based expression, yielding higher intracellular NADH levels and copolymer production due to enhanced genetic stability [60].
Formate Dehydrogenase (FDH) System: FDH catalyzes the oxidation of formate to CO2, reducing NAD+ to NADH. The near-irreversible nature of this reaction and the easy removal of CO2 make it an attractive regeneration system. In the production of (2S,3S)-2,3-butanediol from diacetyl, introducing FDH in E. coli co-expressing 2,3-butanediol dehydrogenase resulted in a final titer of 31.7 g/L, a productivity of 2.3 g/(L·h), and a yield of 89.8% in fed-batch bioconversion. The intracellular NADH concentration increased by 0.43 μmol/g DCW, and the ratio of NADH to NAD+ rose continuously, confirming efficient NADH regeneration. A significant advantage was the minimal byproduct formation, as formate was almost entirely converted to CO2, simplifying downstream purification [61].
Glucose Dehydrogenase (GDH) System: GDH oxidizes glucose to gluconolactone (which hydrolyzes to gluconic acid), regenerating NAD(P)H. This system is highly active and stable, but its use leads to acidification and byproduct formation. When GDH was used for NADH regeneration in the same (2S,3S)-2,3-butanediol production system, it achieved a lower titer of 16.8 g/L and required periodic pH control due to the accumulation of gluconic, acetic, and lactic acids [61].
Xylose Reductase (XR) and Sugar Phosphate Boosting System: A versatile in situ cofactor enhancement system utilizes xylose reductase (XR) with lactose. This system increases the pool of sugar phosphates connected to the biosynthesis of NAD(P)H, FAD, FMN, and ATP. In E. coli, the XR/lactose system increased the productivities of three different metabolically engineered pathways (fatty alcohol biosynthesis, bioluminescence light generation, and alkane biosynthesis) by 2-4-fold. Metabolomic analysis revealed that the system selectively enhanced metabolites involved in relevant cofactor biosynthesis, providing a customized boost according to cellular demand. This approach serves as a generic tool to increase in vivo cofactor generation for synthetic biology applications [62].
Table 1: Performance Comparison of Key NAD(P)H Regeneration Systems
| Regeneration System | Reaction Catalyzed | Key Advantages | Reported Performance |
|---|---|---|---|
| Phosphite Dehydrogenase (PtxD) | Phosphite + NADP+ → Phosphate + NADPH | Irreversible; decouples cofactor supply from growth | Increased lactate fraction in copolymer to 39.0 mol% [60] |
| Formate Dehydrogenase (FDH) | Formate + NAD+ → CO2 + NADH | Irreversible; coproduct easily removed; minimal byproducts | 31.7 g/L (2S,3S)-2,3-butanediol; 89.8% yield [61] |
| Glucose Dehydrogenase (GDH) | Glucose + NAD(P)+ → Gluconolactone + NAD(P)H | Highly active and stable; inexpensive substrate | 16.8 g/L (2S,3S)-2,3-butanediol; requires pH control [61] |
| Xylose Reductase (XR)/Lactose | Increases sugar phosphate pools | Generic booster for multiple cofactors; demand-driven | 2-4 fold productivity increase in diverse pathways [62] |
ATP and Acetyl-CoA Regeneration
Beyond redox cofactors, the regeneration of energy carriers and key precursors is equally critical.
ATP Regeneration: ATP is essential for energy-intensive biosynthetic reactions. Common regeneration strategies involve the use of polyphosphate kinases to regenerate ATP from ADP and polyphosphate [57]. In the context of E. coli-based production of D-pantothenic acid (D-PA), fine-tuning subunits of the ATP synthase in the oxidative phosphorylation pathway, rather than simple overexpression, was employed to optimize intracellular ATP levels. This approach was part of an integrated strategy that also addressed NADPH and one-carbon metabolism, ultimately contributing to a record D-PA titer of 124.3 g/L [58].
Acetyl-CoA Supply: As a central metabolic node, acetyl-CoA supply is frequently enhanced to improve the production of derived compounds. Strategies include modulating the acetate pathway, overexpressing acetyl-CoA synthetase, and engineering pyruvate dehydrogenase complex activity [57]. For instance, in Yarrowia lipolytica, overexpression of ACC and FAS enzymes, which utilize acetyl-CoA, increased lipid content to 25.7% [57]. In E. coli, engineering acetyl-CoA availability led to a 1.9-fold higher yield of 3-hydroxypropionate [57].
Analytical Methods for Cofactor Quantification
Accurate quantification of intracellular cofactor concentrations is fundamental for diagnosing metabolic bottlenecks and assessing the efficacy of engineering strategies. Liquid chromatography/mass spectrometry (LC/MS) has become the preferred platform due to its high sensitivity and specificity for these often-unstable molecules [63].
A systematic study optimized the quantitative analysis of 15 cofactors (including adenosine nucleotides, nicotinamide adenine dinucleotides, and acyl-CoAs) from Saccharomyces cerevisiae [63]. Key findings and recommendations include:
- Chromatography: The porous graphitic carbon stationary phase (Hypercarb column) with reverse-phase elution was identified as the optimal condition for separating a wide range of cofactors in a single run.
- MS Ionization: Analysis in negative ion mode without ion-pairing agents is recommended. Traditional methods using positive mode with ion-pairing agents cause ion suppression, poor stability, and contaminate/damage the mass spectrometer.
- Sample Quenching and Extraction: For S. cerevisiae, fast filtration is the preferred quenching method over conventional cold methanol quenching. Cold methanol damages the cell membrane, causing metabolite leakage and significantly reducing the measured cofactor levels. The optimal extraction solvent was determined to be a mixture of acetonitrile, methanol, and water (4:4:2, v/v/v) with 15 mM ammonium acetate buffer, which minimizes cofactor degradation [63].
Table 2: Essential Reagents and Tools for Cofactor Engineering Research
| Reagent/Tool Category | Specific Examples | Function/Application |
|---|---|---|
| Key Enzymes for Regeneration | Phosphite Dehydrogenase (PtxD), Formate Dehydrogenase (FDH), Glucose Dehydrogenase (GDH), Xylose Reductase (XR) | Catalyze the regeneration of reduced NAD(P)H from NAD(P)+ |
| Genetic Engineering Tools | CRISPR/Cas9, Tet-on Gene Switch, Plasmid Vectors (e.g., pBAD33-Ptrc, pETDuet) | Enable precise gene knock-in, knockout, and tunable gene expression |
| Analytical Standards | AMP, ADP, ATP, NAD+, NADH, NADP+, NADPH, CoA, Acetyl-CoA, Malonyl-CoA | Used for calibration and quantification in LC/MS analysis |
| Chromatography Columns | Hypercarb Porous Graphitic Carbon Column, ZIC-pHILIC, ACQUITY BEH Amide | Separate cofactors prior to mass spectrometric detection |
| Extraction Solvents | Acetonitrile:MeOH:Water (4:4:2) with 15mM Ammonium Acetate | Quench metabolism and extract intracellular cofactors efficiently |
Integrated Engineering and Industrial Applications
Advanced metabolic engineering moves beyond modifying single pathways to integrated, system-wide approaches. A seminal example is the production of D-pantothenic acid (D-PA) in E. coli, which required synchronized optimization of multiple cofactors [58]:
- NADPH Regeneration: Flux Balance Analysis (FBA) and Flux Variability Analysis (FVA) were used to predict optimal carbon flux distributions through the EMP, PPP, and ED pathways. Genetic modifications were implemented to redistribute flux and boost NADPH regeneration.
- Energy Coupling: A heterologous transhydrogenase system from S. cerevisiae was introduced to convert excess NADPH and NADH into ATP, creating an integrated redox-energy coupling strategy.
- One-Carbon Metabolism: The serine-glycine system was modified to enhance the pool of 5,10-methylenetetrahydrofolate (5,10-MTHF), ensuring sufficient one-carbon unit supply for D-PA biosynthesis.
This holistic, multi-module engineering framework resulted in a strain producing 124.3 g/L D-PA with a yield of 0.78 g/g glucose, setting a new benchmark and demonstrating the power of cofactor-centric metabolic design [58].
Similar principles have been successfully applied in other industrial contexts:
- Bioplastic Synthesis: The PtxD system enabled flexible regulation of lactate-based copolymer biosynthesis, paving the way for sustainable bioplastic manufacturing [60].
- Biofuel Production: In n-butanol biosynthesis, cofactor regeneration is a major challenge. Engineering strategies include using alternative host organisms, heterologous enzymes for acid reduction, and protein engineering of pathway enzymes to alter cofactor specificity and drive flux [64].
- Protein Production: In Aspergillus niger, overexpression of genes gndA (6-phosphogluconate dehydrogenase) and maeA (NADP-dependent malic enzyme) increased the intracellular NADPH pool by 45% and 66%, respectively. This enhanced NADPH availability boosted glucoamylase production by 65% and 30%, confirming that cofactor engineering is a valid strategy for improving protein secretion in microbial cell factories [65].
Experimental Protocol: Implementing a Cofactor Regeneration System
This protocol outlines the key steps for integrating and testing a formate dehydrogenase (FDH)-based NADH regeneration system in E. coli for the production of a reduced target metabolite, based on the work detailed in [61].
Objective: To enhance the yield and productivity of an NADH-dependent bioconversion process by co-expressing formate dehydrogenase.
Materials:
- E. coli host strain (e.g., BL21(DE3))
- Expression plasmid(s) containing the gene for the target NADH-dependent enzyme (e.g., 2,3-butanediol dehydrogenase, bdh) and the gene for FDH (e.g., from Candida boidinii).
- Substrate (e.g., Diacetyl)
- Cosubstrate (Sodium formate)
- LB broth and appropriate antibiotics
- Inducer (e.g., IPTG)
Procedure:
Strain Construction:
- Clone the gene encoding the target NADH-dependent enzyme (e.g., bdh) and the gene for FDH (fdh) into a suitable expression vector, such as a pETDuet system, to create a co-expression construct (pETDuet-bdhfdh).
- Transform the constructed plasmid into the E. coli expression host.
Cultivation and Bioconversion:
- Inoculate the recombinant strain into a shake flask containing LB medium with the appropriate antibiotic.
- Grow the culture to the mid-exponential phase (OD600 ≈ 0.6-0.8).
- Induce protein expression by adding a suitable concentration of IPTG (e.g., 0.1-0.5 mM).
- Continue incubation for several hours (e.g., 4-6 h) to allow enzyme production.
- Harvest cells by centrifugation and resuspend in a bioconversion buffer containing the substrate (e.g., 20 g/L diacetyl) and the cosubstrate for regeneration (e.g., 1-2 M sodium formate). The high concentration of formate drives the regeneration cycle.
- Incubate the cell suspension with shaking at an optimal temperature (e.g., 30-37°C). Monitor the reaction over time by sampling.
Process Monitoring and Analysis:
- Metabolite Analysis: Use HPLC or GC to quantify the consumption of the substrate (diacetyl) and the formation of the target product ((2S,3S)-2,3-butanediol) and any byproducts (e.g., acetoin).
- Cofactor Analysis: Quench samples at different time points using a validated method (e.g., fast filtration for E. coli). Extract intracellular metabolites using the optimized solvent (e.g., acetonitrile:methanol:water with buffer) and analyze NADH and NAD+ levels using the LC/MS method described in Section 3 [63].
- pH Control: During bioconversion, the consumption of formate may cause the pH to rise. Monitor pH and maintain it at the optimum (e.g., pH 7.0) by adding HCl as needed.
Visualizing Cofactor Engineering Strategies and Workflows
The following diagram illustrates the core conceptual framework and primary strategies employed in cofactor engineering to enhance metabolic flux.
Cofactor Engineering Conceptual Framework
The experimental workflow for implementing and validating a cofactor engineering strategy, from strain construction to final analysis, is outlined below.
Experimental Workflow for Cofactor Engineering
Metabolic engineering is a discipline that modifies and optimizes metabolic pathways, primarily in microorganisms, to enable the production of valuable compounds such as pharmaceuticals, fuels, and fine chemicals [2] [24]. For beginners in metabolic engineering research, it is crucial to understand that a central challenge in this field involves overcoming the inherent regulation of microbial metabolism to redirect carbon flux toward desired products [1]. A fundamental aspect of this process is precursor optimization—the strategic engineering of central carbon metabolism (CCM) to ensure adequate supply of key metabolic building blocks [66].
Among the most critical precursors for biosynthetic pathways are phosphoenolpyruvate (PEP) and erythrose-4-phosphate (E4P), which serve as direct carbon skeletons entering the shikimate pathway for aromatic amino acid synthesis and numerous valuable derivatives [67] [66]. These metabolites lie at the heart of cellular metabolism, connecting glycolysis, the tricarboxylic acid (TCA) cycle, and the pentose phosphate pathway (PPP) [68]. However, in native microbial systems, only a small fraction (typically less than 2%) of carbon flux is directed toward the shikimate pathway, with the majority channeled toward energy production and biomass formation [67]. This technical guide provides a comprehensive framework for optimizing E4P, PEP, and central metabolite balancing within the broader context of metabolic engineering for therapeutic compound production, offering detailed methodologies and data analysis tools for researchers and drug development professionals.
Theoretical Foundations: The Metabolic Node
The PEP-Pyruvate-Oxaloacetate Node
The PEP-pyruvate-oxaloacetate (PPO) node serves as a critical metabolic junction connecting glycolysis, the TCA cycle, and various biosynthetic pathways [68]. These three metabolites form the core of a network involving at least eleven different types of enzymes, creating remarkable variation across different organisms. PEP contains the highest-energy phosphate bond of all known natural organo-phosphates and serves as a precursor for aromatic amino acids [68]. Pyruvate functions as a precursor for alanine, valine, leucine, isoleucine, and isoleucine, while oxaloacetate is part of the TCA cycle and functions as a precursor for aspartate and nucleotides [68].
The following diagram illustrates the complex interconnections at the PPO node:
PPO Node Metabolic Interconversions. This diagram illustrates the principal enzymes and metabolic flows connecting phosphoenolpyruvate (PEP), pyruvate, and oxaloacetate. Key enzymes include pyruvate kinase (PK), PEP carboxylase (Ppc), PEP carboxykinase (Pck), pyruvate carboxylase (Pyc), and malic enzyme (Mez). The PPO node integrates carbon flow from glycolysis, the pentose phosphate pathway (PPP), and the tricarboxylic acid (TCA) cycle.
E4P in Biosynthetic Pathways
Erythrose-4-phosphate (E4P) is generated through the pentose phosphate pathway and serves as an essential precursor for the shikimate pathway, which leads to the synthesis of aromatic amino acids (phenylalanine, tyrosine, tryptophan) and numerous aromatic compounds with pharmaceutical value [67] [66]. The availability of E4P often limits flux through the shikimate pathway, making its optimization a critical metabolic engineering target [66].
Metabolic Engineering Strategies for Precursor Optimization
Enhancing PEP Availability
Multiple strategic approaches have been successfully employed to increase intracellular PEP pools for enhanced biosynthesis of aromatic compounds:
Weakening Competing Pathways: Deleting or downregulating genes encoding pyruvate kinase (pykA) reduces the flux of PEP to pyruvate, thereby increasing PEP availability for the shikimate pathway [67]. Studies in E. coli have demonstrated that pykA deletion increases tryptophan production.
Enhancing PEP Synthesis: Overexpressing phosphoenolpyruvate synthase (ppsA) reinforces the conversion of pyruvate to PEP [69]. This strategy was successfully applied in dopamine production strains, increasing precursor availability.
Utilizing Alternative Pathways: Introducing heterologous pathways like the phosphoketolase (PHK) pathway can redirect carbon flux from fructose-6-phosphate and xylulose-5-phosphate directly to acetyl-CoA, indirectly increasing E4P availability by shifting metabolic flux toward the PPP [66].
Balancing E4P Supply
PPP Activation: Overexpression of transketolase (tktA) enhances the metabolic flux through the PPP, directly increasing E4P production [69] [66]. In E. coli, tktA overexpression is a established strategy to improve aromatic compound production.
PHK Pathway Integration: The heterologous phosphoketolase pathway catalyzes the conversion of fructose-6-phosphate to acetyl-phosphate, decreasing glycolytic flux while indirectly increasing PPP flux and E4P accumulation [66]. This approach has increased tyrosol production by 135-fold in engineered yeast.
Coordinated Central Metabolism Engineering
Successful precursor optimization requires systems-level approaches that consider the interconnected nature of central metabolism:
Energy and Redox Balancing: Modifications to the PPO node affect cellular energetics. Strategies include introducing NADP+-dependent PDH pathways in S. cerevisiae to increase acetyl-CoA production while conserving ATP [66].
Dynamic Regulation: Implementing dynamic control systems allows cells to automatically adjust pathway expression in response to metabolic status, improving both production and growth [1].
Experimental Protocols and Methodologies
Protocol: Engineering PEP Availability in E. coli
This protocol outlines the key steps for modifying the PPO node to enhance PEP availability for aromatic compound production, based on established metabolic engineering approaches [67]:
Competent Cell Preparation: Grow E. coli W3110 in LB medium at 37°C to mid-exponential phase (OD600 ≈ 0.4-0.6). Harvest cells by centrifugation at 4,000 × g for 10 min at 4°C. Wash twice with sterile ice-cold 10% glycerol and concentrate 100-fold.
Gene Deletion Using CRISPR-Cas9:
- Design sgRNAs targeting pykA and ppc genes using appropriate online tools.
- Transform competent cells with pREDCas9 plasmid and respective pGRB-sgRNA plasmids via electroporation (2.5 kV, 5 ms).
- Select transformants on LB agar with spectinomycin (50 μg/mL) and ampicillin (100 μg/mL).
- Verify deletions by colony PCR and DNA sequencing.
Gene Integration:
- Amplify pck gene with native promoter using primer pairs containing 50-bp homology arms.
- Transform deletion strain with PCR product using λ Red recombinase system.
- Select integrants on LB agar with kanamycin (25 μg/mL).
Fermentation Validation:
- Inoculate single colonies into 5 mL LB medium and grow overnight at 37°C.
- Transfer to 500 mL fermenters with defined mineral medium containing 20 g/L glucose.
- Maintain pH at 6.8 with NH4OH, dissolved oxygen at 30%, and temperature at 37°C.
- Monitor metabolite production via HPLC and assess PEP availability through enzymatic assays.
Protocol: Enhancing E4P Supply via the PHK Pathway
This protocol describes the introduction of the heterologous phosphoketolase pathway in S. cerevisiae to enhance E4P availability [66]:
Pathway Construction:
- Codon-optimize phosphoketolase (PK) and phosphotransacetylase (PTA) genes from Aspergillus nidulans for S. cerevisiae.
- Clone genes into pRS423 vector under control of constitutive TEF1 promoter.
- Transform S. cerevisiae using lithium acetate method with single-stranded carrier DNA.
- Select transformants on synthetic complete medium lacking histidine.
Flux Analysis:
- Grow engineered strains in minimal medium with 20 g/L glucose.
- At mid-exponential phase, harvest cells and perform metabolomic analysis via LC-MS.
- Quantify E4P levels using isotopic labeling with [1-13C]glucose and measure 13C enrichment via GC-MS.
Strain Performance Validation:
- Measure target product (e.g., p-hydroxycinnamic acid) production via HPLC.
- Compare yields between PHK-engineered strains and control strains.
- Assess growth characteristics to ensure engineering does not impose significant fitness costs.
Quantitative Data Analysis and Comparison
Impact of Metabolic Engineering Strategies on Product Yields
Table 1: Comparison of metabolic engineering approaches for precursor optimization and their effects on product yields
| Target Product | Host Organism | Engineering Strategy | Precursor Enhanced | Titer (g/L) | Yield (g/g glucose) | Reference |
|---|---|---|---|---|---|---|
| L-Tryptophan | E. coli TRP03 | Downregulation of pykA, ppc | PEP | 35.0 | Not specified | [67] |
| L-Tryptophan | E. coli TRP07 | Enhanced PEP conversion, upregulated TCA | PEP | 49.0 | 0.186 | [67] |
| Dopamine | E. coli DA-29 | Pathway optimization, cofactor balancing | PEP/E4P (via tyrosine) | 22.58 | Not specified | [69] |
| Mandelic Acid | E. coli | Shikimate pathway enhancement | PEP/E4P | 9.58 | Not specified | [55] |
| p-Hydroxycinnamic Acid | S. cerevisiae | PHK pathway introduction | E4P | 12.5 | 0.155 | [66] |
| Tyrosol/Salidroside | S. cerevisiae | PHK pathway, flux rearrangement | E4P | >10.0 | Not specified | [66] |
Key Enzyme Modifications and Their Effects
Table 2: Key enzyme targets for precursor optimization and their metabolic effects
| Enzyme Target | Gene | Modification | Metabolic Effect | Impact on Precursors | |
|---|---|---|---|---|---|
| Pyruvate kinase | pykA | Deletion/weakening | Reduces PEP to pyruvate flux | Increases PEP availability | [67] |
| PEP carboxylase | ppc | Deletion/weakening | Reduces PEP to OAA conversion | Increases PEP for shikimate | [67] |
| PEP carboxykinase | pck | Overexpression | Enhances OAA to PEP conversion | Increases PEP pool | [67] |
| Transketolase | tktA | Overexpression | Enhances PPP flux | Increases E4P production | [69] |
| Phosphoketolase | xfpk | Heterologous expression | Diverts F6P to acetyl-P | Indirectly increases E4P | [66] |
| Pyruvate carboxylase | pyc | Overexpression | Enhances pyruvate to OAA | Increases TCA intermediates | [67] |
Visualization of Engineering Workflows
Strategic Workflow for Precursor Optimization
The following diagram outlines a systematic approach to optimizing precursor supply in metabolic engineering projects:
Precursor Optimization Workflow. This engineering workflow outlines the systematic process for optimizing precursor supply, from initial pathway analysis through iterative strain improvement. Key phases include comprehensive pathway analysis, strategic selection of engineering interventions, strain construction using modern genetic tools, performance validation, and iterative refinement.
Integrated Metabolic Engineering Approach
Integrated Metabolic Engineering for Precursor Balancing. This diagram illustrates the key metabolic engineering interventions for optimizing E4P and PEP supplies. Strategic modifications include upregulating the pentose phosphate pathway (PPP) via transketolase (tktA) overexpression to enhance E4P, downregulating pyruvate kinase (pykA) to conserve PEP, and introducing heterologous pathways like phosphoketolase (PHK) and PEP synthase (PPS) to redirect carbon flux.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential research reagents and materials for precursor optimization studies
| Reagent/Material | Specifications | Application | Example Use Case |
|---|---|---|---|
| E. coli W3110 | Wild-type strain (ATCC 27325) | Chassis organism | Starting point for genome modifications [67] |
| pREDCas9 Plasmid | SpecR, expresses Cas9 and λ Red recombinase | CRISPR-Cas9 genome editing | Gene deletions (pykA, ppc) [67] |
| pGRB Plasmid | AmpR, sgRNA expression vector | CRISPR guide RNA delivery | Target-specific gene editing [67] |
| Primer STAR HS DNA Polymerase | High-fidelity PCR enzyme | Gene amplification | Amplification of homology arms [67] |
| ClonExpress II Kit | Recombinase-based cloning | Vector construction | One-step plasmid assembly [67] |
| ZYM-5052 Medium | Auto-induction medium | Protein expression | Inducing pathway enzyme expression [55] |
| HPLC System | Reverse-phase C18 column | Metabolite quantification | Tryptophan, dopamine analysis [67] [69] |
| GC-MS System | With isotopic capability | Metabolic flux analysis | 13C-labeling studies for E4P/PEP flux [66] |
| Rauvotetraphylline E | Rauvotetraphylline E, MF:C20H18N2O3, MW:334.4 g/mol | Chemical Reagent | Bench Chemicals |
| 1-O-Methyljatamanin D | 1-O-Methyljatamanin D, MF:C11H16O4, MW:212.24 g/mol | Chemical Reagent | Bench Chemicals |
Optimizing the balance between E4P, PEP, and central metabolites represents a cornerstone of successful metabolic engineering for pharmaceutical production. As demonstrated throughout this technical guide, systematic approaches that combine targeted gene modifications, heterologous pathway implementation, and systems-level metabolic balancing can dramatically improve precursor availability and product yields. The experimental protocols, quantitative data, and visualization tools provided here offer researchers and drug development professionals a comprehensive framework for designing and implementing precursor optimization strategies. Future advances in dynamic regulation, enzyme engineering, and computational modeling will further enhance our ability to precisely control metabolic fluxes for the efficient production of valuable therapeutic compounds.
Metabolic engineering represents a cornerstone of modern industrial biotechnology, leveraging advanced genetic tools to reprogram microbial cellular machinery for the sustainable production of valuable chemicals. This field sits at the intersection of synthetic biology, systems biology, and biochemical engineering, employing a systematic Design-Build-Test-Learn (DBTL) cycle to optimize microbial strains for enhanced production of target compounds [70]. In pharmaceutical applications, metabolic engineering has transformed from a niche research area into an essential platform for producing both small-molecule drug precursors and complex biotherapeutics, offering alternatives to traditional chemical synthesis that are often more sustainable, selective, and compatible with green chemistry principles.
The fundamental premise of metabolic engineering involves modifying an organism's metabolic pathways to redirect flux toward desired products while minimizing competing reactions. This requires a deep understanding of cellular metabolism, sophisticated genetic tools for pathway manipulation, and analytical methods for assessing production outcomes. For pharmaceutical applications, this approach enables the biosynthesis of complex molecules that are challenging to produce economically through chemical synthesis alone. The global metabolic engineering market, valued at $10.2 billion in 2025 and projected to reach $21.4 billion by 2033 with a CAGR of 9.60%, reflects the growing industrial adoption of these technologies, particularly in the pharmaceutical sector [71].
This technical guide explores two critical applications of metabolic engineering in drug development: the production of drug precursors and the creation of innovative biotherapeutics. Through specific case studies, methodological protocols, and practical toolkits, we provide researchers with both the conceptual framework and technical details necessary to implement these approaches in their own work, with content specifically framed for beginners in metabolic engineering research.
Metabolic Engineering of Drug Precursors
Drug precursors are chemical compounds that serve as intermediate substances in the synthesis of active pharmaceutical ingredients (APIs). Their controlled production is essential for both legitimate pharmaceutical manufacturing and prevention of diversion for illicit drug synthesis. Metabolic engineering offers powerful approaches to produce these precursors through sustainable microbial processes.
Case Study: De Novo Biosynthesis of Mandelic Acid
Mandelic acid (MA) is a valuable α-hydroxy acid with applications in pharmaceuticals, cosmetics, and fine chemicals. It serves as a key intermediate in the synthesis of antibiotics, disinfectants, preservatives, and optically pure drugs [55]. A recent study demonstrates the successful metabolic engineering of Escherichia coli for de novo MA biosynthesis through integrated enzyme screening, metabolic flux optimization, and pathway regulation.
The engineered strain incorporated an efficient hydroxymandelate synthase (HMAS) homolog from Actinosynnema mirum for MA synthesis, with enhancements to the shikimate pathway to improve supply of the precursors erythrose-4-phosphate (E4P) and phosphoenolpyruvate (PEP). Researchers employed CRISPR interference (CRISPRi) to repress competing pathways and redirect flux toward MA production [55]. This systematic approach resulted in an MA titer of 9.58 g/L under high-cell-density cultivation conditions—the highest reported for microbial production—demonstrating the potential for industrial-scale application.
Table 1: Key Performance Metrics for Microbial Mandelic Acid Production
| Strain/Approach | Titer | Productivity | Yield | Scale |
|---|---|---|---|---|
| Engineered E. coli (this study) | 9.58 g/L | 0.27 g/L/h | 0.24 g/g glucose | 5 L bioreactor |
| E. coli NST74 (previous study) | 0.76 g/L | N/R | N/R | Shake flask |
| S. cerevisiae (engineered) | 0.236 g/L | N/R | N/R | Shake flask |
| Chemical synthesis | N/A | High | Variable | Industrial |
Regulatory Considerations for Precursor Production
The production of drug precursors through metabolic engineering must account for regulatory frameworks governing these compounds. The European Union, for instance, maintains strict controls on substances that can be used as precursors for illicit drugs. Recent regulatory updates include the addition of 4-piperidone and 1-boc-4-piperidone to Category 1 controlled precursors through Delegated Regulation (EU) 2025/1475, as these compounds are key precursors in the synthesis of fentanyl and its analogues [72].
Economic operators producing such precursors must obtain appropriate licenses, maintain detailed records of transactions, implement suspicious order monitoring, and ensure proper labeling—requirements that extend to biotechnologically produced precursors. These regulatory considerations must be integrated into process development from the earliest stages to ensure compliance while maintaining production efficiency.
Metabolic Engineering of Biotherapeutics
Biotherapeutics represent a rapidly expanding category of pharmaceuticals that includes peptides, proteins, antibodies, and other complex molecules produced through biological systems. Metabolic engineering approaches are increasingly applied to optimize the production of these therapeutics and to create novel treatment modalities.
Case Study: Oral Delivery of Peptide Therapeutics
Peptide therapeutics such as GLP-1 receptor agonists represent an important class of biotherapeutics for treating diabetes, obesity, and other metabolic disorders. However, their delivery has been hampered by the need for injection, resulting in patient compliance challenges. A recent collaboration between BioMed X and Novo Nordisk aims to address this limitation through innovative formulation approaches for oral peptide delivery [73].
The research initiative, titled "Prolonged Retention of Oral Peptide Formulations in the Gut," focuses on developing novel oral formulation technologies that achieve site-specific, prolonged retention of tablets or capsules within the lower small intestine. This approach aims to significantly improve the absorption and bioavailability of peptide-based therapeutics by overcoming limitations of conventional oral peptide formulations, including low intestinal permeability and rapid gastrointestinal transit [73]. The key innovation involves creating dosage forms that remain in the absorption zone longer, allowing continuous release and efficient absorption without compromising gastrointestinal safety or motility.
Case Study: Engineering Microbial Consortia for Complex Molecule Production
Advanced metabolic engineering approaches are now moving beyond single-strain optimization to engineered microbial communities that can division metabolic labor for complex biotherapeutics production. Research at Imperial College London demonstrates how engineered yeast communities can improve bioproduction outcomes through syntrophic relationships [74] [75].
By systematically culturing combinations of auxotrophic yeast mutants, researchers identified pairs that form obligatory cross-feeding relationships, some of which prove stable over time and enable division of metabolic labor for biotechnological applications [75]. This approach allows for more complex biosynthetic pathways to be divided between specialized strains, potentially improving overall productivity and stability compared to single-strain approaches.
The Imperial College team implemented the Reshape Microbiology platform to screen for complex metabolite interactions with fluorescence biosensors, enabling them to scale up their screenings and quantitatively benchmark complex metabolite interactions of various microbial clones [74]. This high-throughput approach allowed them to screen 20x more microbial clones, significantly accelerating their mapping of metabolite interactions and enhancing their understanding of metabolite production, release, and diffusion in yeast.
Table 2: Comparison of Biotherapeutic Production Platforms
| Platform | Advantages | Limitations | Therapeutic Examples |
|---|---|---|---|
| Oral peptide formulations | Improved patient compliance, non-invasive | Low bioavailability, complex formulation | GLP-1 receptor agonists [73] |
| Engineered microbial consortia | Division of labor, stability, complex pathways | Population control challenges, operational complexity | Antibiotics, natural products [74] [75] |
| Single-strain engineering | Well-established tools, easier control | Metabolic burden, limited pathway complexity | Mandelic acid, polymers [55] |
Experimental Protocols and Methodologies
Successful implementation of metabolic engineering strategies requires robust experimental protocols spanning genetic engineering, cultivation, and analytical techniques.
Protocol: CRISPRi-Mediated Pathway Regulation
The mandelic acid case study [55] provides a detailed methodology for implementing CRISPR interference (CRISPRi) for metabolic flux control:
Strain and Plasmid Construction:
- Utilize common E. coli strains (DH5α for cloning, BW25113 for production)
- Employ plasmids pYB1a, pSB1c, and dCas9 from existing laboratory collections
- Assemble all plasmids using Gibson Assembly method with Gibson Assembly Cloning Kit (NEB)
- Introduce plasmids into E. coli competent cells via heat shock transformation
Culture Conditions:
- Inoculate single colonies into 5 mL LB medium with appropriate antibiotics
- Incubate overnight at 37°C with shaking at 220 rpm
- For induction, transfer 1 mL of overnight culture into 100 mL ZYM-5052 medium with 0.2% arabinose
- Cultivate at 30°C with shaking at 220 rpm for 12 hours
- For CRISPRi induction, add IPTG to final concentration of 1 mM
Analytical Methods:
- Monitor cell growth by measuring OD600 using UV-visible spectrophotometer
- For whole-cell biocatalysis, harvest cells by centrifugation at 4200 rpm for 10 minutes
- Analyze metabolite production using appropriate chromatographic methods
Protocol: High-Throughput Screening of Microbial Clones
The Imperial College case study [74] outlines a protocol for high-throughput screening of microbial clones for metabolic engineering:
Strain Engineering:
- Implement fluorescent biosensors for metabolites of interest
- Engineer microbial libraries with pathway variations
Cultivation and Imaging:
- Cultivate clones in multi-well plates under controlled conditions
- Use the Reshape Imaging System for time-lapse imaging in multiple fluorescent spectra
- Track microbial growth and metabolite production simultaneously
Data Analysis:
- Apply quantitative analysis to fluorescence data
- Benchmark complex metabolite interactions
- Map metabolite production, release, and diffusion patterns
This approach generates quantitative data from complex plate-based assays, enabling more accurate modeling of metabolic interactions and identification of superior production strains.
Visualization of Metabolic Engineering Workflows
The following diagrams illustrate key metabolic engineering workflows and pathway relationships described in this guide.
Mandelic Acid Biosynthesis Pathway
High-Throughput Screening Workflow
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of metabolic engineering strategies requires specific reagents, tools, and platforms. The following table summarizes key resources referenced in the case studies.
Table 3: Essential Research Reagents and Tools for Metabolic Engineering
| Tool/Reagent | Function | Example Application | Source/Reference |
|---|---|---|---|
| CRISPR-dCas9 System | Gene repression without cleavage | Downregulation of competing metabolic pathways | [55] |
| Hydroxymandelate Synthase (HMAS) | Key enzyme in MA biosynthesis | Conversion of phenylpyruvate to mandelic acid | [55] |
| Reshape Imaging System | High-throughput fluorescence imaging | Screening microbial clones for metabolite production | [74] |
| Gibson Assembly | Molecular cloning method | Plasmid construction for pathway engineering | [55] |
| ZYM-5052 Medium | Defined cultivation medium | High-cell-density cultivation of engineered strains | [55] |
| Fluorescent Biosensors | Metabolite detection and quantification | Real-time monitoring of metabolic fluxes | [74] |
| Corysamine chloride | Corysamine chloride, MF:C20H16ClNO4, MW:369.8 g/mol | Chemical Reagent | Bench Chemicals |
| Vinaginsenoside R8 | Vinaginsenoside R8, CAS:93376-72-8, MF:C48H82O19, MW:963.2 g/mol | Chemical Reagent | Bench Chemicals |
Metabolic engineering has established itself as a powerful platform for the production of drug precursors and biotherapeutics, offering sustainable, efficient alternatives to traditional chemical synthesis. As demonstrated through the case studies in this guide, successful implementation requires integrated approaches combining enzyme engineering, pathway optimization, and advanced cultivation strategies.
Future developments in the field will likely focus on several key areas: the increasing integration of machine learning and artificial intelligence for pathway prediction and optimization [70]; the application of more sophisticated genome editing tools beyond CRISPRi for multiplexed engineering [75]; and the development of increasingly complex microbial consortia for division of metabolic labor. Additionally, the continued advancement of high-throughput screening technologies, such as the platform implemented at Imperial College [74], will accelerate the design-build-test-learn cycle that underpins metabolic engineering.
For beginners in the field, understanding both the technical methodologies and strategic frameworks presented in this guide provides a foundation for engaging with this rapidly evolving discipline. The integration of engineering principles with biological systems continues to transform pharmaceutical production, enabling more sustainable, efficient, and innovative approaches to drug development.
The transition toward a sustainable bioeconomy necessitates a paradigm shift from petroleum-based feedstocks to alternative, renewable resources. This whitepaper provides an in-depth technical guide for researchers and scientists on leveraging diverse, often underutilized, organic feedstocks for the production of value-added chemicals and fuels through metabolic engineering. Framed within the context of foundational metabolic engineering research, we detail the critical properties of feedstocks, computational and experimental strategies for pathway design, and provide standardized protocols for feedstock evaluation and bioconversion. The integration of these sustainable substrate strategies is essential for developing efficient microbial cell factories, reducing production costs, and advancing cleaner production processes in the pharmaceutical and chemical industries.
A wide variety of wasted or underutilized organic feedstocks are available to build a sustainable future bioeconomy, ranging from crop residues and food processor waste to municipal solid waste [76]. Leveraging these materials is both high-risk and high-reward. While converting mixed, variable, and/or highly contaminated feedstocks can pose significant engineering and economic challenges, their successful bioconversion can divert waste from landfills, reduce fugitive methane emissions, and enable more responsible resource management [76]. For metabolic engineers, especially those new to the field, understanding how to match these diverse feedstocks to appropriate microbial hosts and biochemical conversion processes is a critical first step. The success of these endeavors hinges on the creation of less expensive processes, particularly regarding substrates, which can account for 10–30% of total production costs [77]. By linking production with waste streams, processes can minimize pollution while balancing overall costs, thereby contributing to a circular bioeconomy [77].
Characterizing Alternative Feedstocks
The appropriate selection and characterization of a feedstock are fundamental to designing a successful bioconversion process. Key properties determine the feasibility of downstream processing and the choice of microbial platform.
Critical Feedstock Properties
- Moisture Content: High moisture content (e.g., in fruit and vegetable waste) can favor biochemical conversion without pre-treatment but may increase transportation costs and risk of microbial spoilage.
- Composition: The relative proportions of carbohydrates (cellulose, hemicellulose, starch), lignin, lipids, and proteins dictate the metabolic pathways required in the microbial host and the potential product profile.
- Contamination Level: Feedstocks like municipal solid waste may contain heavy metals or inhibitory compounds that can be toxic to microbial catalysts, necessitating pre-treatment or robust engineering of the host [76].
- Physical Properties: Particle size, density, and viscosity impact handling, mixing, and mass transfer within bioreactors.
Feedstock Categories and Suitability
The table below summarizes major categories of alternative feedstocks and their suitability for different conversion pathways.
Table 1: Characterization of Alternative Feedstocks for Bioconversion
| Feedstock Category | Specific Examples | Key Characteristics | Preferred Conversion Pathway | Compatible Microbial Hosts |
|---|---|---|---|---|
| Agro-Industrial Waste | Cassava wastewater, sugarcane bagasse, corn steep liquor [77] | Rich in starch or other fermentable sugars, often seasonal | Biochemical conversion (e.g., fermentation) [76] | Bacillus subtilis, Bacillus amyloliquefaciens [77] |
| Fruit & Vegetable Waste | Cashew apple juice, banana peel, orange peel [77] | High moisture and sugar content, rapidly degradable | Biochemical conversion | Pseudomonas aeruginosa, Acinetobacter calcoaceticus [77] |
| Lignocellulosic Biomass | Crop residues (e.g., straw, husks), dedicated energy crops | High cellulose/hemicellulose content, rigid structure requires pre-treatment | Gasification or Pyrolysis [76] | Rhodococcus |
| Mixed/Contaminated Waste | Municipal Solid Waste [77] | Highly variable, potentially contaminated with inhibitors | Thermochemical conversion (e.g., gasification, pyrolysis) [76] | Robust hosts or consortia |
Metabolic Engineering Strategies for Feedstock Utilization
Constructing efficient cell factories requires the rational design of metabolic pathways to efficiently convert heterogeneous feedstocks into target products. Pathway yield (YP), the amount of product formed from a substrate, is a crucial metric for designing efficient and atom-economical processes [4].
Computational Pathway Design
Recent advances have enabled a more systematic approach to pathway design. The Quantitative Heterologous Pathway Design algorithm (QHEPath) was developed to evaluate biosynthetic scenarios and identify reactions that can break the native stoichiometric yield limit of a host [4]. In a massive systematic calculation, over 70% of product pathway yields for 300 different chemicals across 5 industrial organisms were found to be improvable by introducing appropriate heterologous reactions [4]. This research identified thirteen core engineering strategies, categorized as carbon-conserving and energy-conserving, with five strategies effective for over 100 different products [4]. For beginners, this underscores that yield improvement is not an ad-hoc process but can be approached systematically.
The following diagram illustrates the computational workflow for designing and validating high-yield pathways using these tools.
Common Engineering Strategies
Key strategies identified for breaking yield barriers include:
- Introducing non-oxidative glycolysis (NOG): This pathway can enhance yields of products like farnesene and poly(3-hydroxybutyrate) (PHB) by conserving carbon atoms that would otherwise be lost as COâ‚‚ [4].
- Reducing equivalent balancing: Engineering pathways to optimize the use of cofactors (NADH, NADPH) can significantly improve flux toward the desired product.
- ATP conservation: Rerouting metabolism to reduce ATP consumption in non-essential pathways frees up energy for product synthesis.
Experimental Protocols for Feedstock Evaluation
For researchers embarking on experimental validation, standardized protocols are essential. Below is a detailed methodology for screening and optimizing biosurfactant production from alternative feedstocks, adaptable for other products.
Protocol: Screening Feedstocks for Biosurfactant Production
Primary Objective: To evaluate the potential of various alternative feedstocks (e.g., banana peel, cassava wastewater) to support the growth of a microbial host (Pseudomonas aeruginosa) and the production of a target biosurfactant (rhamnolipids) [77].
Study Design: This is a prospective, controlled, open-label, laboratory-scale study.
Materials and Reagents Table 2: Research Reagent Solutions for Feedstock Screening
| Reagent/Material | Function in the Experiment | Example Source |
|---|---|---|
| Alternative Feedstocks (e.g., fruit peels, agro-waste) | Acts as the sole carbon source for microbial growth and product synthesis. | Local food processing facilities, agricultural sources. |
| Mineral Salt Medium (MSM) | Provides essential nutrients (N, P, K, trace metals) while forcing the microbe to utilize the feedstock. | Standard laboratory preparation. |
| Reference Strain (e.g., Pseudomonas aeruginosa ATCC 10145) | Model biosurfactant-producing organism. | Culture collection (e.g., ATCC). |
| Analytical Standards (e.g., purified rhamnolipids) | Used for calibration and quantification in HPLC or GC-MS analysis. | Commercial chemical suppliers. |
Inclusion/Exclusion Criteria for Feedstocks:
- Inclusion: All tested feedstocks must be of biological origin and represent a underutilized or waste stream.
- Exclusion: Feedstocks with known high levels of persistent toxins (e.g., pesticides, heavy metals) that cannot be easily removed via pre-treatment.
Experimental Workflow:
- Feedstock Pre-treatment: Dry solid feedstocks (e.g., banana peel) at 60°C for 48 hours and grind to a fine powder (<1 mm). For liquid waste (e.g., cassava wastewater), filter to remove large particulates.
- Media Preparation: Prepare the Mineral Salt Medium (MSM) and supplement it with the pre-treated feedstock as the sole carbon source. A concentration of 2% (w/v or v/v) is a standard starting point.
- Inoculation and Cultivation: Inoculate 100 mL of feedstock-supplemented MSM in a 250 mL Erlenmeyer flask with a 1% (v/v) inoculum of an overnight culture of P. aeruginosa. Incubate at 30°C with shaking at 200 rpm for 96 hours.
- Sample Harvesting: Aseptically withdraw samples at 24-hour intervals for analysis.
The following flowchart visualizes this multi-step experimental process.
Endpoint Analysis:
- Biomass Growth: Measure optical density at 600 nm (OD₆₀₀).
- Biosurfactant Production:
- Qualitative: Oil spread test and drop collapse test.
- Quantitative: Measure surface tension of the cell-free broth using a tensiometer. Quantify rhamnolipid concentration using high-performance liquid chromatography (HPLC).
- Feedstock Utilization: Measure residual sugar content or chemical oxygen demand (COD) in the spent broth.
Statistical Analysis: Perform all experiments in triplicate. Use analysis of variance (ANOVA) with post-hoc tests to determine significant differences in biosurfactant yield between different feedstock types.
The Scientist's Toolkit
This section details essential resources and databases that are indispensable for research in metabolic engineering and feedstock utilization.
Table 3: Essential Resources for Metabolic Engineering Research
| Tool / Database Name | Type | Primary Function in Research |
|---|---|---|
| BiGG Models [4] | Database | A knowledgebase of curated, genome-scale metabolic models used for in-silico simulation of metabolic networks. |
| QHEPath Web Server [4] | Computational Tool | A user-friendly web server to quantitatively calculate and visualize product yields and design heterologous pathways to break yield limits. |
| Springer Nature Experiments [78] | Protocol Repository | A repository of peer-reviewed, reproducible laboratory protocols and methods for the life sciences. |
| Protocols.io [78] | Protocol Repository | An open-access repository for sharing and collaborating on scientific methods, featuring group discussion features. |
| JoVE Unlimited [78] | Video Repository | A complete video library showcasing research experiments and techniques, aiding in understanding complex procedures. |
| Brachyoside B | Brachyoside B, CAS:86764-12-7, MF:C36H60O10, MW:652.9 g/mol | Chemical Reagent |
| Lobetyol | Lobetyol | Lobetyol is a natural product for research into anti-cancer and anti-inflammatory pathways. This product is for Research Use Only. Not for human or veterinary diagnostic or therapeutic use. |
The strategic utilization of alternative feedstocks represents a cornerstone of the future sustainable bioeconomy. Success in this domain requires an integrated approach that combines a deep understanding of feedstock properties with advanced metabolic engineering techniques. As detailed in this guide, computational tools like the QHEPath algorithm and CSMN models enable the rational design of high-yield pathways, while robust experimental protocols allow for their practical validation. For researchers, mastering the interplay between feedstock selection, pathway design, and bioprocess optimization is key to transforming waste streams into valuable products, thereby driving innovation in cleaner production and drug development. The journey from a raw, variable feedstock to a purified chemical is complex, but the tools and strategies outlined herein provide a foundational roadmap for this critical endeavor.
Overcoming Metabolic Challenges and Optimization Frameworks
In the pursuit of engineering robust microbial cell factories, metabolic engineers often encounter critical bottlenecks that limit titers, rates, and yields (TRY) of desired compounds. Two of the most pervasive challenges are substrate inhibition and flux imbalances, which consistently constrain bio-production efficiency across diverse host organisms and metabolic pathways [79] [80]. Substrate inhibition occurs when elevated concentrations of a substrate—whether an initial carbon source like glucose or an intermediate metabolite—paradoxically suppress enzymatic activity and cellular growth [79]. Flux imbalances emerge when mismatched reaction rates within engineered pathways cause metabolic traffic jams, leading to the accumulation of intermediate metabolites that often divert carbon toward byproducts or trigger regulatory toxicity [80]. For researchers entering metabolic engineering, understanding these bottlenecks is fundamental, as they represent recurring obstacles that must be systematically addressed to advance sustainable biomanufacturing and therapeutic production [79].
Substrate Inhibition: Mechanisms and Identification
Fundamental Mechanisms
Substrate inhibition manifests through several biochemical mechanisms. At high concentrations, substrate molecules may bind simultaneously to both the active site and a non-catalytic site on the enzyme, creating non-productive complexes that effectively reduce catalytic turnover [79]. Alternatively, excess substrate can promote the formation of dead-end complexes or interfere with essential cofactor regeneration cycles. In many industrial fermentation processes utilizing lignocellulosic hydrolysates, inhibitors such as furfural derivatives, phenolic compounds, and organic acids exacerbate these effects by synergistically impairing microbial growth and metabolism [79].
Experimental Detection and Analysis
Identifying substrate inhibition requires carefully designed experiments to characterize kinetic parameters and establish operational thresholds. The following protocol outlines a systematic approach for quantifying substrate inhibition:
Protocol 1: Assessing Substrate Inhibition in Microbial Cultures
Culture Preparation: Inoculate duplicate series of shake flasks containing minimal medium with varying concentrations of the target substrate (e.g., 5, 10, 20, 50, 100 g/L glucose). Use identical inoculum size and cultivation conditions (pH, temperature, agitation).
Growth Kinetics Monitoring: Measure optical density (OD600) at regular intervals (e.g., every 2 hours) throughout the exponential growth phase. Calculate maximum growth rates (μmax) for each substrate concentration.
Metabolite Profiling: Collect supernatant samples at mid-exponential phase and analyze via HPLC or GC-MS to quantify substrate consumption rates and metabolic byproducts (e.g., acetate, lactate, ethanol).
Inhibitor Threshold Determination: Identify the substrate concentration at which μmax decreases by ≥15% compared to the maximum observed rate. This represents the inhibition threshold.
Data Analysis: Fit growth kinetics data to substrate inhibition models (e.g., Haldane equation) to quantify inhibition constants (Ki).
Table 1: Characteristic Inhibition Thresholds for Common Substrates
| Substrate | Microbial Host | Inhibition Threshold | Observed Impact |
|---|---|---|---|
| Glucose | E. coli | 50-100 g/L [79] | Reduced growth rate, acetate accumulation |
| Xylose | S. cerevisiae | 30-50 g/L [79] | Extended lag phase, reduced yield |
| Lignocellulosic hydrolysate | Various | Varies by pretreatment [79] | Complete growth arrest at high concentrations |
| Lactic acid | K. oxytoca | pH-dependent [79] | Growth inhibition, reduced productivity |
Flux Imbalances: Analysis and Engineering Solutions
Pathway Flux Principles
Flux imbalances occur when enzymatic capabilities within a pathway become mismatched, creating metabolic bottlenecks that reduce overall efficiency [80]. In engineered pathways, heterogeneous expression levels of biosynthetic enzymes often lead to intermediate metabolite accumulation, which can trigger feedback inhibition or shunt metabolic carbon toward competing pathways [79]. The tricarboxylic acid (TCA) cycle and its associated pathways for organic acid production (e.g., succinic, fumaric, malic acids) represent prime examples where precise flux balancing is essential for optimal performance [79].
Diagnostic Approaches
Several experimental methodologies enable quantitative assessment of flux distributions in engineered strains:
Protocol 2: Metabolic Flux Analysis Using Isotopic Tracers
Tracer Preparation: Prepare minimal medium with [1-13C] or [U-13C] labeled glucose as the sole carbon source.
Steady-State Cultivation: Grow engineered strains in controlled bioreactors under defined environmental conditions until metabolic steady state is achieved.
Sampling and Quenching: Rapidly collect cell aliquots and quench metabolism using cold methanol (-40°C).
Intracellular Metabolite Extraction: Employ appropriate extraction buffers to recover polar and non-polar metabolite fractions.
Mass Spectrometry Analysis: Utilize LC-MS or GC-MS to determine mass isotopomer distributions of central carbon metabolites.
Flux Calculation: Apply computational modeling (e.g., constraint-based reconstruction and analysis) to infer intracellular flux distributions from labeling patterns.
The diagnostic diagram below illustrates the logical workflow for identifying and addressing flux imbalances in engineered pathways:
Integrated Engineering Strategies
Advanced Intervention Approaches
Contemporary metabolic engineering employs sophisticated strategies to simultaneously address substrate inhibition and flux imbalances. Dynamic metabolic control represents a particularly powerful approach, enabling autonomous flux adjustment in response to metabolic status [80]. These systems typically incorporate biosensors that detect key metabolites or stress signals, coupled with genetic circuits that modulate expression of bottleneck enzymes. For example, substrate-responsive promoters can trigger expression of transporter proteins or detoxification enzymes when inhibitory concentrations are detected [80].
Table 2: Molecular Tools for Addressing Metabolic Bottlenecks
| Tool Category | Specific Examples | Application Context | Key Limitations |
|---|---|---|---|
| Gene Expression Tuners | Promoter libraries, RBS variants, CRISPRi [80] | Fine-tuning pathway enzyme levels | Limited dynamic range, context-dependent performance |
| Biosensor-Regulated Systems | Transcription factor-based biosensors, riboswitches [80] | Dynamic control in response to metabolites | Need for sensor characterization, cross-talk issues |
| Pathway Engineering | Byproduct knockout (Δldh, Δpdc) [79], alternative carbon routes [79] | Reducing carbon loss to competing pathways | Metabolic burden, reduced fitness |
| Enzyme Engineering | Site-directed mutagenesis, directed evolution [79] | Improving enzyme kinetics, reducing inhibition | Requires structural knowledge, high-throughput screening |
| Cofactor Balancing | NOX expression (NAD+ regeneration) [79] | Addressing redox imbalances | May alter cellular energy status |
Pathway Optimization Workflow
The complex relationships between substrate utilization, pathway engineering, and flux control can be visualized through the following comprehensive pathway diagram:
Research Reagents and Experimental Toolkit
Successfully implementing the described protocols requires specific research reagents and materials. The following table details essential components for conducting metabolic bottleneck analyses and engineering solutions:
Table 3: Essential Research Reagents for Metabolic Bottleneck Investigations
| Reagent/Material | Specification | Experimental Function | Example Application |
|---|---|---|---|
| 13C-labeled substrates | [1-13C]glucose, [U-13C]glucose >99% purity | Metabolic flux analysis using isotopic tracing | Quantifying pathway flux distributions [80] |
| HPLC/GC-MS standards | Succinic, malic, fumaric, lactic acids | Metabolite quantification and validation | Measuring extracellular metabolite concentrations [79] |
| Chromosomal integration vectors | pKD46, pET system variants | Gene knockout/knockin applications | Deleting byproduct pathways (ldh, pfl) [79] |
| Biosensor plasmids | TF-based reporter systems | Dynamic regulation implementation | Connecting metabolite sensing to gene expression [80] |
| Promoter libraries | Synthetic promoter sets with varying strengths | Fine-tuning gene expression levels | Optimizing enzyme expression to balance flux [80] |
| Antibiotics | Kanamycin, chloramphenicol, spectinomycin | Selection pressure maintenance | Maintaining plasmid stability during cultivation |
| Enzyme assays kits | Lactate dehydrogenase, pyruvate dehydrogenase | Direct enzyme activity measurement | Confirming functional knockout or overexpression [79] |
| Endolide F | Endolide F, MF:C25H32N4O6, MW:484.5 g/mol | Chemical Reagent | Bench Chemicals |
| Levinoid C | Levinoid C, MF:C15H22O3, MW:250.33 g/mol | Chemical Reagent | Bench Chemicals |
Substrate inhibition and flux imbalances represent fundamental challenges that transcend specific pathways or host organisms in metabolic engineering. Addressing these bottlenecks requires integrated approaches combining systematic experimental characterization (Protocols 1-2) with advanced engineering strategies ranging from targeted gene knockouts to sophisticated dynamic control systems. The continued development of biosensors, enzyme engineering platforms, and computational models promises to enhance our capability to preemptively identify and resolve these limitations. For researchers beginning in metabolic engineering, mastering the principles and methodologies outlined in this guide provides a critical foundation for developing efficient microbial cell factories capable of sustainable production of biofuels, therapeutics, and platform chemicals.
Dynamic Regulation Strategies for Pathway Optimization
Metabolic engineering aims to construct efficient microbial cell factories for the sustainable production of chemicals, materials, and pharmaceuticals. A significant challenge in this field is the inherent trade-off between cell growth and product synthesis. Static engineering approaches often lead to metabolic burden, unbalanced pathway fluxes, and accumulation of toxic intermediates, ultimately limiting product titers, yields, and productivity [81] [82].
Dynamic regulation has emerged as a powerful strategy to overcome these limitations. Instead of maintaining constant gene expression, dynamic control systems modulate the expression of pathway enzymes in response to intracellular or extracellular stimuli. This allows microbial hosts to autonomously manage metabolic resources, first prioritizing growth and then switching to production, thereby optimizing pathway performance across different fermentation stages [81].
This guide explores the core principles and applications of dynamic regulation strategies, focusing on their implementation for pathway optimization within the Design-Build-Test-Learn (DBTL) cycle framework, providing both theoretical foundations and practical methodologies for researchers and scientists [82] [41].
Core Concepts and Classification of Strategies
Dynamic regulation strategies can be systematically classified based on their design and triggering mechanisms. The two primary categories are pathway-dependent and pathway-independent control.
Pathway-Dependent Control utilizes biosensors that respond specifically to pathway metabolites, such as intermediates, products, or cellular redox states. These systems induce expression of downstream enzymes only when a critical metabolite concentration is reached, preventing intermediate toxicity and balancing flux. Examples include transcription factor-based biosensors for myo-inositol or fatty acids [81] [82].
Pathway-Independent Control triggers expression changes in response to generic cellular signals unrelated to the pathway. This includes:
- Quorum Sensing Systems: Exploit cell-to-cell communication molecules to coordinate population-wide behavior, enabling a growth-to-production switch at high cell density [81].
- Time-Based Controllers: Use endogenous circadian rhythms or delayed genetic circuits to program expression after a set duration.
- Stress-Response Systems: Leverage promoters induced by metabolic stress (e.g., substrate limitation, oxygen starvation) to redirect fluxes [81].
The most advanced approaches involve layering multiple orthogonal dynamic regulation systems to independently control different pathway modules, achieving superior coordination compared to single-loop control [81].
Quantitative Data and Performance Comparison
The performance of different dynamic regulation strategies is quantitatively assessed using key metrics. The table below summarizes data from selected case studies, highlighting the effectiveness of these approaches.
Table 1: Performance Comparison of Dynamic Regulation Strategies in Model Microbes
| Host Organism | Target Product | Regulation Strategy | Key Features | Reported Titer (g/L) | Fold Improvement |
|---|---|---|---|---|---|
| E. coli K-12 | D-Glucaric Acid | Layered Dynamic Control [81] | Quorum Sensing (pathway-independent) + myo-inositol biosensor (pathway-dependent) | ~2.0 | 4.0 vs. base strain |
| E. coli | 2,3-Butanediol | Promoter Replacement [82] | Static promoter engineering to modulate enzyme expression levels | 73.8 | 4.2 vs. previous study |
| E. coli P2 | L-Tyrosine | gTME [82] | Global transcriptional machinery engineering for system-wide changes | Information Missing | 1.14 (114% increase) |
| S. cerevisiae | Rubusoside | Systematic Engineering [82] | Combination of static pathway optimization strategies | 1.37 | Not Specified |
Table 2: Characteristics of Common Induction and Sensing Systems for Dynamic Control
| System Type | Inducer/Signal | Orthogonality | Tunability | Ease of Implementation | Best Use Case |
|---|---|---|---|---|---|
| Chemical Inducer | e.g., IPTG, aTc | High | High | Straightforward | Bench-scale validation, multi-layer logic |
| Quorum Sensing | Acyl-Homoserine Lactone (AHL) | Moderate | Moderate | Moderate | Population-level control, growth-to-production switch |
| Metabolite Biosensor | Pathway Intermediate | Low (pathway-specific) | Moderate | Complex (requires sensor development) | Preventing intermediate toxicity, auto-balancing flux |
| Stress-Response Promoter | e.g., Oxygen, Glucose | Low | Low | Straightforward | Exploiting fermentation phases and nutrient shifts |
Integrated Workflows and Experimental Protocols
Implementing dynamic regulation requires an iterative workflow that integrates design, building, testing, and learning. Machine learning (ML) is increasingly used to analyze complex biological datasets and identify optimal design rules, thereby accelerating these DBTL cycles [82].
Workflow for Developing Dynamically Regulated Pathways
The following diagram illustrates the general workflow for designing and implementing a dynamic regulation system.
Protocol: Sensor-Response System Characterization
This protocol provides a detailed methodology for characterizing the performance of a dynamic sensor-response system, such as a metabolite biosensor, in a microbial host like E. coli [83] [84].
1. Rationale and Background: Before characterizing any sensor system, comprehensively review the literature on the biosensor's core components (e.g., transcription factor, promoter). Define the expected input range (metabolite concentration) and the dynamic output range (gene expression level). The primary objective is to quantify the transfer function between the input signal and the output response [84].
2. Study Design:
- Type: In vitro laboratory experiment.
- Strains: The strain harboring the sensor-response genetic construct. A control strain with a constitutive promoter driving the reporter gene is essential.
- Culture Conditions: Perform characterization in a controlled bioreactor or using microtiter plates with controlled shaking and temperature.
- Variables:
- Independent Variable: Concentration of the target metabolite (or chemical analogue) added to the medium.
- Dependent Variables: Fluorescence intensity (reporter output), cell density (OD₆₀₀), and metabolite concentration over time.
3. Experimental Procedure: 1. Pre-culture: Inoculate a single colony of the sensor strain into liquid medium with appropriate antibiotics. Grow overnight. 2. Main culture: Dilute the pre-culture into fresh medium and grow until mid-exponential phase (OD₆₀₀ ≈ 0.5-0.6). 3. Induction: Divide the culture into separate flasks or wells. Add the target metabolite at a range of predefined concentrations (e.g., 0, 0.1, 0.5, 1, 5, 10 mM). Include a negative control (no inducer). 4. Monitoring: Incubate the cultures and sample every 30-60 minutes for at least 6-8 hours. - Measure OD₆₀₀ for cell growth. - For each sample, measure fluorescence (excitation/emission appropriate for the reporter, e.g., GFP) and normalize to OD₆₀₀. - Optionally, use HPLC or LC-MS to quantify the actual intracellular concentration of the metabolite.
4. Data Analysis: - Transfer Function: Plot the normalized reporter expression (output) against the final measured metabolite concentration (input). Fit a sigmoidal function (Hill equation) to determine key parameters: - Leakiness: Expression level in the absence of inducer. - Dynamic Range: Ratio between maximum and minimum expression. - Induction Threshold & ECâ‚…â‚€: The metabolite concentration required for half-maximal activation. - Cooperativity (Hill coefficient): Steepness of the response. - Kinetics: Plot normalized expression over time for each induction level to assess response time and stability.
5. Expected Results and Interpretation: A well-characterized biosensor will show a clear, dose-dependent response with low leakiness and a high dynamic range. The resulting transfer function is critical for informing how the sensor will perform when integrated into a larger metabolic pathway [81] [84].
Essential Research Reagents and Tools
A successful dynamic regulation project relies on a toolkit of molecular biology reagents, genetic parts, and analytical equipment. The following table details key components.
Table 3: Research Reagent Solutions for Dynamic Pathway Optimization
| Reagent / Material | Category | Function / Description | Example Application |
|---|---|---|---|
| Plasmid Vectors | Genetic Part | Backbone for hosting genetic circuits; varying copy numbers and origins of replication. | Tunable expression of pathway enzymes and regulatory proteins [81]. |
| Biosensor Modules | Genetic Part | Pre-characterized transcription factor-promoter pairs responsive to specific metabolites. | Detecting intermediate buildup (e.g., myo-inositol) to induce downstream enzymes [81] [82]. |
| Quorum Sensing Modules | Genetic Part | Genes for AHL synthases (luxI family) and AHL-responsive promoters (luxR family). | Implementing population-density-dependent regulation [81]. |
| Fluorescent Reporters | Reporter | Genes for proteins like GFP, mCherry. Serve as proxies for gene expression levels. | Characterizing promoter strength and sensor response curves in vivo [84]. |
| DNA Assembly Kit | Molecular Biology | Enzymatic kits for seamless assembly of multiple DNA fragments (e.g., Gibson Assembly). | Rapid construction of complex genetic circuits combining regulators and pathway genes. |
| HPLC / LC-MS | Analytical Equipment | High-Performance Liquid Chromatography / Mass Spectrometry for precise quantification. | Measuring extracellular and intracellular concentrations of metabolites and products [82]. |
| Microplate Reader | Analytical Equipment | Instrument for high-throughput measurement of absorbance and fluorescence in cell cultures. | Monitoring growth (OD) and reporter gene expression during sensor characterization. |
Future Perspectives and Integration with Machine Learning
The future of dynamic regulation is tightly coupled with the rise of machine learning (ML) and advanced modeling. ML can address several key challenges [82]:
- Predicting System Behavior: ML models trained on multi-omics data (transcriptomics, proteomics, metabolomics) can predict how a genetic perturbation will affect the entire system, guiding the design of more effective controllers.
- Optimizing Multi-Layer Control: Algorithms like Bayesian Optimization can efficiently navigate the vast design space of layered systems (e.g., tuning promoter strengths, ribosome binding sites, and regulator expression) to find optimal combinations that maximize titer, yield, and productivity.
- De Novo Design of Parts: ML models, particularly deep learning, are being used to design novel biosensors, enzymes, and regulatory elements with desired properties, expanding the toolbox for metabolic engineers [82].
The integration of dynamic regulation, ML, and automation through the DBTL cycle represents the cutting edge of metabolic engineering, paving the way for the next generation of intelligent microbial cell factories.
Metabolic engineering is a scientific field fundamentally concerned with the manipulation of metabolic networks for the cost-effective production of fuels, chemicals, and pharmaceuticals [85]. At the heart of this discipline lies the concept of the metabolic network model, a mathematical representation of cellular metabolism that enables researchers to predict and optimize cellular behavior for industrial and therapeutic applications [85]. Metabolic fluxes—the rates at which metabolites flow through biochemical pathways—represent among the most important determinants of cell physiology in metabolic engineering [85]. By quantifying these fluxes under various conditions, scientists can identify bottlenecks in metabolic networks, quantify metabolic control, and establish predictive models that guide the design of strategies to improve desired outputs [85].
The field has evolved significantly over the past three decades, with genome-scale metabolic models (GEMs) emerging as comprehensive knowledge bases of cellular metabolism [86]. These mathematical formulations encapsulate the complete set of metabolic reactions occurring in a cell, tissue, organ, or organism, providing a systems-level framework for investigating metabolic function and dysfunction [86]. For beginners in metabolic engineering research, understanding the fundamental principles, methods, and applications of metabolic flux analysis and GEMs is essential for contributing to advancements in biotechnology, biomedicine, and synthetic biology.
Theoretical Foundations of Metabolic Models
Metabolic Network Models and Stoichiometric Matrix
At the core of all metabolic modeling approaches lies the concept of the metabolic network model, which is mathematically represented by the stoichiometry matrix S [85]. This matrix tabulates the stoichiometric coefficients for all known metabolic reactions and transport processes within a cell, essentially defining the biochemical transformation network that constitutes cellular metabolism [85]. The stoichiometric matrix provides a structured representation of metabolic connectivity, where rows typically represent metabolites and columns represent biochemical reactions. This mathematical framework enables researchers to systematically analyze metabolic capabilities and constraints using computational approaches.
The development of genome-scale metabolic models has been facilitated by advances in genome annotation and computational biology [85]. These comprehensive models integrate genomic information with biochemical knowledge to reconstruct the entire metabolic network of an organism. The stoichiometric matrix serves as the foundation for constraint-based modeling approaches, which leverage mass-balance, energy-balance, and capacity constraints to define the set of possible metabolic behaviors available to a cell [85]. For metabolic engineers, these models provide invaluable blueprints for understanding and manipulating cellular metabolism toward desired outcomes.
Fundamental Computational Approaches
Several computational techniques have been developed to analyze metabolic networks and estimate flux distributions:
Table 1: Fundamental Metabolic Modeling Approaches
| Method | Key Principle | Data Requirements | Primary Applications |
|---|---|---|---|
| Flux Balance Analysis (FBA) | Optimizes an objective function (e.g., biomass) subject to stoichiometric constraints | Stoichiometric matrix, exchange rates, objective function | Prediction of optimal metabolic behavior, strain design [85] [86] |
| Metabolic Flux Analysis (MFA) | Estimates fluxes from measured extracellular rates subject to stoichiometric constraints | Experimentally measured uptake/secretion rates, stoichiometric matrix | Quantification of metabolic fluxes without optimality assumptions [85] |
| 13C-Metabolic Flux Analysis (13C-MFA) | Integrates isotopic tracer data with stoichiometric models | 13C-labeled substrates, mass spectrometry data, isotopomer measurements | Highly precise determination of intracellular metabolic fluxes [85] |
| Flux Space Sampling | Generates random points in the feasible flux space to explore possible metabolic states | Stoichiometric matrix, constraints | Analysis of metabolic network capabilities without objective functions [86] |
Flux Balance Analysis (FBA) operates on the principle of optimizing a cellular objective—typically biomass production or ATP generation—within the constraints imposed by the stoichiometric matrix and measured exchange fluxes [86]. This approach does not require detailed kinetic information about enzymes, making it particularly valuable for systems where such data are unavailable. However, FBA's predictions depend critically on the assumed objective function, which may not always accurately reflect cellular priorities in engineered systems [86].
In contrast, Metabolic Flux Analysis (MFA) employs stoichiometric models in conjunction with experimentally measured extracellular fluxes to quantify intracellular metabolic rates without assuming optimal cell performance [85]. This makes MFA particularly valuable for analyzing cells under non-ideal or stressed conditions often encountered in industrial bioprocesses. The method relies on solving systems of linear equations derived from mass balances around intracellular metabolites.
Methodologies and Protocols
13C-Metabolic Flux Analysis (13C-MFA)
13C-Metabolic Flux Analysis has emerged as the gold standard in metabolic engineering for accurate and precise flux quantification in living cells [85]. This powerful methodology involves performing tracer experiments where 13C-labeled substrates (e.g., [1,2-13C]glucose) are fed to growing cells until the 13C-labeled carbons are fully incorporated into intracellular metabolites and macromolecules [85]. The resulting labeling patterns are then measured using techniques such as mass spectrometry, and these data are integrated with stoichiometric models and extracellular flux measurements to determine precise intracellular metabolic fluxes.
The experimental workflow for 13C-MFA involves several critical steps: (1) designing and performing isotopic tracer experiments with appropriate labeling patterns; (2) cultivating cells under well-controlled conditions at metabolic steady state; (3) measuring extracellular substrate consumption and product formation rates; (4) quantifying isotopic labeling in intracellular metabolites; and (5) applying computational algorithms to estimate fluxes that best fit the experimental data [85]. For beginners, it is essential to recognize that 13C-MFA requires metabolic and isotopic steady state, which may limit its application in certain dynamic systems or complex microbial communities.
Flux Sampling and Comparative Analysis
For large-scale metabolic models, particularly human GEMs where selecting appropriate objective functions is challenging, flux space sampling provides a valuable alternative to optimization-based approaches [86]. Sampling methods generate uniformly distributed random points within the feasible flux space defined by stoichiometric and capacity constraints, thereby characterizing the range of possible metabolic states without requiring assumption of cellular objectives [86].
The ComMet (Comparison of Metabolic states) methodology exemplifies a modern approach to comparing metabolic states in large GEMs [86]. This eight-step pipeline includes: (1) specification of constraints for different metabolic conditions; (2) preprocessing to remove blocked reactions; (3) analytical approximation of flux distributions; (4) principal component analysis of flux spaces; (5) basis rotation; (6) identification of condition-specific modules; (7) comparison of metabolic conditions; and (8) visualization of distinguishing features [86]. This approach enables researchers to identify metabolic differences between conditions (e.g., healthy vs. diseased states) without a priori assumptions about cellular objectives.
Comparative Flux Sampling Analysis (CFSA) represents another advanced methodology specifically designed for strain engineering applications [87]. CFSA performs extensive comparison of complete metabolic spaces corresponding to maximal or near-maximal growth and production phenotypes, using statistical analysis to identify reactions with altered flux as potential targets for genetic interventions [87]. This method has been successfully applied to improve lipid production in Cutaneotrichosporon oleaginosus and naringenin production in Saccharomyces cerevisiae [87].
Protocols for Constraint-Based Modeling
For researchers beginning with constraint-based metabolic modeling, the following protocol provides a foundational workflow:
Model Acquisition or Reconstruction: Obtain a genome-scale metabolic model for your organism of interest from databases such as BiGG Model or MetaNetX, or reconstruct a new model using genomic annotation and biochemical databases [88].
Model Validation and Curation: Verify model functionality by testing known metabolic capabilities and comparing simulations with experimental data.
Constraint Definition: Specify constraints based on experimental conditions, including substrate uptake rates, oxygen availability, and byproduct secretion rates.
Objective Function Selection: Choose an appropriate objective function relevant to the biological context, such as biomass production for growth or ATP synthesis for energy maintenance.
Simulation and Analysis: Perform FBA or related analyses to predict flux distributions, and validate predictions with experimental data where possible.
Iterative Refinement: Update the model based on discrepancies between predictions and experimental observations to improve model accuracy.
For 13C-MFA studies, additional specialized protocols are required for tracer experiment design, mass spectrometric measurement of isotopic labeling, and computational flux estimation using software such as INCA or OpenFLUX [85].
Advanced Applications
Host-Microbe Interactions
Metabolic modeling has emerged as a powerful framework for investigating host-microbe interactions at a systems level [89]. By simulating metabolic fluxes and cross-feeding relationships, GEMs enable exploration of metabolic interdependencies and emergent community functions in host-associated microbial communities [89]. These approaches can be applied independently or in conjunction with experimental data to generate hypotheses and provide systems-level insights into host-microbe dynamics.
Integrated host-microbe metabolic models have been developed to study interactions in the human gut, revealing how microbial metabolism influences host health and disease states [89]. These multi-species models capture the complex metabolic exchanges between host cells and microbial communities, providing insights into how diet, probiotics, and antibiotics affect overall system metabolism [89]. For pharmaceutical researchers, these models offer opportunities to understand how drug metabolism is influenced by host-microbiome interactions and to identify novel therapeutic targets.
Strain Design for Bioproduction
Metabolic models have become indispensable tools for designing microbial cell factories for chemical production [87]. Algorithms such as Comparative Flux Sampling Analysis (CFSA) enable identification of genetic engineering targets—including gene knockouts, downregulations, and overexpressions—that redirect metabolic flux toward desired products [87]. This approach has been successfully applied to improve production of various compounds, including lipids in oleaginous yeast and flavonoids in S. cerevisiae [87].
A significant advantage of modern strain design algorithms is their ability to propose growth-uncoupled production strategies, where product formation is decoupled from biomass accumulation, allowing continued production during stationary phase and often resulting in higher overall titers [87]. These approaches generate a reduced list of high-probability engineering targets that can be implemented and validated in a step-wise manner, increasing the efficiency of the metabolic engineering design-build-test-learn cycle [87].
Food Science and Microbiome Engineering
In food science, metabolic modeling approaches are being applied to design and optimize fermented food microbiomes [90]. Genome- and metagenome-scale metabolic models help rationalize and predict microbial interactions in complex food communities, supporting the development of new fermented foods and improvement of traditional products [90]. Both bottom-up (designing defined consortia) and top-down (engineering natural communities) strategies benefit from metabolic modeling insights.
These approaches face unique challenges in food systems, including the need to integrate dynamics of microbial succession, spatial organization in food matrices, and complex physicochemical parameters [90]. However, the potential applications are significant, ranging from optimizing starter cultures for cheese and yogurt production to developing novel fermented foods with enhanced nutritional profiles or extended shelf life [90].
Computational Tools and Software
Table 2: Essential Software Tools for Metabolic Modeling
| Tool/Resource | Primary Function | License | Developer/Provider |
|---|---|---|---|
| COBRA Toolbox | Constraint-based reconstruction and analysis | Open Source | Various [86] |
| ModelSEED | Genome-scale model reconstruction | Open Source | University of Chicago |
| Bioconductor | R-based bioinformatics toolkit | Artistic 2.0 | Fred Hutchinson Cancer Research Center [91] |
| Biopython | Python tools for computational biology | Biopython License | Open Bioinformatics Foundation [91] |
| PathVisio | Biological pathway drawing and analysis | Apache 2.0 | Maastricht University [91] |
| Nextflow | Workflow management for bioinformatics | Apache 2.0 | Nextflow Team [91] |
| Galaxy | Scientific workflow and data integration | Academic Free | Collaborative project [91] |
| Orange | Component-based visual programming for data mining | GPL | University of Ljubljana [91] |
| Sampling Algorithms | Flux space sampling for large GEMs | Varies | Multiple research groups [86] |
Databases and Knowledgebases
Critical to metabolic modeling efforts are comprehensive databases that provide curated biochemical and genomic information:
- BiGG Models: A knowledgebase of curated genome-scale metabolic models [88]
- KEGG: Kyoto Encyclopedia of Genes and Genomes, providing pathway information [88]
- MetaCyc: A database of non-redundant, experimentally elucidated metabolic pathways [88]
- BioModels: A repository of computational models of biological processes [88]
- Gene Expression Omnibus (GEO): A public functional genomics data repository [88]
These resources provide essential data for model reconstruction, refinement, and contextualization with experimental data. For beginners, starting with existing curated models from BiGG Models or MetaNetX is recommended before attempting de novo model reconstruction.
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Metabolic Flux Studies
| Reagent/Resource | Function/Application | Examples/Specifications |
|---|---|---|
| 13C-Labeled Substrates | Tracer experiments for 13C-MFA | [1,2-13C]glucose, [U-13C]glutamine; ≥99% isotopic purity [85] |
| Mass Spectrometry Kits | Sample preparation for isotopic analysis | Derivatization reagents for GC-MS; quenching solutions for intracellular metabolites |
| Cell Culture Media | Defined media for flux experiments | Chemically defined formulations with precise carbon sources |
| Enzyme Assay Kits | Validation of key metabolic activities | Spectrophotometric or fluorometric assays for pathway enzymes |
| Metabolite Standards | Quantification of extracellular fluxes | HPLC/MS standards for organic acids, amino acids, sugars |
| DNA/RNA Extraction Kits | Multi-omics integration | High-quality isolation for transcriptomic and genomic analyses |
| CRISPR/Cas9 Systems | Genetic manipulation of flux targets | Gene knockouts, knockdowns, or regulatory modifications [87] |
Future Perspectives and Challenges
The field of metabolic modeling continues to evolve with several emerging trends and persistent challenges. A significant frontier is the development of methods for non-standard biological systems, including heterogeneous cultures, systems where isotopic steady state cannot be easily reached, and systems with dynamic metabolic fluxes [85]. Methodologies such as INST-MFA (Isotopically Non-Stationary MFA) and systems for analyzing co-cultures are addressing these challenges [85].
Another important direction is the integration of machine learning approaches with traditional constraint-based modeling [92]. This synergy leverages the pattern recognition capabilities of machine learning with the mechanistic insights provided by metabolic models, potentially enabling more accurate predictions of metabolic behavior across diverse conditions [92].
For beginners entering the field, the future will likely involve increasingly accessible tools and standardized workflows for metabolic modeling. However, fundamental challenges remain, including the need for more comprehensive annotation of enzyme functions in less-characterized organisms, improved methods for integrating multi-omics data into metabolic models, and better approaches for predicting metabolic regulation [85]. Despite these challenges, metabolic flux analysis and GEMs continue to provide indispensable frameworks for understanding and engineering biological systems across biotechnology, biomedicine, and synthetic biology.
In metabolic engineering, a fundamental challenge is the inherent conflict between a microorganism's natural objective—often rapid growth and proliferation—and the engineer's goal of high-yield production of a target compound. This competition for finite cellular resources, such as energy, carbon, and reducing equivalents, creates a critical trade-off between growth and production [93]. Understanding and managing this trade-off is essential for developing efficient microbial cell factories. Cells cannot optimize all physiological tasks simultaneously due to limitations in resource allocation, a concept well-studied in evolutionary biology and formalized in models like the Y-model of resource allocation [93] [94]. In this model, a common resource pool (Y) is partitioned among competing tasks, such that increasing investment in one (e.g., product synthesis) necessarily diminishes resources available for another (e.g., growth) [93].
The assumption that rapidly proliferating cells like microbes or cancer cells universally prioritize biomass maximization is an oversimplification [93]. Cellular objectives are nuanced and can shift based on environment, functional state, and genetic makeup. For instance, while cancer cells typically prioritize growth, the presence of the Warburg effect (aerobic glycolysis) suggests a complex metabolic strategy that may prioritize other functions in certain contexts [93]. Similarly, non-proliferative cells like neurons or muscle cells prioritize tasks like tissue maintenance and energy dynamics over growth [93]. This complexity necessitates a systematic approach to reprogramming cellular objectives for bioproduction, moving beyond simple growth maximization to carefully balanced, multi-objective optimization.
Fundamental Principles of Metabolic Trade-offs
The Conceptual Framework of Resource Allocation
The Y-model provides a foundational theory for understanding growth-production trade-offs. It depicts how a central, limited resource (Y)—frequently metabolic energy from substrates like glucose—is allocated to competing physiological tasks [93] [94]. The branching structure of the "Y" visually represents the splitting of this resource stream. In a production context, the model can be extended, where resources are divided among growth, reproduction, maintenance, and physical activity, or in a bioprocess, among growth, product synthesis, cellular maintenance, and stress responses [94].
This partitioning leads to the establishment of a Pareto front, a concept borrowed from economics and engineering. On this front, any improvement in one objective (e.g., increasing production yield) inevitably leads to a deterioration in another (e.g., reducing growth rate) [93]. An organism's phenotype represents a specific point on this front, reflecting its particular resource allocation strategy. The role of the metabolic engineer is to shift this Pareto front outward through strain engineering and to identify the optimal operating point on the front for a given bioprocess.
Mathematical Modeling of Trade-offs: From FBA to Advanced Formulations
Constraint-based modeling, particularly Flux Balance Analysis (FBA), is a cornerstone for quantitatively analyzing these trade-offs. FBA uses genome-scale metabolic models (GEMs) to predict metabolic flux distributions by assuming the cell optimizes a specified objective function, most often biomass production [93] [4]. While this growth-centric objective is reasonable for many microbes, it often fails to predict behavior in non-proliferating or production-oriented states [93] [3].
To better capture trade-offs, methods like Flux Variability Analysis (FVA) are employed. Frameworks such as FluTO (Flux Trade-off) leverage FVA to mathematically describe trade-offs among metabolic reactions. FluTO identifies invariant reaction fluxes under specific conditions and designates a weighted sum of fluxes equal to an invariant flux, formalizing the concept of a shared resource pool (Y) [93]. The model hypothesizes that trade-offs can be described by the equation ( Y = \sum \alphai xi ), where ( Y ) is the common resource, ( xi ) represents metabolic traits (fluxes), and ( \alphai ) are weighting coefficients determining the allocation [93]. An adaptive version, FluTOr, was later developed to identify relative trade-offs where the resource Y is variable, accounting for phenotypic plasticity [93].
Table 1: Key Computational Methods for Analyzing Metabolic Trade-offs
| Method/Tool | Primary Function | Application in Trade-off Analysis |
|---|---|---|
| Flux Balance Analysis (FBA) [93] [4] | Predicts metabolic flux distributions by optimizing an objective function. | Establishes baseline growth-versus-production predictions. |
| Flux Variability Analysis (FVA) [93] | Determines the range of possible fluxes for each reaction in a network. | Identifies invariant fluxes and rigid trade-offs in the network. |
| FluTO [93] | Identifies absolute trade-offs between metabolic fluxes under fixed resource constraints. | Mathematically formalizes the Y-model for metabolic reactions. |
| FluTOr [93] | Identifies relative trade-offs, allowing for variable resource pools. | Captures phenotypic plasticity and adaptive responses. |
| QHEPath [4] | Quantitative heterologous pathway design algorithm. | Systematically identifies heterologous reactions to break stoichiometric yield limits of the host. |
Computational Strategies for Breaking Trade-offs
A primary goal of computational metabolic engineering is to design strains that "break" the fundamental growth-production trade-off, achieving high yields without sacrificing fitness. This often involves introducing heterologous pathways that create more efficient, direct routes from substrates to products.
Cross-Species Metabolic Modeling and Pathway Design
The QHEPath algorithm and the underlying Cross-Species Metabolic Network (CSMN) model represent a significant advance in this area [4]. This approach involves constructing a high-quality, multi-species metabolic network that integrates reactions from diverse organisms. A critical step is rigorous quality control to eliminate thermodynamically infeasible cycles, such as those that allow infinite generation of reducing equivalents (NADPH) or energy (ATP), which would otherwise lead to over-optimistic yield predictions [4].
The QHEPath algorithm systematically evaluates the potential of introducing heterologous reactions to surpass the native host's stoichiometric yield limit (YP0). It calculates the maximum pathway yield (YmP) and identifies the specific set of heterologous reactions needed to achieve it, distinguishing them from the minimal reactions required merely to make a non-native product [4]. A systematic evaluation of 12,000 biosynthetic scenarios for 300 products across 5 industrial organisms revealed that over 70% of product pathway yields could be improved by introducing appropriate heterologous reactions [4].
Identified Engineering Strategies
This large-scale computational analysis identified thirteen recurring engineering strategies, categorized as carbon-conserving and energy-conserving, with five strategies effective for over 100 different products [4]. These strategies often involve bypassing native, inefficient segments of metabolism or providing alternative routes to generate essential cofactors.
Diagram: Generic strategy of using a heterologous bypass to avoid an inefficient native metabolic route that leads to carbon or energy loss, thereby breaking the stoichiometric yield limit.
Experimental Design for Mapping and Optimizing Trade-offs
Once computational strategies are identified, experimental methods are required to map the trade-off landscape and find the optimal balance in a real biological system.
Design of Experiments for Combinatorial Pathway Optimization
A key challenge is finding the optimal expression level for each gene in a heterologous pathway. Combinatorial optimization, which tests multiple genes simultaneously, is superior to sequential optimization because it captures gene-gene interactions (epistasis) [95]. For example, the effect of optimizing one enzyme's expression may depend on the expression levels of other enzymes in the pathway. However, testing all possible combinations (a full factorial design) becomes experimentally intractable as the number of genes increases.
Statistical Design of Experiments (DoE) addresses this by minimizing the number of strains to be constructed while maximizing the information gained [95]. In a DBTL cycle, genes are considered "factors," and their expression levels (e.g., low vs. high) are "levels." Different factorial designs offer a trade-off between experimental workload and informational resolution:
- Resolution V: Can clearly identify main effects and two-factor interactions but requires more strains.
- Resolution IV: Confounds two-factor interactions with each other but requires fewer strains.
- Resolution III/Plackett-Burman (PB): Confounds main effects with two-factor interactions; useful for screening many factors but can be misleading if interactions are significant [95].
A study using a kinetic model of a seven-gene pathway found that Resolution IV designs offer a good balance, enabling the identification of optimal strains and providing valuable guidance for subsequent DBTL cycles without the excessive burden of a full factorial or Resolution V design [95].
Diagram: A DBTL cycle for pathway optimization, highlighting the use of DoE to efficiently design a strain library and a learning phase to model the results.
Protocol for DoE-based Pathway Optimization
This protocol outlines a DBTL cycle for balancing gene expression in a heterologous pathway.
Design Phase:
- Define Factors and Levels: Select the genes (factors) in the pathway to be optimized. Choose two expression levels (e.g., low and high) for each, which could correspond to weak vs. strong promoters or low vs. high copy number plasmids.
- Choose Experimental Design: Select a factorial design (e.g., Resolution IV) using software like the
FrF2package in R, JMP, or Minitab [95]. This generates a list of strain genotypes to be constructed. - Define Response: Specify the primary measurable output, such as the final product titer (g/L), yield (mol/mol substrate), or a combined metric.
Build Phase:
- Use high-throughput genetic engineering techniques (e.g., Golden Gate assembly, CRISPR-Cas, automated DNA assembly) to construct the specified strain library in the chosen microbial host (e.g., E. coli or S. cerevisiae).
Test Phase:
- Cultivate all strains in a controlled, parallel fermentation system (e.g., microtiter plates or mini-bioreactors).
- Measure the defined response (product titer/yield) for each strain. Include analytical techniques like HPLC or GC-MS for quantification.
- Perform replicates (e.g., biological triplicates) to account for experimental noise.
Learn Phase:
- Model Fitting: Fit a linear model to the experimental data using ordinary least squares regression. The model has the form:
y = β₀ + Σ(ME_i * F_i) + Σ(2FI_i:j * F_i * F_j)whereyis the response,β₀is the intercept,ME_iis the main effect of factor i, and2FI_i:jis the two-factor interaction between factors i and j [95]. - Statistical Analysis: Perform an Analysis of Variance (ANOVA) to determine which main effects and interactions significantly influence production.
- Identification of Optimum: Use the model to predict the gene expression profile (combination of levels) that maximizes the response. This may be a strain that was already built or a new combination to test in the next DBTL cycle.
- Model Fitting: Fit a linear model to the experimental data using ordinary least squares regression. The model has the form:
Table 2: The Scientist's Toolkit: Essential Reagents and Resources
| Category/Item | Specific Examples | Function in Trade-off Analysis |
|---|---|---|
| Model Host Organisms | Escherichia coli, Saccharomyces cerevisiae (baker's yeast) [24] [2] | Well-characterized, genetically tractable chassis for heterologous pathway expression. |
| Genetic Engineering Tools | Promoter libraries, RBS libraries, CRISPR-Cas systems, plasmid vectors. | To precisely modulate the expression level of pathway genes (factors in DoE). |
| Analytical Software | R (with FrF2 package), JMP, Minitab [95] |
To generate factorial designs and perform statistical analysis (ANOVA) on the results. |
| Analytical Techniques | HPLC, GC-MS, LC-MS | To accurately quantify product titers, yields, and metabolic fluxes. |
| Metabolic Modeling Suites | COBRA Toolbox, QHEPath Web Server [4] | For in silico prediction of metabolic fluxes, trade-offs, and heterologous pathway design. |
| Cultivation Systems | Microtiter plates, mini-bioreactors, controlled lab-scale fermenters. | For high-throughput, reproducible cultivation of strain libraries under defined conditions. |
Effectively addressing the growth-production trade-off is not a singular task but an iterative process that integrates computational prediction with experimental validation. The conceptual framework of resource allocation, embodied by the Y-model and Pareto optimality, provides a lens through which to understand the fundamental problem. Advanced computational methods, including the CSMN and QHEPath algorithm, now enable the systematic identification of engineering strategies—particularly carbon- and energy-conserving heterologous pathways—that can break the stoichiometric yield limits of native hosts. Finally, rigorous experimental frameworks like Design of Experiments are essential for efficiently navigating the combinatorial complexity of pathway optimization in the laboratory, transforming the theoretical Pareto front into a practical, high-performing microbial cell factory. For researchers beginning in this field, mastery of these complementary concepts and tools is the key to success in metabolic engineering.
Enhancing Strain Tolerance and Productivity Through Adaptive Evolution
In the field of metabolic engineering, the development of robust microbial cell factories is paramount for efficient biomanufacturing. Adaptive Laboratory Evolution (ALE) stands as a powerful technique to enhance strain tolerance, fitness, and productivity under industrial-relevant conditions. This guide frames ALE within the iterative Design-Build-Test-Learn (DBTL) cycle, a framework proven to accelerate strain engineering by combining systematic genetic manipulation with data-driven learning [96]. ALE leverages the natural selection process to evolve microbial populations with improved phenotypes, such as higher product yields or greater resilience to inhibition, which are critical for reducing development costs and time to market in the pharmaceutical and biotechnology sectors [96]. For beginners in metabolic engineering research, understanding ALE is crucial as it provides a practical approach to solve complex physiological challenges that are often difficult to address through rational design alone.
Adaptive Evolution in the Metabolic Engineering Workflow
The Design-Build-Test-Learn (DBTL) Cycle
The DBTL cycle is a cornerstone of modern strain engineering. In this framework, adaptive evolution primarily operates within the "Test" and "Learn" phases, generating valuable phenotypic and genotypic data to inform subsequent engineering cycles [96].
- Design: This initial phase involves planning the evolution experiment. For ALE, this includes selecting the initial strain, defining the selective pressure (e.g., high product concentration, inhibitory compounds, or temperature), and designing the cultivation regime (e.g., serial passaging in batch or continuous culture) [96].
- Build: This stage involves constructing the initial strain(s) to be subjected to evolution. While ALE often starts with a wild-type or base engineered strain, it can be combined with rational design by using strains pre-engineered with specific pathways. Modern genome editing tools like CRISPR can also be used to introduce diversity before evolution begins [96].
- Test: In ALE, this is the execution of the long-term cultivation under the selected selective pressure. The evolved populations and isolated clones are then phenotyped. This involves measuring key performance indicators like growth rate, substrate consumption, product yield, and tolerance to stressors [96].
- Learn: This critical phase involves sequencing the genomes of evolved strains to identify causative mutations. The generated data is analyzed to uncover new genetic targets and understand tolerance mechanisms. This new knowledge then directly informs the "Design" phase of the next DBTL cycle, where these mutations can be reverse-engineered into new strains for validation and further improvement [96].
The following diagram illustrates how adaptive evolution is integrated into this iterative framework.
Comparison of Strain Engineering Approaches
A key strength of ALE is its complementarity with rational and semi-rational design methods. The table below contrasts these primary strain engineering approaches.
Table 1: Comparison of Strain Engineering Strategies
| Feature | Rational Design | Random Mutagenesis (e.g., ALE) | Semi-Rational Design |
|---|---|---|---|
| Basis | Prior knowledge of pathway and regulation [96] | Random mutations selected by fitness [96] | Hypothesis-driven from omics data or literature [97] |
| Throughput | Low to medium | High | Medium to high |
| Edit Precision | High (specific edits) | Low (random SNPs, indels) [96] | High (targeted libraries) |
| Key Advantage | Predictable, targeted changes | Accesses unexplored genetic space; complex phenotypes [96] | Balances breadth and specificity |
| Primary Challenge | Limited by current biological understanding | Requires deconvolution; potential mutational burden [96] | Requires robust phenotyping capacity |
| Ideal Use Case | Pathway insertion, gene knockouts | Improving tolerance, fitness, and complex traits [96] | Optimizing pathway flux, enzyme engineering |
Experimental Protocol for Adaptive Laboratory Evolution
A typical ALE experiment follows a structured workflow to ensure meaningful and reproducible results. The core process involves serial passaging of a microbial population over many generations in a controlled environment, with periodic analysis to monitor progress.
Detailed Methodology
This section provides a step-by-step protocol for conducting an ALE experiment to enhance tolerance to inhibitory compounds, a common challenge in bioprocessing.
Materials:
- Microbial strain (e.g., Escherichia coli, Saccharomyces cerevisiae)
- Appropriate liquid growth medium (e.g., M9, LB)
- Inhibitory compound of interest (e.g., furfural, acetate, high ethanol)
- Erlenmeyer flasks or bioreactors
- Incubator shaker
- Spectrophotometer for measuring optical density (OD)
Procedure:
Initial Setup and Inoculation:
- Prepare a base culture of your chosen strain. Grow it overnight in a standard medium to reach the mid-exponential phase.
- Centrifuge the cells and resuspend them in the evolution medium containing a low, sub-inhibitory concentration of the target stressor (e.g., 10-30% of the ICâ‚…â‚€). This is the passage 0 (P0) culture.
Serial Passaging:
- Incubate the culture under appropriate conditions (temperature, shaking). Monitor growth by periodically measuring the OD.
- Once the culture reaches the stationary phase (or a predetermined OD threshold), transfer a small aliquot (typically 1-2% v/v, resulting in a 1:50 to 1:100 dilution) into fresh medium containing the same concentration of the stressor [96].
- This transfer marks the first passage (P1). Repeat this serial passaging for multiple cycles. The number of generations can be estimated from the dilution factor.
Increasing Selective Pressure:
- Periodically (e.g., every 20-30 generations), assess the fitness of the evolving population. A simple method is to compare its growth rate in the presence of the stressor to that of the unevolved ancestor.
- Once the evolved population shows robust growth (e.g., a growth rate similar to the ancestor in a non-stress condition), increase the concentration of the inhibitory compound in the fresh medium for subsequent passages. Incrementally increase the stress level in a step-wise fashion to drive further adaptation.
Archiving and Isolation:
- At regular intervals (e.g., every 25-50 generations), archive 1 mL of culture mixed with cryoprotectant (e.g., 15% glycerol) by storing it at -80°C. This creates a frozen "fossil record" of the evolution experiment.
- After a sufficient number of generations (often 100+), plate the evolved culture to obtain single colonies. Isolate multiple clones for further analysis.
Accelerating Adaptive Evolution
Traditional ALE can be time-consuming. The table below summarizes strategies to accelerate the evolutionary process.
Table 2: Methods for Accelerating Adaptive Laboratory Evolution
| Method | Description | Key Advantage |
|---|---|---|
| Chemical Mutagenesis | Use of mutagens like ethyl methanesulfonate (EMS) to increase mutation rate [96] | Increases genetic diversity, shortening time to isolate beneficial mutants |
| UV Mutagenesis | Exposure to UV light to induce DNA damage and mutations [96] | Simple, cost-effective method for generating genetic diversity |
| Enhanced Recombination | Engineering strains with enhanced homologous recombination systems [96] | Facilitates incorporation of a wider range of genetic changes |
| Mismatch Repair Deletion | Deletion of genes (e.g., mutS) involved in DNA mismatch repair to elevate mutation frequency [96] | Creates a hypermutable strain background, accelerating evolution |
Analysis and Validation of Evolved Strains
Genomic Analysis and Reverse Engineering
The "Learn" phase of the DBTL cycle is critical. Genome sequencing of evolved isolates is performed to identify the mutations responsible for the improved phenotype.
- Sequencing and Variant Calling: Sequence the genomes of several evolved clones and the ancestral strain. Use bioinformatics tools to align sequences and identify single nucleotide polymorphisms (SNPs), insertions/deletions (indels), and copy number variations.
- Prioritizing Mutations: Focus on mutations that appear repeatedly in independent evolved clones (parallel evolution), as they are strong candidates for causal mutations [96]. Also, prioritize mutations in genes with known functions related to the selective pressure.
- Reverse Engineering: The final step is to validate the causality of identified mutations. This is done by introducing the specific mutation(s) into the genome of the unevolved ancestor strain using precise genome editing tools like CRISPR-Cas9. If the reconstituted strain recovers the improved phenotype, the mutation is confirmed [96].
The following diagram outlines this validation workflow.
Quantitative Outcomes of Adaptive Evolution
ALE has been successfully applied to enhance a wide range of phenotypes in industrial microorganisms. The table below summarizes quantitative results from published studies.
Table 3: Exemplary Outcomes of Adaptive Evolution Experiments
| Phenotype Target | Organism | Evolution Strategy | Key Outcome |
|---|---|---|---|
| Tolerance to Multiple Inhibitors | Escherichia coli | Separate populations evolved in 11 different inhibitory compounds [96] | Isolates tolerated concentrations 60%–400% higher than initial toxic levels |
| Thermotolerance | Saccharomyces cerevisiae | Serial passaging at progressively higher temperatures | Evolved strains capable of growth at 40°C+, improving fermentation robustness |
| Sugar Co-utilization | Corynebacterium glutamicum | Evolution on mixed sugar substrates | Achieved simultaneous consumption of glucose and L-arabinose [97] |
| Reduced Metabolic Burden | Various | Dynamic regulation of essential genes based on metabolic sensors [98] | Improved yields; e.g., 10% increase in isopropanol titer from dynamic control [98] |
Integration with Advanced Metabolic Engineering Strategies
Alleviating Metabolic Burden
A common challenge in metabolic engineering is "metabolic burden," where the rewiring of metabolism for production impairs cell growth and productivity [99]. ALE can be used directly to alleviate this burden by evolving strains to re-optimize resource allocation after introduction of a heterologous pathway. Furthermore, insights from ALE can inform dynamic metabolic engineering strategies. For instance, using sensors to dynamically control pathway expression only after a growth phase can help manage the trade-off between biomass formation and product synthesis, a strategy that has shown theoretical productivity improvements of over 30% in some cases [98].
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Reagents and Materials for Adaptive Evolution
| Item | Function in ALE Experiments |
|---|---|
| Chemical Mutagens (e.g., EMS) | To increase genetic diversity and accelerate the emergence of beneficial mutations [96]. |
| CRISPR-Cas9 System | For reverse engineering of identified mutations into a clean genetic background for validation [96]. |
| Next-Generation Sequencing Kits | For whole-genome sequencing of evolved isolates to identify causal mutations [96]. |
| Specialized Growth Media | To apply precise selective pressure (e.g., containing inhibitors, substrate mixtures, or limiting nutrients). |
| Cryopreservation Reagents (e.g., Glycerol) | For long-term storage of intermediate evolutionary populations, creating a "fossil record" [96]. |
Adaptive Laboratory Evolution is a powerful and versatile tool within the metabolic engineering DBTL cycle. By harnessing the power of natural selection, it allows researchers to generate robust industrial strains with enhanced tolerance and productivity, tackling complex physiological challenges that are difficult to solve through rational design alone. The true strength of ALE is realized when it is integrated with other methods: the causal mutations identified in evolved strains provide invaluable, experimentally validated targets that expand our understanding of cellular metabolism and directly inform the rational design of next-generation cell factories. For the beginner researcher, mastering ALE provides a practical and effective strategy to address some of the most persistent challenges in developing efficient microbial cell factories for drug development and sustainable biomanufacturing.
Metabolic engineering has emerged as a pivotal discipline for rewiring cellular metabolism to enhance the production of valuable chemicals, biofuels, and pharmaceuticals from renewable resources. This technical guide provides a comprehensive overview of systematic approaches spanning five hierarchical levels: enzyme, pathway, network, genome, and cell. By integrating methodologies from enzyme engineering with computational tools for network redesign, metabolic engineering enables the development of efficient microbial cell factories. This whitepaper details experimental protocols, presents quantitative performance data, and visualizes key workflows, serving as an introductory resource for researchers and drug development professionals entering the field.
Metabolic engineering is the science of improving product formation or cellular properties through targeted modification of specific biochemical reactions or introduction of novel genes using recombinant DNA technology [100]. The field has evolved through three distinct waves of innovation: the first wave in the 1990s focused on rational pathway analysis and flux optimization; the second wave in the 2000s incorporated systems biology and genome-scale metabolic models; and the current third wave leverages synthetic biology tools to design and construct complete metabolic pathways for producing both natural and non-natural compounds [100]. This progression has expanded the scope of metabolic engineering from simple pathway optimization to comprehensive cellular redesign.
Within the context of drug development, metabolic engineering offers powerful alternatives to traditional chemical synthesis for complex natural products, which constitute over 52% of new chemical entities approved by the FDA from 1981-2006 [101]. These compounds often contain multiple chiral centers and labile connectivities that make chemical synthesis challenging, but can be produced biosynthetically through engineered organisms [101]. This guide systematically addresses the core hierarchical strategies employed in modern metabolic engineering, providing both theoretical frameworks and practical methodologies for implementation.
Enzyme Engineering: Molecular Foundations
Enzyme engineering forms the molecular foundation of metabolic engineering, focusing on optimizing individual catalytic components within biological systems. This process involves modifying existing enzymes or designing novel biocatalysts to enhance catalytic efficiency, substrate specificity, stability, and compatibility with industrial process conditions.
Physics-Based Modeling Approaches
Physics-based modeling using molecular mechanics (MM) and quantum mechanics (QM) provides a fundamental approach to enzyme engineering that complements laboratory-directed evolution. These computational methods enable researchers to investigate enzyme mechanisms, interpret the origins of catalytic efficiency and selectivity, and calculate transition state barriers through atomically resolved three-dimensional structures [102]. The stabilization of transition states through pre-organized electrostatic effects, a concept pioneered by Linus Pauling and quantitatively demonstrated by Ariel Warshel using multiscale simulations, represents a key design principle for enhancing enzymatic activity [102].
Experimental Protocol: Electric Field Optimization for Transition State Stabilization
- Structure Preparation: Obtain high-resolution crystal structure of the enzyme-substrate complex or generate using AlphaFold2 for soluble proteins [102]
- Reaction Mapping: Identify key reactive conformations and transition state geometries using quantum mechanics/molecular mechanics (QM/MM) simulations
- Electric Field Calculation: Compute electric field strength using Coulomb's law based on atomic charges derived from MM, polarizable MM, or QM methods [102]
- Site Identification: Identify mutation sites that optimize electric field alignment with the reaction coordinate using vibrational Stark shift spectroscopy principles [102]
- Mutant Validation: Express and purify selected variants, then measure enzymatic kinetics to quantify improvements in catalytic efficiency (kcat/KM)
Structure- and Topology-Informed Engineering
Structure-informed enzyme engineering leverages protein structural information to identify beneficial mutations. This approach focuses on enhancing shape complementarity between the enzyme active site and substrate, optimizing tunnel accessibility for reactant and product diffusion, and modifying surface residues to adjust pH optimality [102]. For example, conserved guanine binding sites drive ribozyme selectivity, while single residues in catechol O-methyl transferase position its SAM cofactor to achieve preferred donor-acceptor distances [102].
Table 1: Enzyme Engineering Techniques and Applications
| Engineering Approach | Key Methodologies | Primary Applications | Typical Efficiency Gains |
|---|---|---|---|
| Structure-Based Design | AlphaFold2 prediction, Molecular docking, Tunnel engineering | Substrate specificity, Product release | 2-10 fold improvement in substrate specificity [102] |
| Electrostatic Optimization | QM/MM simulations, Electric field calculations, Vibrational Stark spectroscopy | Catalytic rate enhancement, Transition state stabilization | 3-100 fold increase in kcat/KM [102] |
| Directed Evolution | High-throughput screening, Random mutagenesis, Gene shuffling | Thermostability, Solvent tolerance, pH optimum adjustment | Varies widely; can hit evolutionary dead ends [102] |
| De Novo Enzyme Design | Rosetta, Protein sequence space exploration, Artificial scaffold creation | Novel reactivities, Non-natural chemical transformations | Requires optimization but provides unique scaffolds [102] |
Pathway Engineering: DBTL Framework
Pathway engineering operates at the intermediate scale, focusing on the design, construction, and optimization of multi-enzyme pathways for target compound production. The Design-Build-Test-Learn (DBTL) cycle provides a systematic framework for iterative pathway optimization [41].
Metabolic Pathway Design Principles
Computational pathway design begins with selecting appropriate enzymes and enumerating possible metabolic routes from available substrates to desired products. The ET-OptME framework exemplifies advanced approaches that integrate enzyme efficiency and thermodynamic feasibility constraints into genome-scale metabolic models, achieving at least 70% increase in precision and 47% increase in accuracy compared to previous methods [103]. This protein-centered workflow layers multiple constraints to deliver more physiologically realistic intervention strategies.
Experimental Protocol: Heterologous Pathway Implementation
- Host Selection: Choose industrially proven hosts like Escherichia coli or Saccharomyces cerevisiae based on growth characteristics, genetic tractability, and precursor availability [24]
- Gene Assembly: Clone pathway genes into appropriate expression vectors with compatible promoters and ribosomal binding sites
- Precursor Balancing: Modify host metabolism to ensure adequate precursor supply, potentially adding transporter engineering or deleting competing pathways [100]
- Cofactor Engineering: Balance redox cofactors (NADH/NAD+, ATP/ADP) through enzyme engineering or supplementary pathway expression [100]
- Pathway Optimization: Fine-tune gene expression levels using promoter engineering, ribosomal binding site modification, or codon optimization [100]
The following diagram illustrates the hierarchical metabolic engineering workflow, progressing from enzyme-level engineering to full cellular network redesign:
Hierarchical Metabolic Engineering Workflow
Pathway Engineering Applications
Successful pathway engineering applications include the production of artemisinin, a potent antimalarial compound, in engineered microorganisms. Complete metabolic pathways were designed, constructed, and optimized using synthetic nucleic acid elements, demonstrating the power of synthetic biology in metabolic engineering [100]. Similarly, the reconstruction of the erythromycin polyketide pathway in E. coli required not only expression of the three massive polyketide synthase modules but also introduction of a phosphopantetheine transferase for posttranslational modification and genes for producing the necessary (2S)-methylmalonyl-CoA extender unit [101].
Table 2: Representative Metabolic Engineering Achievements in Bioproduction
| Product | Host Organism | Titer/Yield/Productivity | Key Engineering Strategies |
|---|---|---|---|
| Lysine | Corynebacterium glutamicum | 223.4 g/L, 0.68 g/g glucose [100] | Cofactor engineering, Transporter engineering, Promoter engineering |
| 3-Hydroxypropionic acid | C. glutamicum | 62.6 g/L, 0.51 g/g glucose [100] | Substrate engineering, Genome editing engineering |
| Lactic acid | C. glutamicum | l-lactic acid: 212 g/L, d-lactic acid: 264 g/L [100] | Modular pathway engineering |
| Succinic acid | E. coli | 153.36 g/L, 2.13 g/L/h [100] | Modular pathway engineering, High-throughput genome engineering, Codon optimization |
| Butanol | Engineered Clostridium spp. | 3-fold yield increase [36] | CRISPR-Cas genome editing, De novo pathway engineering |
| Biodiesel | Microalgae | 91% conversion efficiency from lipids [36] | Lipid pathway engineering, Transesterification optimization |
Network Redesign: Systems-Level Optimization
Network-level metabolic engineering adopts a systems perspective, considering the interconnected nature of cellular metabolism rather than focusing on isolated pathways. This approach utilizes genome-scale metabolic models and computational frameworks to identify strategic intervention points that optimize flux toward desired products while maintaining cellular viability.
Computational Tools for Network Analysis
Genome-scale metabolic reconstructions bridge genotype-phenotype relationships, enabling researchers to explore the metabolic potential of cell factories and identify target genes for engineering interventions [100]. The ET-OptME framework represents a recent advancement that integrates enzyme efficiency and thermodynamic feasibility constraints into these models, significantly improving prediction accuracy and precision compared to purely stoichiometric methods [103]. This integration is crucial because thermodynamic bottlenecks and enzyme usage costs fundamentally constrain metabolic flux distributions.
Experimental Protocol: Constraint-Based Network Redesign
- Model Reconstruction: Develop a genome-scale metabolic model incorporating all known biochemical reactions, gene-protein-reaction associations, and thermodynamic parameters [104]
- Constraint Implementation: Apply physiological constraints (substrate uptake rates, maintenance requirements) and thermodynamic constraints (reaction directionality) [103]
- Flux Analysis: Use flux balance analysis or flux variability analysis to predict metabolic behavior under different genetic and environmental conditions
- Intervention Identification: Apply optimization algorithms (OptForce, FSEOF) to pinpoint gene knockouts, additions, or regulatory modifications that enhance product formation [103]
- Implementation and Validation: Construct engineered strains and characterize metabolic performance using flux analysis, metabolomics, and product quantification
Network-Level Applications
Network redesign has successfully enhanced production of numerous valuable compounds. For example, metabolic modeling of Saccharomyces cerevisiae and E. coli has predicted strategies for bioethanol production [100], while multiobjective memetic algorithms have identified key gene knockout targets for cubebol, L-threonine, and L-valine production [100]. These approaches often reveal non-intuitive engineering targets that would be difficult to identify through pathway-focused analysis alone.
The following diagram illustrates the iterative Design-Build-Test-Learn (DBTL) cycle that forms the core framework for modern metabolic engineering:
Design-Build-Test-Learn (DBTL) Cycle
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of metabolic engineering strategies requires carefully selected research reagents and host systems. The following table details essential materials and their applications in metabolic engineering workflows.
Table 3: Essential Research Reagent Solutions for Metabolic Engineering
| Reagent Category | Specific Examples | Function in Metabolic Engineering |
|---|---|---|
| Host Organisms | Escherichia coli, Saccharomyces cerevisiae, Bacillus subtilis, Streptomyces coelicolor [24] | Platform strains for heterologous expression; chosen for genetic tractability, growth characteristics, and precursor availability |
| Expression Systems | Plasmid vectors, Inducible promoters (T7, pBAD), Ribosomal binding site libraries | Enable controlled gene expression and fine-tuning of metabolic pathway enzymes |
| Genome Editing Tools | CRISPR-Cas systems, TALENs, ZFNs [36] | Facilitate precise gene knockouts, insertions, and regulatory modifications |
| Enzyme Engineering Tools | Site-directed mutagenesis kits, Directed evolution platforms, Structural biology resources | Enable optimization of individual enzymes for enhanced activity, specificity, and stability |
| Analytical Reagents | LC-MS standards, Metabolomics kits, Enzyme activity assays | Allow quantification of metabolic fluxes, pathway intermediates, and final products |
| Bioinformatics Tools | Genome-scale modeling software, Pathway enumeration algorithms, Thermodynamic calculation packages | Support in silico design and prediction of metabolic network behavior |
The systematic integration of enzyme engineering with network redesign represents the cutting edge of metabolic engineering. This hierarchical approach enables researchers to optimize biological systems across multiple scales, from individual catalytic components to entire metabolic networks. Future advances will likely involve increased incorporation of machine learning and artificial intelligence for enzyme and pathway discovery, expanded use of non-food feedstocks, and enhanced integration of metabolic engineering with circular economy frameworks [36].
For drug development professionals, metabolic engineering offers increasingly viable pathways to complex natural products and novel compounds that are difficult to access through traditional chemical synthesis. As computational tools continue to improve in predictive accuracy and genetic manipulation technologies become more sophisticated, the scope and efficiency of metabolic engineering will expand further, solidifying its role as a cornerstone of sustainable bioproduction in the pharmaceutical industry and beyond.
Analytical Methods and Cross-Disciplinary Validation Approaches
Metabolic engineering is a specialized field that combines biology and chemistry, focusing on the modification and optimization of metabolic pathways, primarily in microorganisms, to improve the productivity and yield of essential compounds [24]. Emerging in the 1990s, it enables scientists to design new biochemical pathways and enhance existing ones through genetic engineering [24]. Within this framework, metabolomics and fluxomics serve as critical analytical pillars. Metabolomics provides a wholistic and comprehensive quantification of the complete set of small-molecule metabolites, which are the end products of cellular regulatory processes, thereby offering a direct snapshot of physiological status [105]. Fluxomics, its dynamic counterpart, aims to quantify the rates of metabolic reactions through these pathways. Together, these techniques provide the data necessary to inform and validate metabolic engineering strategies, from the production of biofuels and pharmaceuticals to the creation of novel organisms [24].
The core objective of integrating these techniques is to move from a static picture of the metabolic network to a dynamic model that can predict the outcome of genetic or environmental perturbations. This guide details the advanced analytical techniques that underpin metabolomics and fluxomics, providing a technical foundation for researchers, scientists, and drug development professionals engaged in metabolic engineering.
Core Analytical Techniques in Metabolomics
Metabolomics involves the comprehensive analysis of metabolites, typically leveraging separation techniques coupled with mass spectrometry (MS) or nuclear magnetic resonance (NMR) spectroscopy. The field is broadly divided into untargeted and targeted approaches. Untargeted metabolomics aims to profile as many metabolites as possible in a biological system without prior bias, while targeted methods focus on the accurate quantification of a predefined set of metabolites [106].
Measurement Technologies and Separation Techniques
The two primary analytical platforms in metabolomics are Mass Spectrometry (MS) and Nuclear Magnetic Resonance (NMR) Spectroscopy [105]. MS-based methods are highly sensitive and can detect thousands of metabolite features in a single run, but they often require chromatographic separation to reduce sample complexity [106]. NMR, while less sensitive, is highly reproducible, quantitative, and provides structural information non-destructively [105].
- Liquid Chromatography-Mass Spectrometry (LC-MS/MS): LC-MS/MS is the workhorse of untargeted metabolomics [106]. The liquid chromatography (LC) step separates metabolites based on their chemical properties (e.g., hydrophobicity in reversed-phase LC) before they enter the mass spectrometer. The tandem MS (MS/MS) capability fragments selected ions, generating spectral patterns that are crucial for metabolite identification [106].
- Nuclear Magnetic Resonance (NMR) Spectroscopy: NMR is used for both structural elucidation and quantification. It measures the absorption of radiofrequency radiation by atomic nuclei (e.g., ^1H, ^13C) in a strong magnetic field. The resulting chemical shifts provide detailed information about the molecular structure of metabolites [105]. NMR is complemented by advanced methods like hyperpolarized NMR, which dramatically enhances signal sensitivity for real-time metabolic flux analysis [105].
- Gas Chromatography-Mass Spectrometry (GC-MS): GC-MS is highly robust and reproducible, making it ideal for the analysis of volatile metabolites or those that can be made volatile through chemical derivatization. It boasts extensive, well-curated spectral libraries for confident metabolite identification.
Table 1: Comparison of Major Analytical Platforms in Metabolomics
| Platform | Key Strength | Key Limitation | Primary Application in Metabolomics |
|---|---|---|---|
| LC-MS/MS | High sensitivity, broad metabolite coverage | Susceptible to matrix effects, complex data | Untargeted profiling, biomarker discovery [106] |
| NMR Spectroscopy | Highly quantitative, non-destructive, provides structural info | Lower sensitivity compared to MS | Targeted quantification, structural elucidation, in vivo metabolomic imaging [105] |
| GC-MS | Highly reproducible, extensive libraries | Requires derivatization for many metabolites | Profiling of central carbon metabolism, volatile compounds |
Untargeted Metabolomics Workflow and Data Visualization
The untargeted workflow is a multi-step process that generates complex, high-dimensional data. Data visualization is a crucial step at every stage, providing core components of data inspection, evaluation, and sharing capabilities [106]. The sheer number of available tools makes it challenging to select the right visual strategy.
A generalized workflow for LC-MS/MS-based untargeted metabolomics includes [106]:
- Sample Preparation: Extraction of metabolites from biological matrices (cells, tissues, biofluids).
- Data Acquisition: Running samples on the LC-MS/MS instrument.
- Data Pre-processing: This includes peak picking, feature extraction, retention time alignment, and normalization. Tools like XCMS, MS-DIAL, and OpenMS are commonly used.
- Statistical Analysis & Visualization: Multivariate statistics (PCA, PLS-DA) are applied to identify significant features. Visualization through volcano plots, cluster heatmaps, and other graphs is essential for interpretation [106].
- Metabolite Annotation & Identification: Using MS/MS spectral matching to databases (e.g., GNPS, HMDB) or NMR libraries to putatively identify metabolites.
- Pathway & Integration Analysis: Mapping significant metabolites onto biochemical pathways to derive biological insight.
The following diagram illustrates the key decision points and processes in a standard untargeted metabolomics workflow:
Effective visualization strategies are paramount for making sense of the data at each stage. As noted in a 2025 review, "Visualizations are used as a means to augment researchers decision-making capabilities by summarizing data, extracting and highlighting patterns within the data, and organizing and showcasing relations between data" [106]. For instance, volcano plots provide a snapshot view of treatment impacts by displaying statistical significance versus the magnitude of change, while cluster heatmaps visualize patterns in metabolite abundance across sample groups [106]. Network visualizations are increasingly used to organize and interpret complex MS/MS spectral relationships [106].
Determining Metabolic Flux with Fluxomics
While metabolomics reveals the snapshot concentrations of metabolites, fluxomics aims to quantify the dynamic flow of metabolites through metabolic pathways—the in vivo reaction rates. This information is critical for metabolic engineers to identify rate-limiting steps and optimize metabolic networks for production.
Core Principles and Methodologies
The fundamental principle of fluxomics is the use of isotopic tracers, most commonly ^13C-labeled carbon sources. By introducing a labeled substrate (e.g., [1-^13C]glucose) into a growing culture, the label propagates through the metabolic network. Measuring the resulting labeling patterns in intracellular metabolites using techniques like GC-MS or LC-MS allows for the calculation of metabolic flux.
The two primary computational approaches are:
- ^13C Metabolic Flux Analysis (^13C-MFA): This is the gold standard for quantifying fluxes in central carbon metabolism. It combines isotopic labeling data with a stoichiometric model of the metabolic network and a constraint-based optimization algorithm to compute the most probable flux map.
- Stable Isotopic Labeling and Kinetic Flux Profiling: This approach is used for non-steady-state conditions to measure flux changes over time.
Experimental Protocol: ^13C-Based Metabolic Flux Analysis
A detailed protocol for a standard ^13C-MFA experiment in a microbial system like E. coli or S. cerevisiae is as follows [24]:
Strain and Culture Preparation:
- Transform the host organism (e.g., E. coli) with the desired genetic construct.
- Inoculate a single colony into a minimal medium with unlabeled glucose and grow overnight.
Tracer Experiment:
- Sub-culture the cells into fresh minimal medium containing a defined mixture of ^13C-labeled glucose (e.g., 100% [U-^13C]glucose or a mixture with unlabeled glucose).
- Grow the cells in a controlled bioreactor to ensure steady-state growth conditions (exponential phase).
- Harvest cells rapidly (e.g., via fast filtration) when the culture reaches a mid-exponential optical density.
Metabolite Extraction and Derivatization:
- Quench metabolism immediately using cold methanol or other quenching solutions.
- Extract intracellular metabolites using a solvent system like cold methanol/water.
- For GC-MS analysis, derivatize the polar metabolites in the extract (e.g., using MSTFA [N-Methyl-N-(trimethylsilyl)trifluoroacetamide] to form trimethylsilyl derivatives).
Mass Spectrometry Measurement:
- Analyze the derivatized sample via GC-MS.
- Acquire data in selected ion monitoring (SIM) or scan mode to detect the mass isotopomer distributions (MIDs) of key metabolic fragments from amino acids, sugars, and organic acids.
Computational Flux Analysis:
- Compile the measured MIDs and extracellular uptake/secretion rates.
- Input the data into a flux analysis software package (e.g., INCA, OpenFlux).
- The software fits the experimental data to a stoichiometric network model by iteratively adjusting fluxes until the simulated MIDs match the measured ones, resulting in a quantitative flux map.
The following diagram outlines the logical flow of a ^13C-MFA experiment, from tracer introduction to flux map generation:
The Scientist's Toolkit: Key Reagents and Materials
Successful execution of metabolomics and fluxomics experiments relies on a suite of specialized reagents and materials. The following table details essential items for the featured experiments.
Table 2: Key Research Reagent Solutions for Metabolomics and Fluxomics
| Item Name | Function / Application | Technical Specification / Example |
|---|---|---|
| ^13C-Labeled Glucose | Tracer substrate for ^13C-MFA to track carbon fate. | [U-^13C]glucose (uniformly labeled); 99% atomic purity [24]. |
| MSTFA (N-Methyl-N-(trimethylsilyl)trifluoroacetamide) | Derivatization agent for GC-MS; makes polar metabolites volatile and stable. | Used in sample preparation prior to GC-MS injection. |
| LC-MS Grade Solvents | Mobile phases for chromatography; high purity is critical for signal-to-noise. | Acetonitrile, methanol, and water with low LC-MS grade purity. |
| Deuterated Solvent for NMR | Lock signal and field frequency stabilization for NMR spectroscopy. | Deuterium oxide (Dâ‚‚O) for aqueous samples; chloroform-d for lipid extracts [105]. |
| Internal Standards (IS) | Correction for sample preparation and instrument variability. | Stable isotope-labeled IS (e.g., ^13C, ^15N) for targeted MS; internal chemical shift reference (e.g., TSP) for NMR. |
| Host Organism | Genetically tractable chassis for metabolic engineering. | Escherichia coli or Saccharomyces cerevisiae (baker's yeast) [24]. |
Data Analysis and Integration
The final challenge in metabolomics and fluxomics is the analysis and integration of the complex, multi-dimensional data generated.
Data Processing and Multi-Omics Integration
Raw data from MS instruments must be processed to extract meaningful biological information. This involves using software tools for peak detection, alignment, and normalization to create a data matrix of features (metabolites) × samples × intensities [106]. Following this, statistical analysis identifies significantly altered metabolites between conditions. A major frontier is the integration of metabolomic data with other omics data layers (genomics, transcriptomics, proteomics) to build a more comprehensive model of cellular physiology. This "multi-omics" approach allows researchers to connect genetic modifications to changes in metabolic flux and end-product yield [106].
The Role of Artificial Intelligence and Machine Learning
The field is increasingly leveraging artificial intelligence (AI) and machine learning (ML) to handle data complexity. A 2025 review highlights the use of AI and ML for advanced metabolomic data analysis [105]. Applications include:
- Deep Learning: Used for the de novo interpretation of MS/MS spectra to improve the speed and accuracy of metabolite identification.
- Neural Topic Modeling: As demonstrated in a 2025 preprint, models like PubMedBERT and GPT-4o can be used to map the entire metabolomics research landscape, revealing emerging trends and clusters from over 80,000 publications [107].
- Predictive Modeling: ML algorithms can predict optimal genetic manipulation sites or culture conditions to maximize the production of a desired compound, directly informing metabolic engineering strategies [105].
AI and Machine Learning in Metabolic Prediction and Design
The convergence of artificial intelligence (AI) and metabolic science is revolutionizing our ability to predict complex diseases and engineer biological systems. Machine learning (ML) models have demonstrated exceptional capability in identifying subtle, non-linear patterns within high-dimensional biomedical data, enabling early risk prediction and informing metabolic design strategies. This technical guide explores the application of ML in two key domains: predicting metabolic dysfunction-associated fatty liver disease (MAFLD) in clinical populations and rewiring cellular metabolism for biochemical production in engineered systems. For clinical prediction, ML models leverage body composition metrics and routine clinical biomarkers to achieve robust, non-invasive risk stratification, with Gradient Boosting Machines (GBM) achieving area under the curve (AUC) values of 0.879 in validation cohorts [108]. In metabolic engineering, hierarchical approaches systematically optimize production at multiple biological levels, from individual enzymes to genome-scale networks, facilitating the sustainable manufacturing of valuable chemicals [100]. This whitepaper provides an in-depth technical examination of the methodologies, experimental protocols, and visualization frameworks essential for implementing these AI-driven approaches within metabolic research and development.
Machine Learning for Predicting Metabolic Dysfunction-Associated Fatty Liver Disease
Methodological Framework and Model Performance
The predictive framework for MAFLD employs a structured pipeline from data acquisition to model interpretation. Research by Hong et al. utilized data from the 2017-2018 National Health and Nutrition Examination Survey (NHANES), initially comprising 9,254 participants, with 2,007 individuals meeting the final inclusion criteria after rigorous exclusion for missing data [108]. MAFLD was diagnosed based on 2020 international expert consensus criteria, requiring hepatic steatosis (assessed via FibroScan with a Controlled Attenuation Parameter (CAP) ≥274 dB/m) plus at least one of three conditions: overweight/obesity, type 2 diabetes, or evidence of metabolic dysregulation [108].
The study implemented six machine learning algorithms: Decision Tree (DT), Support Vector Machine (SVM), Generalized Linear Model (GLM), Gradient Boosting Machine (GBM), Random Forest (RF), and XGBoost. The Boruta algorithm was used for feature selection to identify the most relevant predictors from anthropometric, demographic, lifestyle, and clinical variables [108]. Model performance was evaluated using cross-validation and a separate validation set, with the GBM algorithm emerging as the top performer, achieving AUC values of 0.875 (training) and 0.879 (validation) while maintaining consistent sensitivity and specificity [108].
Table 1: Performance Metrics of Machine Learning Models for MAFLD Prediction
| Algorithm | Training AUC | Validation AUC | Sensitivity | Specificity |
|---|---|---|---|---|
| GBM | 0.875 | 0.879 | High | High |
| XGBoost | - | 0.784* | - | - |
| Random Forest | - | 0.798* | - | - |
| Logistic Regression | - | 0.831* | - | - |
| MLP | - | 0.823* | - | - |
*External validation results from Zhu et al. [109]
To enhance clinical interpretability, researchers employed SHapley Additive exPlanations (SHAP), a game theory-based approach that quantifies the contribution of each feature to individual predictions. SHAP analysis identified visceral adipose tissue (VAT), body mass index (BMI), and subcutaneous adipose tissue (SAT) as the most influential predictors, with VAT demonstrating the highest feature importance value [108]. This interpretability framework provides transparency into model decision-making, a critical requirement for clinical adoption.
Experimental Protocol for MAFLD Prediction Studies
Data Collection and Preprocessing Protocol:
- Participant Recruitment: Recruit a representative cohort through established study frameworks (e.g., NHANES protocol) or clinical populations.
- MAFLD Assessment: Diagnose MAFLD using transient elastography (FibroScan) with CAP ≥274 dB/m as the steatosis threshold, coupled with assessment of metabolic criteria per international consensus guidelines [108].
- Body Composition Measurement: Quantify total abdominal fat area (TAFA), visceral adipose tissue (VAT), and subcutaneous adipose tissue (SAT) using dual-energy X-ray absorptiometry (DXA) with automated analysis software (e.g., Hologic APEX) [108].
- Clinical Covariate Collection: Record demographic characteristics (age, gender, race, education), lifestyle factors (alcohol consumption, smoking status), and clinical measures (HbA1c, fasting plasma glucose, HDL, LDL, blood pressure) following standardized protocols [108].
- Data Cleansing: Implement exclusion criteria for participants under 20 years old and those with missing hepatic steatosis assessment, obesity-related measurements, or incomplete covariate data.
Machine Learning Implementation Protocol:
- Feature Selection: Apply the Boruta algorithm or similar wrapper method to identify statistically significant predictors from the complete feature set.
- Data Partitioning: Split the dataset into training (70-80%) and validation (20-30%) sets, ensuring proportional representation of MAFLD cases in each partition.
- Model Training: Implement multiple ML algorithms (GBM, XGBoost, RF, SVM, DT, GLM) using cross-validation (e.g., 10-fold) on the training set to optimize hyperparameters.
- Model Validation: Evaluate final model performance on the held-out validation set using AUC, sensitivity, specificity, and accuracy metrics.
- Model Interpretation: Apply SHAP analysis to quantify feature importance and generate individual explanations for model predictions.
Figure 1: Experimental workflow for developing ML models to predict MAFLD risk, from cohort selection to model interpretation.
Machine Learning in Metabolic Engineering and Cellular Design
Hierarchical Metabolic Engineering Framework
The third wave of metabolic engineering leverages synthetic biology and machine learning to systematically rewire cellular metabolism across multiple biological hierarchies. This structured approach enables the creation of efficient microbial cell factories for sustainable chemical production [100]. The hierarchical framework operates across five distinct levels:
- Part Level: Engineering individual biological components, including enzyme optimization through directed evolution, codon optimization, and promoter engineering to fine-tune expression levels [100].
- Pathway Level: Constructing and balancing complete biosynthetic pathways using modular pathway engineering strategies, with ML algorithms predicting optimal enzyme combinations and expression levels [100].
- Network Level: Optimizing genome-scale metabolic networks using constraint-based modeling and flux balance analysis, with ML identifying key knockout targets to redirect metabolic flux [100].
- Genome Level: Implementing multiplex genome editing and regulatory circuit engineering to globally rewire cellular metabolism and regulatory networks [100].
- Cell Level: Engineering cellular properties including stress tolerance, substrate utilization, and product secretion capabilities to enhance overall bioprocess efficiency [100].
Table 2: Metabolic Engineering Strategies and Representative Production Examples
| Engineering Hierarchy | Key Strategies | Target Products | Host Organisms |
|---|---|---|---|
| Part Level | Enzyme engineering, promoter engineering, codon optimization | 3-hydroxypropionic acid, valine | S. cerevisiae, E. coli |
| Pathway Level | Modular pathway engineering, cofactor engineering, transporter engineering | Lysine, lactic acid, muconic acid | C. glutamicum, E. coli |
| Network Level | Flux balance analysis, gene knockout identification, regulatory network modeling | Bioethanol, adipic acid, lycopene | S. cerevisiae, E. coli |
| Genome Level | Genome-scale editing, regulatory circuit engineering, CRISPRi/a | Succinic acid, 1,4-butanediol | E. coli, C. glutamicum |
| Cell Level | Tolerance engineering, chassis engineering, signaling transplant | Butyric acid, glycolic acid | E. coli, Y. lipolytica |
Machine learning accelerates this hierarchical engineering process by predicting enzyme performance, optimizing pathway flux, identifying gene knockout targets, and guiding strain optimization strategies. For example, ML models can predict the functional expression of heterologous enzymes or identify non-obvious gene targets to overcome metabolic bottlenecks [100].
Experimental Protocol for Hierarchical Metabolic Engineering
Strain Construction and Optimization Protocol:
- Pathway Design: Identify or design biosynthetic pathways to target compounds using genome-scale metabolic models and pathway databases.
- DNA Assembly: Synthesize and assemble genetic constructs using standardized biological parts (promoters, RBS, terminators) and advanced DNA assembly techniques.
- Host Transformation: Introduce genetic constructs into suitable microbial chassis (e.g., E. coli, S. cerevisiae, C. glutamicum) via transformation or electroporation.
- Screening and Selection: Implement high-throughput screening methods (colorimetric assays, FACS, HPLC) to identify high-performing variants from libraries.
- Iterative Engineering: Apply the Design-Build-Test-Learn (DBTL) cycle, using ML to analyze screening data and inform subsequent engineering iterations.
Fermentation and Bioprocess Optimization:
- Culture Conditions: Optimize media composition, pH, temperature, and aeration to maximize product titers, rates, and yields (TRY).
- Fed-Batch Fermentation: Implement controlled fed-batch processes with substrate feeding strategies to maintain optimal growth and production conditions.
- Product Quantification: Analyze metabolite concentrations using HPLC, GC-MS, or LC-MS to determine TRY metrics.
- Data Integration: Combine multi-omics data (genomics, transcriptomics, metabolomics) with fermentation performance data to train ML models for further strain improvement.
Figure 2: The five hierarchies of metabolic engineering, from individual parts to whole-cell optimization.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagent Solutions for Metabolic Prediction and Engineering Studies
| Reagent/Material | Function/Application | Example Use Cases |
|---|---|---|
| FibroScan 502 V2 Touch | Non-invasive assessment of hepatic steatosis via Controlled Attenuation Parameter (CAP) | MAFLD diagnosis in clinical cohorts [108] |
| Dual-energy X-ray Absorptiometry (DXA) | Quantitative measurement of body composition (VAT, SAT, TAFA) | Obesity-related metabolic risk assessment [108] |
| Hologic APEX Software | Automated analysis of DXA scan data | Body composition quantification for ML feature set [108] |
| Genome-scale Metabolic Models | Computational representation of cellular metabolism | Prediction of metabolic fluxes and gene knockout targets [100] |
| CRISPR-Cas9 Systems | Precision genome editing for metabolic pathway engineering | Gene knockouts, regulatory element insertion in microbial hosts [100] |
| RNA-seq Reagents | Transcriptomic profiling of engineered strains | Systems-level analysis of metabolic perturbations [100] |
| LC-MS/GС-MS Systems | Metabolite quantification and identification | Measurement of product titers and metabolic fluxes [100] |
| High-throughput Screening Platforms | Rapid evaluation of microbial variant libraries | Identification of optimal enzyme variants or pathway configurations [100] |
The integration of AI and machine learning with metabolic science is creating powerful frameworks for both predicting human metabolic diseases and designing engineered biological systems. In clinical applications, ML models leveraging body composition data and routine biomarkers can achieve high predictive accuracy for conditions like MAFLD, offering non-invasive screening tools for early intervention. In metabolic engineering, hierarchical approaches combined with ML optimization are accelerating the development of microbial cell factories for sustainable chemical production. The experimental protocols, visualization frameworks, and research tools outlined in this whitepaper provide a foundation for researchers to implement these advanced methodologies in their own work, contributing to the growing intersection of artificial intelligence and metabolic science.
The selection of an appropriate microbial host is a foundational decision in metabolic engineering, directly influencing the success of research and development projects aimed at producing novel compounds, biologics, and sustainable chemicals. For researchers, scientists, and drug development professionals entering the field, navigating the landscape of potential host organisms can be daunting. This technical guide provides an in-depth comparison of three central categories of microbial workhorses: the bacterium Escherichia coli, yeast (primarily Saccharomyces cerevisiae), and a selection of specialized microbes gaining prominence for unique applications. Framed within the context of metabolic engineering for beginner researchers, this whitepaper synthesizes current knowledge and recent advances to offer a structured framework for host selection, supported by quantitative data, experimental protocols, and visual guides to core concepts.
Core Characteristics and Comparative Analysis
A fundamental understanding of the inherent strengths and limitations of each host system is crucial for initial screening and selection. The choice between them often involves trade-offs between simplicity, cost, speed, and the biochemical complexity required for the target molecule.
Escherichia coli
As a prokaryotic model organism, E. coli remains one of the most widely used hosts for metabolic engineering due to its rapid growth, well-understood genetics, and cost-effectiveness [110] [111]. It can quickly reach high cell densities in inexpensive, simple media, enabling the production of large quantities of protein or metabolic product in a short time [112]. Its genetic manipulation is highly standardized, with a vast collection of readily available expression vectors and strains [110]. However, as a prokaryote, it lacks the cellular machinery for many eukaryotic post-translational modifications (PTMs), such as complex glycosylation [111]. This makes it unsuitable for producing many complex eukaryotic proteins, including certain therapeutic antibodies. Furthermore, it often misfolds complex eukaryotic proteins, leading to their deposition in insoluble inclusion bodies that require cumbersome refolding procedures [111] [112]. Its environment is also unsuitable for producing large, multi-domain membrane proteins, such as many human G-protein coupled receptors (GPCRs) [110].
Yeast Systems
Yeast, particularly Saccharomyces cerevisiae, offers a compelling compromise between microbial simplicity and eukaryotic complexity. As a eukaryote, it performs many PTMs, including glycosylation, which are essential for the biological activity and stability of many eukaryotic proteins [111]. It also generally folds proteins correctly and can secrete them directly into the culture medium, significantly simplifying downstream purification [111]. Compared to mammalian cells, yeast systems are relatively simple, fast, and inexpensive to culture. A significant limitation, however, is that its glycosylation pattern is of the high-mannose type, which differs from the complex glycans produced by mammalian cells and can be immunogenic in therapeutic applications [110] [111]. Yeast also typically has a slower growth rate and lower overall protein yield than E. coli [111]. Recent research highlighted at the 38th International Specialized Symposium on Yeasts (ISSY38) underscores the trend toward using unconventional yeast species like Scheffersomyces stipitis and Ogataea polymorpha for specific processes, such as lignocellulosic biomass conversion, due to their native superior capabilities [113].
Specialized Microbes
Beyond the two primary workhorses, several specialized microbes are engineered for specific niches. This category includes other bacteria (e.g., Vibrio natriegens, Pseudomonas putida, Bacillus subtilis), algae, and fungal systems [110]. These hosts may offer unique benefits such as unique substrate utilization (e.g., C1 gases like CO2), exceptional stress tolerance, superior secretion capabilities, or specialized metabolism that aligns with a target product [110] [113]. For instance, P. putida is known for its resilience to solvents, while B. subtilis is an excellent secretor of enzymes. The green algae Chlamydomonas reinhardtii is explored for photosynthetic production [110]. The main drawback is that these systems are generally less characterized and have fewer readily available genetic tools, often requiring expert knowledge to implement effectively [110].
Table 1: High-Level Comparison of Key Microbial Hosts for Metabolic Engineering
| Feature | E. coli | Yeast (S. cerevisiae) | Specialized Microbes |
|---|---|---|---|
| Phylogeny | Prokaryote | Eukaryote | Varies (Prokaryote/Eukaryote) |
| Growth Speed | Very High (Rapid) [111] | Moderate [111] | Variable |
| Cost | Low [112] | Low to Moderate | Variable |
| Genetic Tools | Extensive & Mature [110] | Extensive & Mature | Limited, Developing |
| Post-Translational Modifications | Limited or None [111] | Yes, but non-human glycosylation [110] | Varies |
| Membrane Protein Production | Limited, especially for eukaryotes [110] | Good | Varies (can be excellent for some) |
| Typical Yield | High for simple proteins | Moderate | Variable, can be high for specific products |
| Key Advantage | Speed, cost, yield | Eukaryotic folding & secretion, PTMs | Specialized metabolism, stress tolerance |
Table 2: Quantitative Performance in Recent Metabolic Engineering Case Studies (2024-2025)
| Host | Target Product | Titer/ Yield | Key Engineering Strategy | Citation |
|---|---|---|---|---|
| E. coli | Dopamine | 22.58 g/L | Promoter optimization, FADH2-NADH cofactor module, two-stage pH fermentation [69] | [69] |
| E. coli | Biliverdin & Phycoerythrobilin | Successful Synthesis | Heterologous expression of ApHO1 and PebS genes from algae and phage [114] | [114] |
| Yeast | Short Branched-Chain Fatty Acids, Retinaldehyde | Substantially Improved | Rewiring central metabolism by manipulating sugar phosphorylation to create a Crabtree-attenuated chassis [115] | [115] |
| Scheffersomyces stipitis (Yeast) | Glycolic Acid (from PET plastic) | Several-fold better than engineered S. cerevisiae | Exploitation of native biodiversity and metabolic pathways [113] | [113] |
Decision Framework and Experimental Methodology
Selecting the optimal host is a systematic process that begins with a detailed analysis of the target protein or molecule's characteristics.
Host Selection Decision Workflow
A rational decision scheme for host selection, as outlined in a 2023 guide, can be visualized as a logical workflow. This structured approach ensures that the biological requirements of the product drive the decision [110].
Detailed Experimental Protocol: Engineering a High-Yield Dopamine Strain in E. coli
The following protocol, adapted from a 2025 study achieving a landmark dopamine titer of 22.58 g/L, illustrates the multi-faceted approach of modern metabolic engineering in a prokaryotic host [69].
Objective: To construct a plasmid-free, high-yield dopamine-producing E. coli strain (DA-29) and optimize its fermentation.
Key Reagent Solutions:
- Chassis Strain: E. coli W3110 [69].
- Key Genes: hpaBC from E. coli BL21 (for L-DOPA synthesis) and DmDdc from Drosophila melanogaster (for decarboxylation to dopamine) [69].
- Promoters: A set of promoters with varying strengths (e.g., T7, trc, M1-93) for balancing expression of pathway genes [69].
- Fermentation Supplements: Fe²⺠and Ascorbic Acid (to mitigate dopamine oxidation) [69].
Methodology:
Strain Construction and Pathway Assembly:
- Knockout Degradation Pathway: Delete the gene encoding tyramine oxidase (tynA) in E. coli W3110 to prevent dopamine degradation [69].
- Integrate Biosynthesis Module: Constitutively integrate the hpaBC and DmDdc genes into the genome. The hpaBC gene encodes a monooxygenase system that hydroxylates tyrosine to form L-DOPA, while DmDdc decarboxylates L-DOPA to dopamine [69].
- Promoter Optimization: Systematically test different promoters to control the expression levels of hpaBC and DmDdc. The goal is to balance metabolic flux to avoid accumulation or depletion of the intermediate L-DOPA, thereby maximizing dopamine yield [69].
Metabolic Engineering for Enhanced Flux:
- Increase Precursor Availability: Engineer the central metabolism to increase carbon flux towards the aromatic amino acid pathway, enhancing the supply of the precursor tyrosine.
- Cofactor Regeneration: Construct an FADH2-NADH supply module to ensure sufficient cofactors are available for the enzymatic reactions, particularly for the HpaB monooxygenase [69].
- Gene Dosage Amplification: Increase the copy number of key enzyme genes in the pathway to further boost flux towards dopamine [69].
Fermentation Process Optimization:
- Two-Stage pH Strategy: Implement a fed-batch fermentation in a bioreactor with a two-stage pH control.
- Stage 1: Maintain an optimal pH for robust cell growth and biomass accumulation.
- Stage 2: Shift to a lower pH to chemically suppress dopamine degradation [69].
- Co-feeding Strategy: Co-feed Ferrous ions (Fe²âº) and Ascorbic Acid as antioxidants to directly prevent the oxidation of dopamine in the culture medium [69].
- Two-Stage pH Strategy: Implement a fed-batch fermentation in a bioreactor with a two-stage pH control.
Detailed Experimental Protocol: Rewiring Yeast Central Metabolism
A 2025 study demonstrated a novel approach to fundamentally alter the metabolic phenotype of S. cerevisiae from fermentative to respiratory, creating a more efficient chassis for certain products [115].
Objective: To engineer a Crabtree-attenuated S. cerevisiae strain for improved synthesis of mitochondrial products like short branched-chain fatty acids (SBCAs) and retinaldehyde.
Key Reagent Solutions:
- Strain: S. cerevisiae CEN.PK2-1C or BY4741 [115].
- Genetic Parts: Catabolite-regulated promoters and/or sugar kinase genes for dynamic control of sugar phosphorylation [115].
Methodology:
Dynamic Regulation of Sugar Phosphorylation:
- Rationale: The native Crabtree effect (preference for fermentation over respiration, even in oxygen) is driven by high glycolytic flux. Manipulating the initial step of sugar phosphorylation is a key leverage point.
- Implementation: Replace the native, constitutively strong promoters of hexokinase genes (e.g., HXK2) with synthetic, catabolite-repressed promoters. Alternatively, engineer the proteins themselves. This creates a dynamic regulatory network that restricts sugar phosphorylation when sugar levels are high, preventing carbon overflow to ethanol [115].
Multidimensional Phenotyping:
- Characterization: Confirm the successful metabolic switch using several methods:
- Physiology: Measure significantly reduced ethanol production and increased respiratory flux.
- Transcriptomics: Analyze genome-wide expression changes to verify a shift towards respiratory gene programs.
- Morphology: Observe enlarged, more developed mitochondria consistent with a respiratory phenotype [115].
- Characterization: Confirm the successful metabolic switch using several methods:
Biochemical Production Validation:
- Test Platform Performance: Use the engineered Crabtree-attenuated strain for the synthesis of target compounds that benefit from a respiratory metabolism, such as:
- SBCAs: Feed amino acids and measure the improved yield from mitochondrial catabolism.
- Retinaldehyde: Demonstrate the accumulation of the aldehyde product instead of its further reduction to retinol, leveraging the altered redox state of the cell [115].
- Test Platform Performance: Use the engineered Crabtree-attenuated strain for the synthesis of target compounds that benefit from a respiratory metabolism, such as:
The metabolic pathway and engineering strategy for this approach can be visualized as follows:
The Scientist's Toolkit: Essential Research Reagents
Successful metabolic engineering relies on a suite of standard and specialized reagents. The following table details key materials and their functions as featured in the cited research and the broader field.
Table 3: Key Research Reagent Solutions for Metabolic Engineering
| Reagent / Material | Function / Application | Examples / Notes |
|---|---|---|
| Expression Vectors | Carry the gene of interest and control its expression in the host. | Varies by host (e.g., pET vectors for E. coli, YEplac series for yeast). Available from institutional/non-profit repositories [110]. |
| Chassis Strains | The host organism engineered for production. | E. coli: W3110, BL21(DE3) [69]. Yeast: CEN.PK2-1C, BY4741 [115]. Specialized strains with auxotrophies or specific deletions are common. |
| Gene Editing Tools | Enables precise genomic modifications (knock-in, knockout, mutation). | CRISPR-Cas systems, MAGE for E. coli [69] [116]. Essential for pathway engineering and eliminating competing reactions. |
| Promoter Libraries | Fine-tune the expression levels of multiple genes in a pathway. | A set of promoters with known, varying strengths (e.g., T7, trc, M1-93 for E. coli) [69]. Critical for metabolic balance. |
| Cofactor Supplements | Provide essential cofactors for enzymatic reactions or prevent product degradation. | Fe²âº, Ascorbic Acid (as antioxidant), FAD, NADH [69]. |
| Biosensors | Link production of a target molecule to a detectable output (e.g., fluorescence). | Engineered transcription factors or GPCRs in yeast [113]. Enable high-throughput screening of optimized strains. |
| Analytical Standards | Quantify target product and intermediates during fermentation. | Pure dopamine, biliverdin, retinaldehyde, etc. Used for calibration in HPLC, GC-MS, LC-MS. |
The comparative analysis presented in this guide underscores that there is no single "best" microbial host for metabolic engineering. The optimal choice is a strategic decision contingent upon the specific characteristics of the target product and the project's constraints. E. coli excels in cost-effective, high-titer production of simple proteins and metabolites. Yeast provides a powerful eukaryotic platform for proteins requiring basic PTMs and for complex pathway engineering where its subcellular organization is beneficial. Specialized microbes offer tailored solutions for unique challenges, from consuming alternative feedstocks to producing exotic natural products.
The future of the field, as highlighted in recent literature, points toward increased sophistication and integration. Key trends include the rise of "next-generation metabolic engineering," which leverages omics data from diverse natural strains to identify optimal engineering strategies [113], and the growing integration of artificial intelligence to predict pathway efficiency and design optimal enzymes and regulatory networks [56] [116]. Furthermore, the creation of highly engineered non-conventional chassis—such as Crabtree-attenuated yeast or E. coli strains with custom cofactor systems—will continue to expand the boundaries of what is possible [69] [115]. For the beginner researcher, mastering the foundational knowledge of host systems laid out in this guide provides the essential platform from which to engage with these exciting future developments.
For metabolic engineers, particularly those new to the field, the transition from shake flasks to bioreactor systems represents a critical validation step in bioprocess development. This scale-up is not merely an increase in volume but a fundamental test of whether a metabolically engineered pathway, often optimized in a microtiter plate or shake flask, can function with the same efficiency and yield under the controlled yet dynamic conditions of a bioreactor [117] [24]. In the context of metabolic engineering for the production of pharmaceuticals, biofuels, or other biochemicals, successful scale-up is the gateway to industrial relevance, confirming that the engineered host organism, such as E. coli or S. cerevisiae, can maintain its optimized metabolic fluxes and produce the target compound at a commercially viable scale [24].
The shake flask has been a cornerstone of biological cultivations for nearly a century due to its simple handling and versatility for screening media compositions and parameters [117]. However, the environment within a shake flask is fundamentally different from that of a stirred-tank bioreactor. Scaling up a process, therefore, involves addressing key engineering principles to ensure the metabolic performance of the engineered organism translates effectively to the new system [118]. This guide provides an in-depth technical framework for this essential validation, ensuring that the promise of metabolic engineering at the bench can be realized in pilot and production-scale systems.
Fundamental Differences Between Shake Flasks and Bioreactors
Understanding the distinct physical and chemical environments of shake flasks and bioreactors is the first step in designing a successful scale-up strategy. The table below summarizes the core differences that impact process performance and microbial physiology.
Table 1: Key differences between shake flask and stirred-tank bioreactor systems.
| Parameter | Shake Flask | Stirred-Tank Bioreactor |
|---|---|---|
| Mixing Mechanism | Shaking (orbital or linear) [117] | Mechanical agitation via impellers [119] |
| Oxygen Supply | Surface aeration from shaking [119] | Sparged air/oxygen and agitation [119] |
| Parameter Control | Limited (temperature only, typically) [120] | Tight, automated control of pH, DO, temperature [119] [118] |
| Power Input (P/V) | Calculated from shaking frequency, diameter, and fill volume [117] [121] | Directly related to impeller design and agitation speed [118] |
| Shear Stress | Generally low [121] | Can be significant, depends on impeller type and speed [119] [120] |
| Heat Transfer | Passive through flask walls [119] | Active via thermal jacketing or internal coils [119] |
| Feeding Strategies | Typically batch [119] | Batch, fed-batch, or continuous [119] [122] |
| Surface Area to Volume Ratio | Relatively high | Decreases significantly with scale [118] |
A critical concept in scale-up is the distinction between scale-dependent and scale-independent parameters [118]. Scale-independent parameters, such as pH, temperature, dissolved oxygen (DO) concentration, and media composition, are typically optimized in small-scale bioreactors and can be kept constant during scale-up. In contrast, scale-dependent parameters, including impeller rotational speed, gas-sparging rates, and power input per unit volume (P/V), are inherently affected by the bioreactor's geometric configuration and operating parameters and must be re-optimized at each scale [118].
Core Scale-Up Parameters and Methodologies
The goal of scale-up is not to keep all physical parameters identical, which is physically impossible, but to define an operating range that maintains the physiological state and productivity of the culture across scales [118]. The following parameters are central to this effort.
Oxygen Mass Transfer
In aerobic bioprocesses, the Oxygen Transfer Rate (OTR) must meet the cellular demand to prevent oxygen limitation, which can alter metabolism and reduce yield [119] [120]. The key parameter is the volumetric oxygen mass transfer coefficient, kLa. In shake flasks, OTR is influenced by shaking frequency, flask diameter, and liquid fill volume [117]. In bioreactors, kLa is controlled by adjusting the agitation speed and gas flow rate [119]. Sufficient dissolved oxygen tension must be maintained, often through cascade control systems that sequentially increase agitation, gas flow, and oxygen concentration in the sparged gas [119].
Volumetric Power Input (P/V)
Power input is a crucial engineering parameter for comparing cultivation conditions. It influences mixing, shear stress, and gas dispersion. A successful scale-up case for the production of 6-pentyl-α-pyrone by Trichoderma harzianum demonstrated that maintaining a constant P/V of 0.4 kW/m³ from 500 mL shake flasks to 10 L bioreactors resulted in equivalent maximum product concentration [121]. However, the study also highlighted that physiological responses like growth rate and productivity can still differ between the systems due to different shear environments, even at the same P/V [121]. The average volumetric power input in an unbaffled shake flask can be calculated using the following engineering correlation [117]:
Where Ne' is the modified Newton number, Ï is the liquid density, n is the shaking frequency, d is the maximum flask diameter, and VL is the liquid filling volume.
Mixing and Agitation
Adequate mixing ensures homogeneity of nutrients, metabolites, and cells, preventing the formation of gradients in pH, substrate, or dissolved oxygen [118]. Mixing time increases with bioreactor scale, and large-scale bioreactors can develop zones with varying substrate concentrations, which can affect the metabolism and product quality of the culture [118]. Impeller selection is critical; for example, Rushton-type impellers are common for high oxygen demand but generate high shear, while pitched-blade impellers are better for shear-sensitive cells like mammalian or filamentous fungal cultures [119].
Table 2: Common scale-up criteria and their implications. Adapted from [118].
| Scale-Up Criterion | Primary Objective | Key Impact & Consideration |
|---|---|---|
| Constant Power per Unit Volume (P/V) | Maintain similar shear and mixing energy. | Increases mixing time at larger scales; may not protect against shear stress. |
| Constant Oxygen Transfer Coefficient (kLa) | Maintain equivalent oxygen supply capacity. | Common for aerobic processes; may require different P/V and gas flow at different scales. |
| Constant Impeller Tip Speed | Maintain similar maximum shear forces. | Results in a significant decrease in P/V, potentially compromising mixing. |
| Constant Mixing Time | Maintain homogeneity. | Results in a dramatic, often infeasible, increase in P/V at large scale. |
A Practical Scale-Up Validation Protocol
The following workflow provides a structured, experimental approach to validate the scale-up of a metabolically engineered process from a shake flask to a bench-scale (e.g., 5 L) bioreactor.
Diagram 1: Experimental scale-up validation workflow.
Pre-Scale-Up Shake Flask Characterization
Before moving to a bioreactor, the shake flask process must be thoroughly characterized. This goes beyond final titer and includes online monitoring of dissolved oxygen tension (DOT) using specialized flasks to determine the maximum oxygen transfer rate (OTRmax) and the point of oxygen limitation [117]. The metabolic profile (e.g., substrate consumption, by-product formation) should be established. The power input (P/V) should be calculated based on the shaking frequency, diameter, and fill volume to provide a baseline for scale-up [117] [121]. A common pitfall is using inappropriate flask sealing, which can severely limit oxygen transfer; sterile barriers must allow sufficient gas exchange [117].
Bench-Scale Bioreactor Experimental Setup
- Bioreactor Configuration: For a 5 L system, standard configuration includes two Rushton impellers for microbial systems or pitched-blade impellers for shear-sensitive organisms, a ring sparger for aeration, and calibrated probes for pH and DO [119] [122].
- Scale-Up Calculation: Using constant
kLaas the primary criterion is common for aerobic processes. Correlations forkLaas a function of operating conditions are used to determine the initial agitation and aeration setpoints for the 5 L vessel to match thekLaachieved in the characterized shake flasks [118]. - Process Control Strategy: Set up control loops for pH (using acid/base addition), temperature (via thermal jacket), and dissolved oxygen (via agitation speed/air flow cascade) [119] [122]. Define a feeding strategy if moving from a batch to a fed-batch process.
Execution, Monitoring, and Analysis
Run the bioreactor experiment, collecting samples at regular intervals. Compare the following against the shake flask control:
- Growth Kinetics: Maximum cell density, specific growth rate, and viability.
- Metabolic Data: Substrate consumption rate, production of target metabolite, and formation of by-products (e.g., lactate, acetate).
- Process Performance: Final product titer, yield, and productivity [121].
Crucially, analyze the product quality. As demonstrated in a perfusion process scale-up case, changes in scale can alter critical quality attributes, such as charge distribution, even if titer is maintained [123]. This may necessitate further adjustments to perfusion rates or pH control to match the quality profile of the small-scale product [123].
The Scientist's Toolkit: Essential Research Reagent Solutions
The following table lists key materials and equipment essential for conducting rigorous scale-up validation experiments.
Table 3: Key reagents and equipment for scale-up experiments.
| Item | Function in Scale-Up Validation |
|---|---|
| Baffled and Unbaffled Shake Flasks | Baffled flasks can increase OTRmax for high-density cultures, but unbaffled are standard for most applications and are better characterized [117]. |
| Gas-Permeable Seals | Sterile seals (e.g., foam plugs, specialized caps) that allow sufficient CO2/O2 exchange are critical to prevent oxygen limitation in flasks [117]. |
| Respiration Activity Monitoring System (RAMOS) | A specialized flask system that allows online monitoring of OTR, CTR, and RQ in shake flasks, providing crucial data for kLa-based scale-up [117]. |
| Single-Use Bioreactors | Practical for pharma/food industries, reducing cross-contamination risk and cleaning validation; may have reduced OTR for high-oxygen-demand processes [119]. |
| Internal or External Spin Filters | Essential for perfusion processes to retain cells; filter mesh type and mechanical strength are critical scale-up factors to prevent fouling or deformation [123]. |
| Anti-Foam Agents | Chemicals used to control foam formation induced by aeration and agitation, though their addition can affect process performance and product purity [119]. |
| Cell Retention Devices | For perfusion processes, reliable devices are needed. Performance consistency across scales is a major challenge [123]. |
Visualization of Oxygen Transfer Principles
The core challenge of scaling aerobic processes is understanding and matching oxygen supply. The following diagram illustrates the key components of oxygen transfer in a stirred-tank bioreactor.
Diagram 2: Oxygen transfer dynamics in a bioreactor.
The Oxygen Transfer Rate (OTR) is the product of the mass transfer coefficient (kLa) and the driving force, which is the difference between the saturation concentration of oxygen (C*) and the actual dissolved oxygen concentration in the bulk liquid (Câ‚—) [119]. The kLa value is increased by higher agitation (which breaks bubbles into smaller ones, increasing surface area) and a higher aeration rate. The controller maintains Câ‚— at a defined setpoint by manipulating these parameters, ensuring the OTR meets the cellular oxygen demand [119].
Economic and Sustainability Assessment of Engineered Pathways
Metabolic engineering is devoted to the directed modulation of metabolic pathways for metabolite overproduction or the improvement of cellular properties, including native pathway engineering and the synthesis of heterologous pathways for converting microorganisms into microbial cell factories [124]. The field employs experimental, computational, and modelling approaches for the elucidation of metabolic pathways and their manipulation by genetic, media, or other environmental means [124]. An economic and sustainability assessment provides a critical framework for evaluating the viability of these engineered pathways, balancing technical performance with cost-effectiveness and environmental impact. For beginners in metabolic engineering research, understanding this balance is essential for directing research toward solutions that are not only scientifically innovative but also practically implementable on an industrial scale.
The core challenge in this field lies in designing pathways that achieve high yields while minimizing resource consumption and environmental footprint. Computational tools and quantitative assessments enable researchers to predict metabolic behavior, identify bottlenecks, and select the most promising engineering strategies before committing to extensive laboratory work. This guide provides a comprehensive technical foundation for conducting such assessments, covering computational design, economic evaluation, sustainability metrics, and experimental validation protocols relevant to researchers, scientists, and drug development professionals.
Computational Pathway Design and Yield Analysis
The rational design of efficient metabolic pathways requires sophisticated computational tools to predict performance and identify optimal genetic modifications. The development of a high-quality cross-species metabolic network model (CSMN) and a quantitative heterologous pathway design algorithm (QHEPath) has enabled systematic evaluation of biosynthetic scenarios [4]. These tools address the critical challenge of quantitatively predicting potential pathways for breaking stoichiometric yield limits in host organisms, a fundamental aspect of improving the economic viability of microbial cell factories.
Computational Methods for Pathway Design
The QHEPath algorithm was developed to explore heterologous reactions for enhancing product yield (YP) to break the native yield limit (YP0) of host organisms [4]. This method evaluates whether YP0 can be surpassed by introducing heterologous reactions by comparing the increase in maximum pathway yield (Ym^P) relative to Y_P0. The algorithm systematically identifies specific heterologous reactions that contribute to breaking the yield limit, distinguishing them from reactions merely required for basic producibility [4]. The methodology involves:
- Model Reconstruction and Quality Control: A quality-control workflow refines reactions from biochemical databases to construct a high-quality CSMN by automatically eliminating various types of errors, including infinite generation of reducing equivalents, energy, or metabolites that would otherwise lead to incorrect yield calculations [4].
- Pathway Yield Calculation: Using flux balance analysis (FBA) on the curated metabolic model to compute the maximum theoretical yield of target products from specified substrates [4].
- Heterologous Reaction Identification: Systematically identifying heterologous reactions from a universal biochemical database that, when integrated into the host metabolism, can enhance product yield beyond native limits [4].
Through systematic calculations using CSMN and QHEPath across 12,000 biosynthetic scenarios involving 300 products and 4 substrates in 5 industrial organisms, researchers revealed that over 70% of product pathway yields can be improved by introducing appropriate heterologous reactions [4]. This comprehensive analysis identified thirteen distinct engineering strategies, categorized as carbon-conserving and energy-conserving, with 5 strategies effective for over 100 products [4].
Key Engineering Strategies for Yield Enhancement
Table 1: Classification and Prevalence of Metabolic Engineering Strategies for Yield Enhancement
| Strategy Category | Number of Products Affected | Primary Mechanism | Representative Examples |
|---|---|---|---|
| Carbon-Conserving Strategies | >100 products | Reduces carbon loss during conversion | Non-oxidative glycolysis (NOG) |
| Energy-Conserving Strategies | >100 products | Improves ATP/energy efficiency | Energy-coupled transport systems |
| Combined Carbon & Energy Approaches | Multiple products | Simultaneous carbon and energy optimization | Integrated redox-cofactor balancing |
The systematic computational analysis revealed that the non-oxidative glycolysis (NOG) pathway exemplifies a carbon-conserving strategy that has been experimentally validated to enhance yields of products such as farnesene and poly(3-hydroxybutyrate) (PHB) in E. coli beyond native stoichiometric limits [4]. These findings demonstrate the power of computational approaches to identify universally applicable engineering principles that can guide experimental efforts across diverse products and host systems.
Computational Pathway Design Workflow
Economic Assessment Framework
The economic viability of engineered metabolic pathways must be evaluated through comprehensive assessment frameworks that account for both capital and operational expenditures alongside potential revenues. Advanced economic modeling for bioprocesses incorporates life-cycle cost-benefit analyses that evaluate financial viability, energy efficiency, and policy relevance of technological innovations [125]. These analyses are particularly crucial when assessing emerging technologies such as carbon capture and storage (CCS), AI-driven emissions monitoring, and nanotechnology-enhanced filtration, which present varying economic profiles [125].
Key Economic Metrics for Pathway Evaluation
Table 2: Economic Assessment Metrics for Engineered Metabolic Pathways
| Economic Metric | Calculation Method | Acceptance Threshold | Application in Metabolic Engineering |
|---|---|---|---|
| Return on Investment (ROI) | (Net Benefits / Total Costs) × 100 | >15-20% for biotech | Evaluates profitability of engineering efforts |
| Capital Expenditure (CAPEX) | Sum of equipment, installation, engineering costs | Project-specific | Bioreactor systems, downstream processing |
| Operational Expenditure (OPEX) | Raw materials, utilities, labor, maintenance | Minimized relative to output | Substrate costs, energy consumption |
| Cost-Benefit Ratio | Total Benefits / Total Costs | >1.0 (positive) | Ranges from $30-40:$1 for high-impact projects [125] |
| Payback Period | Initial Investment / Annual Cash Inflow | <5-7 years | Time to recoup strain development costs |
Economic analyses repeatedly demonstrate that proactive investments in advanced technologies generate substantial long-term returns. Research indicates that economies can save $30–40 in health and productivity-related expenses for every $1 invested in air pollution mitigation technologies [125]. While this specific example addresses environmental technologies, similar economic principles apply to metabolic engineering, where initial investments in strain development and process optimization can yield substantial returns through reduced production costs and increased yields.
Cost-Benefit Analysis of Engineering Strategies
The economic assessment of metabolic engineering strategies must account for both direct costs and broader economic externalities. For example, carbon capture and storage presents significant capital expenditure (up to $500 million per facility) but offers long-term returns through carbon credits and enhanced oil recovery, yielding up to $30–40 in economic benefits for every $1 invested [125]. Similarly, AI-based monitoring systems demonstrate strong economic efficiency by reducing energy consumption in industrial operations by up to 15% and improving regulatory compliance at a larger scale [125]. These principles translate to metabolic engineering through:
- Strain Development Costs: Including DNA synthesis, pathway assembly, screening, and optimization efforts
- Process Integration Expenses: Modifications to fermentation and downstream processing requirements
- Operational Savings: Reduced substrate costs, higher productivity, and decreased waste treatment
- Environmental Compliance: Potential carbon credit generation and regulatory advantage
Sustainability and Environmental Impact Assessment
Sustainability assessment in metabolic engineering extends beyond economic considerations to encompass environmental impacts, resource efficiency, and social dimensions. The integration of advanced pollution control technologies with renewable energy systems, such as hydrogen-powered pollution control units and solar-driven filtration, amplifies both environmental and economic benefits [125]. This alignment of metabolic engineering with broader sustainability goals creates opportunities for developing processes that simultaneously achieve economic resilience and environmental stewardship.
Life Cycle Assessment Methodology
Life cycle assessment provides a standardized framework for evaluating the environmental impacts of engineered metabolic pathways across their complete value chain. The methodology includes:
- Goal and Scope Definition: Clearly defining system boundaries, functional units, and impact categories
- Life Cycle Inventory: Quantifying energy and material inputs and environmental releases throughout the product life cycle
- Impact Assessment: Evaluating potential environmental and human health impacts using established impact categories
- Interpretation: Analyzing results to identify significant issues, evaluate completeness, and draw conclusions
For metabolic engineering applications, LCA typically focuses on metrics such as greenhouse gas emissions, fossil energy consumption, water usage, and land use compared to conventional production methods. The systematic review process following PRISMA guidelines, including structured literature selection, inclusion/exclusion criteria, and data extraction, provides a robust foundation for comprehensive sustainability assessments [125].
Sustainability Metrics for Engineered Pathways
Table 3: Key Sustainability Indicators for Metabolic Engineering Projects
| Sustainability Dimension | Key Performance Indicators | Measurement Methods | Benchmark Values |
|---|---|---|---|
| Environmental | Carbon footprint (COâ‚‚eq/kg product) | Life Cycle Assessment | <2.0 kg COâ‚‚eq/kg for chemicals |
| Environmental | Water consumption (L/kg product) | Water footprint accounting | Minimize relative to conventional processes |
| Environmental | Non-renewable energy input (MJ/kg) | Cumulative energy demand | <50% of conventional processes |
| Economic | Production cost ($/kg) | Techno-economic analysis | Competitive with petrochemical routes |
| Social | Toxicity/hazard potential | Risk assessment | Reduced hazard classification |
The sustainability profile of engineered pathways is significantly influenced by substrate selection, energy source, and downstream processing requirements. The use of renewable feedstocks, integration with waste streams, and implementation of energy-efficient operations can dramatically improve the environmental performance of bioprocesses. Furthermore, innovations such as AI-driven monitoring can reduce energy consumption in industrial operations by up to 15% [125], demonstrating how digital technologies can contribute to sustainability goals in metabolic engineering.
Experimental Protocols and Validation
Experimental validation of computationally predicted pathways requires rigorous methodologies to quantify metabolic fluxes, pathway activities, and product yields. A comprehensive methods guide for studying different aspects of cell and tissue metabolism provides a framework for selecting appropriate techniques based on research goals [126]. These methods enable researchers to move from in silico predictions to empirical validation, closing the design-build-test cycle essential for metabolic engineering advancement.
Metabolomics and Flux Analysis Protocols
Untargeted Metabolomics Protocol:
- Sample Harvesting: Rapid quenching of metabolism using cold organic solvents (e.g., acetonitrile at -40°C) to stop all enzymatic activity immediately [126]
- Metabolite Extraction: Using cold organic solvents that precipitate proteins while extracting metabolites of interest [126]
- Chromatographic Separation:
- Liquid Chromatography for polar metabolites
- Gas Chromatography for volatile metabolites [126]
- Mass Spectrometry Analysis: Detection of mass-to-charge ratios (m/z) for metabolite identification and quantification [126]
- Data Analysis: Using platforms like MetaboAnalyst 6.0 for statistical analysis, pathway enrichment, and visualization [126]
Metabolic Flux Analysis using Isotope Tracing:
- Isotope Labeling: Feeding cells with (^{13})C-labeled substrates (e.g., [U-(^{13})C]glucose)
- Time-course Sampling: Collecting samples at multiple time points after isotope introduction
- Mass Spectrometry Analysis: Measuring isotopic enrichment in intracellular metabolites
- Flux Calculation: Using computational models to infer metabolic reaction rates from isotopic labeling patterns
Experimental Validation Workflow
Pathway Simulation and MGWAS Integration
Simulations of metabolic pathway models can enhance the interpretation of metabolome genome-wide association studies (MGWAS) by investigating the influence of genetic variants on metabolite concentrations [20]. This approach involves:
- Model Selection: Using established metabolic models with differential equations, initial metabolite concentrations, and enzyme reaction rates derived from experimental data [20]
- Parameter Adjustment: Systematically adjusting enzyme reaction rates to simulate the effects of genetic variations [20]
- Concentration Prediction: Simulating changes in metabolite levels resulting from altered enzyme activities [20]
- Experimental Correlation: Comparing simulation results with empirical MGWAS data to validate predictions [20]
This integrated approach allows researchers to distinguish true genetic associations from false positives, identify enzymes with minimal metabolic impact, and prioritize genetic variants for experimental investigation [20].
Research Reagent Solutions and Essential Materials
Table 4: Key Research Reagents for Metabolic Engineering Studies
| Reagent/Material | Function/Application | Technical Specifications | Sustainability Considerations |
|---|---|---|---|
| (^{13})C-Labeled Substrates | Metabolic flux analysis | >99% isotopic purity; [U-(^{13})C]glucose common | Potential recycling of labeling patterns |
| Cold Organic Solvents | Metabolic quenching & extraction | HPLC-grade acetonitrile, methanol | Recycling programs for solvent waste |
| Mass Spectrometry Standards | Instrument calibration & quantification | Stable isotope-labeled internal standards | Reduced consumption through method optimization |
| DNA Assembly Kits | Pathway construction | Gibson Assembly, Golden Gate Shuffling kits | Temperature reduction for energy savings |
| Growth Media Components | Cell cultivation & maintenance | Defined chemical composition | Agricultural sustainability of carbon sources |
| Chromatography Columns | Metabolite separation | HILIC, reversed-phase, GC columns | Extended lifespan through proper maintenance |
The selection of research reagents significantly influences both the experimental outcomes and the sustainability profile of metabolic engineering research. Proper management of these materials, including recycling programs for solvents and optimization of reagent usage, can substantially reduce the environmental impact of research activities while maintaining scientific rigor. Furthermore, the trend toward miniaturization and automation in laboratory workflows offers opportunities to reduce reagent consumption and waste generation while increasing experimental throughput.
Integrating Multi-Omics Data for Comprehensive System Validation
Metabolic engineering relies on the rational redesign of biological systems to achieve specific production goals, such as the efficient synthesis of natural products or biofuels. For beginners in the field, it is crucial to understand that this process is fundamentally guided by the Design-Build-Test-Learn (DBTL) cycle [41]. Within this framework, multi-omics data integration serves as the critical link between the "Test" and "Learn" phases, enabling a systems-level validation of engineered strains. The core challenge in metabolic engineering is that perturbations—such as gene knockouts or heterologous pathway insertion—create complex, system-wide ripple effects. Analyzing only a single molecular layer (e.g., transcriptomics) provides an incomplete picture. Integrating data from transcriptomics, proteomics, and metabolomics offers a comprehensive view, from genetic instruction (transcriptome) through functional machinery (proteome) to ultimate chemical activity (metabolome) [127] [128]. This holistic validation is essential for moving beyond simple hits to developing robust, high-titer bioprocesses.
Core Methodologies for Multi-Omics Data Integration
Several computational strategies have been developed to integrate disparate omics data types. These methods can be broadly categorized, each with distinct strengths and applications in metabolic engineering.
Combined Omics and Correlation-Based Integration
This approach involves merging different omics datasets to identify direct or indirect relationships between molecular entities.
- Gene–Metabolite Network Analysis: This method visualizes interactions between genes and metabolites in a biological system. Researchers first collect gene expression and metabolite abundance data from the same biological samples. These data are then integrated using statistical methods, like the Pearson correlation coefficient (PCC), to identify genes and metabolites that are co-regulated. The resulting network, which can be visualized using software like Cytoscape, helps identify key regulatory nodes and pathways involved in metabolic processes [127].
- Gene Co-Expression Analysis Integrated with Metabolomics: This powerful approach identifies groups of genes (modules) with similar expression patterns that may participate in the same biological pathways. These gene modules are then linked to metabolite profiles from metabolomics data. By calculating the correlation between the representative expression profile of a module (its eigengene) and metabolite intensity patterns, researchers can identify which metabolic pathways are co-regulated with specific gene sets [127]. This provides critical insight into the regulation of metabolic pathways for target compounds.
Machine Learning and Network-Based Integration
These more advanced methods are essential for handling the high dimensionality and heterogeneity of multi-omics data.
- Network-Based Approaches: These methods provide a holistic view of relationships among biological components. For instance, Similarity Network Fusion (SNF) builds a similarity network for each omics data type separately (e.g., transcriptomics, proteomics), which are subsequently merged. Edges with high associations in each omics network are highlighted, revealing underlying biological structures that are not apparent from single-omics analyses [127] [128]. These networks are invaluable for identifying critical interaction hubs that represent potential metabolic engineering targets.
- Machine Learning Strategies: These techniques utilize one or more types of omics data to comprehensively understand responses at the classification and regression levels. Methods include matrix factorization (e.g., MOFA+) and neural network-based approaches (e.g., variational autoencoders) [127] [129]. They can integrate matched data (from the same cell) or unmatched data (from different cells), and are particularly useful for patient stratification and predicting metabolic behavior under different genetic or environmental perturbations [128] [129].
Table 1: Summary of Primary Multi-Omics Integration Methods
| Integration Approach | Example Methods | Key Principle | Primary Application in Metabolic Engineering |
|---|---|---|---|
| Correlation-Based | Gene–Metabolite Networks, Pearson Correlation Coefficient (PCC) | Applies statistical correlations to uncover relationships between different omics layers. | Identifying co-regulated genes and metabolites; hypothesis generation. |
| Network-Based | Similarity Network Fusion (SNF), Interactome Analysis | Builds and merges similarity networks from each omics type to reveal system-level interactions. | Discovering non-obvious interaction hubs and key regulatory targets. |
| Machine Learning | MOFA+ (Factor Analysis), Variational Autoencoders | Uses statistical and deep learning models to find latent factors or features that explain variance across omics data. | Predictive modeling of metabolic flux; classifying strain performance. |
Experimental Protocols for Multi-Omics Studies
A typical workflow for a multi-omics study in metabolic engineering involves sample preparation, data generation, and integrated data analysis.
Protocol for an Integrated Transcriptomics and Metabolomics Study
This protocol is designed to validate the systemic impact of a metabolic engineering intervention, such as the expression of a heterologous pathway.
Sample Preparation:
- Strain Design: Engineer your host strain (e.g., E. coli or S. lividans) with the desired genetic modifications (e.g., gene knockout, promoter swap, heterologous pathway insertion). Include a wild-type or empty-vector control strain.
- Cultivation: Grow biological replicates (n ≥ 4) of both engineered and control strains under defined, controlled conditions (e.g., in a bioreactor to ensure consistency).
- Harvesting: Simultaneously quench metabolism and harvest cells from all cultures at the same physiological stage (e.g., mid-exponential phase) to ensure data comparability. Rapidly cool samples using liquid nitrogen or cold methanol to instantly halt enzymatic activity.
Data Generation:
- Transcriptomics:
- Extract total RNA using a kit designed to preserve RNA integrity (RIN > 8.0).
- Prepare sequencing libraries (e.g., using Illumina kits) and perform RNA sequencing (RNA-seq) on a suitable platform (e.g., Illumina NovaSeq) to a minimum depth of 20 million reads per sample.
- Process raw sequencing data: perform quality control (FastQC), align reads to a reference genome (HISAT2, STAR), and quantify gene-level counts (featureCounts).
- Metabolomics:
- Extract metabolites from the quenched cell pellets using a solvent system like methanol:acetonitrile:water.
- Analyze samples using Liquid Chromatography-Mass Spectrometry (LC-MS) in both positive and negative ionization modes.
- Process raw LC-MS data: perform peak picking, alignment, and annotation using software (e.g., XCMS, CAMERA) and reference databases (e.g., HMDB, KEGG).
- Transcriptomics:
Integrated Data Analysis:
- Preprocessing: Normalize and scale the transcript count data and metabolite intensity data separately.
- Correlation Analysis: Conduct a Pearson or Spearman correlation analysis between all significantly changing transcripts and metabolites (e.g., p-value < 0.05, fold-change > 2).
- Network Construction: Input the significant gene-metabolite correlation pairs (e.g., |r| > 0.8) into Cytoscape.
- Validation: Overlay the network with pathway information from databases like KEGG to identify enriched metabolic pathways. This helps validate if the engineered pathway is active and reveals any unexpected systemic responses.
Diagram 1: Integrated transcriptomics and metabolomics workflow.
Protocol for Multi-Omics Data Integration Using Machine Learning
For projects involving multiple engineered strains and high-dimensional data, machine learning provides a powerful integration framework.
Data Compilation and Preprocessing:
- Compile your transcriptomics, proteomics, and/or metabolomics data matrices from all tested strains and conditions.
- Perform rigorous normalization, log-transformation, and batch-effect correction on each dataset individually. Handle missing values appropriately (e.g., imputation).
Model Training and Application:
- For Matched Data (from the same sample): Use a tool like MOFA+, which is a factor analysis model.
- Input your normalized, multi-omics data matrices.
- MOFA+ will identify a set of latent factors that capture the common variance across all omics datasets.
- Interpret the factors by examining the loadings for each omics view to see which features (genes, proteins, metabolites) drive each factor.
- For Unmatched Data (from different samples): Use a tool like GLUE (Graph-Linked Unified Embedding), which uses graph variational autoencoders and prior biological knowledge (e.g., pathway databases) to align the different omics spaces into a common embedding [129].
- For Matched Data (from the same sample): Use a tool like MOFA+, which is a factor analysis model.
Interpretation for Strain Validation:
- Correlate the inferred latent factors (from MOFA+) or the cell embeddings (from GLUE) with strain performance metrics (e.g., product titer, yield, productivity).
- Identify which combinations of molecular features across the omics layers are predictive of high performance, providing a validated, multi-layered signature of a successful engineering outcome.
Success in multi-omics integration depends on both computational tools and high-quality experimental reagents.
Table 2: Key Research Reagent Solutions for Multi-Omics Studies
| Item | Function | Example Use Case |
|---|---|---|
| Heterologous Host Strains | Genetically tractable production chassis with precursor pathways. | S. lividans K4-114 (clean background, native antibiotic pathways knocked out) for polyketide production [101]. |
| RNA Stabilization Kits | Preserve RNA integrity instantly upon cell harvest, preventing degradation. | Ensuring transcriptomics data accurately reflects the in vivo state at the time of sampling. |
| LC-MS Grade Solvents | High-purity solvents for metabolomics to minimize background noise and ion suppression. | Extracting and analyzing intracellular metabolites with high sensitivity and reproducibility. |
| Pathway Databases (KEGG, MetaCyc) | Curated knowledge bases of metabolic pathways and enzyme functions. | Annotating metabolites and constructing enzyme-metabolite networks for data interpretation [127]. |
| Cytoscape Software | Open-source platform for visualizing complex molecular interaction networks. | Visualizing and analyzing gene-metabolite correlation networks [127]. |
Visualization of Multi-Omics Integration Concepts
The following diagram illustrates the logical relationship between different omics layers and the computational methods used to integrate them, highlighting how this process feeds back into the DBTL cycle.
Diagram 2: Multi-omics data integration and the DBTL cycle.
The integration of multi-omics data is no longer an optional advanced technique but a core component of rigorous system validation in modern metabolic engineering. By moving beyond single-omics analyses, researchers can achieve a holistic understanding of their engineered systems, identifying not only the intended effects but also compensatory mechanisms and unforeseen bottlenecks. The methodologies outlined—from correlation-based networks to machine learning—provide a versatile toolkit for beginners to start deciphering the complex interactions between genomic perturbations and phenotypic outcomes. As the field progresses, the ability to effectively integrate and interpret these rich, multi-layered datasets will be paramount to accelerating the design of next-generation microbial cell factories for sustainable chemical and therapeutic production.
Conclusion
Metabolic engineering has evolved from single-gene manipulations to sophisticated systems-level approaches that integrate computational modeling, AI-driven design, and multi-omics validation. The field continues to transform biomedical research through sustainable production of drug precursors, therapeutic compounds, and valuable biomolecules. Future advancements will likely focus on integrating non-canonical cofactors, developing more predictive AI models, and creating specialized chassis organisms for clinical applications. As metabolic engineering bridges with precision medicine, it promises to enable novel diagnostic biomarkers, personalized therapeutics, and more efficient drug development pipelines, ultimately accelerating the translation of engineered metabolic systems into clinical solutions.
References
- 1. Metabolic Engineering - an overview [https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/metabolic-engineering]
- 2. MANUFACTURING MOLECULES THROUGH METABOLIC ... [https://www.ncbi.nlm.nih.gov/books/NBK84444/]
- 3. Questions, data and models underpinning metabolic ... [https://www.frontiersin.org/journals/systems-biology/articles/10.3389/fsysb.2022.998048/full]
- 4. Unveiling Metabolic Engineering Strategies by Quantitative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11615770/]
- 5. mfapy: An open-source Python package for 13C-based ... [https://www.sciencedirect.com/science/article/pii/S2214030121000171]
- 6. Fluxer: a web application to compute, analyze and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7319574/]
- 7. Visualizing regulatory interactions in metabolic networks - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2216428/]
- 8. Visualizing regulatory interactions in metabolic networks [https://bmcsystbiol.biomedcentral.com/articles/10.1186/1752-0509-1-S1-P5]
- 9. 1953 Nobel Prize in Physiology or Medicine [https://www.rockefeller.edu/our-scientists/fritz-a-lipmann/2517-nobel-prize/]
- 10. The Nobel Prize in Physiology or Medicine 2016 - Press ... [https://www.nobelprize.org/prizes/medicine/2016/press-release/]
- 11. The Nobel Prize in Physiology or Medicine 2019 - Press ... [https://www.nobelprize.org/prizes/medicine/2019/press-release/]
- 12. Frances H. Arnold – Facts – 2018 - NobelPrize.org [https://www.nobelprize.org/prizes/chemistry/2018/arnold/facts/]
- 13. Frances Arnold [https://en.wikipedia.org/wiki/Frances_Arnold]
- 14. Protocol for analyzing energy metabolic pathway ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10960080/]
- 15. Low-Cost Algorithms for Metabolic Pathway Pairwise ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8883897/]
- 16. What is Central Carbon Metabolism? [https://www.creative-proteomics.com/resource/what-is-central-carbon-metabolism.htm]
- 17. Central Metabolic Pathway Guide [https://www.creative-proteomics.com/resource/central-metabolic-pathway-guide.htm]
- 18. A Guide to Metabolic Network Modeling for Plant Biology [https://www.mdpi.com/2223-7747/14/3/484]
- 19. Citric acid cycle [https://en.wikipedia.org/wiki/Citric_acid_cycle]
- 20. Simulating metabolic pathways to enhance interpretations ... [https://www.nature.com/articles/s41598-025-01634-7]
- 21. GEMsembler: consensus model assembly and structural ... [https://pubmed.ncbi.nlm.nih.gov/40919938/]
- 22. Metadag: a web tool to generate and analyse metabolic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11776228/]
- 23. Modeling for understanding and engineering metabolism [https://pubmed.ncbi.nlm.nih.gov/40070847/]
- 24. Metabolic Engineering | Research Starters [https://www.ebsco.com/research-starters/biology/metabolic-engineering]
- 25. Adenosine triphosphate [https://en.wikipedia.org/wiki/Adenosine_triphosphate]
- 26. Metabolic Energy - The Cell - NCBI Bookshelf - NIH [https://www.ncbi.nlm.nih.gov/books/NBK9903/]
- 27. The Mechanism of Oxidative Phosphorylation - The Cell - NCBI [https://www.ncbi.nlm.nih.gov/books/NBK9885/]
- 28. An Integrated Qualitative and Quantitative Biochemical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4675806/]
- 29. Identifying Bayesian optimal experiments for uncertain ... [https://www.nature.com/articles/s41598-024-65196-w]
- 30. An Emerging Star for Therapeutic and Catalytic Protein ... [https://www.bioprocessintl.com/process-development/an-emerging-star-for-therapeutic-and-catalytic-protein-production]
- 31. Engineering a Robust Escherichia coli W Platform for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12508785/]
- 32. Yeast vaccine production platform for human and animal ... [https://www.frontiersin.org/articles/10.3389/fimmu.2025.1697177/full]
- 33. Escherichia Coli Strain Market Size and Analysis, 2025-2032 [https://www.coherentmarketinsights.com/market-insight/escherichia-coli-strain-market-6132]
- 34. Yeast Market Size & Demand Forecast 2025-2035 [https://www.futuremarketinsights.com/reports/yeast-market]
- 35. McKinsey technology trends outlook 2025 [https://www.mckinsey.com/capabilities/tech-and-ai/our-insights/the-top-trends-in-tech]
- 36. Synthetic biology and metabolic engineering paving the way ... [https://pubs.rsc.org/en/content/articlehtml/2025/ya/d5ya00118h]
- 37. Network biology bridges the gaps between quantitative ... [https://www.sciencedirect.com/science/article/abs/pii/S136759312100140X]
- 38. Advances in large-scale DNA engineering with the ... [https://www.nature.com/articles/s12276-025-01530-0]
- 39. Expanding genetic engineering capabilities in Vibrio ... [https://pubmed.ncbi.nlm.nih.gov/40650977/]
- 40. Biotechnology systems engineering: preparing the next ... [https://www.frontiersin.org/journals/systems-biology/articles/10.3389/fsysb.2025.1583534/full]
- 41. Metabolic Pathway Design: A practical guide [https://research.manchester.ac.uk/en/publications/metabolic-pathway-design-a-practical-guide/]
- 42. Synthetic biology and metabolic engineering strategies in ... [https://www.sciencedirect.com/science/article/pii/S0926669025006065]
- 43. Machine learning for metabolic engineering: A review [https://www.sciencedirect.com/science/article/pii/S109671762030166X]
- 44. CRISPR/Cas9 technology in tumor research and drug ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1552741/full]
- 45. Review CRISPR-based metabolic pathway engineering [https://www.sciencedirect.com/science/article/abs/pii/S1096717620301592]
- 46. Applications of CRISPR/Cas System to Bacterial Metabolic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5979482/]
- 47. Predictive CRISPR-mediated gene downregulation for ... [https://www.sciencedirect.com/science/article/pii/S1096717625001740]
- 48. Application of Gene Knockout and Heterologous Expression ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9699552/]
- 49. Combinatorial metabolic engineering using an orthogonal ... [https://www.nature.com/articles/s41467-017-01695-x]
- 50. Beyond Cutting: CRISPR-Driven Synthetic Biology Toolkit for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12347677/]
- 51. Biosynthesis Strategies and Application Progress of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12388565/]
- 52. Metabolic engineering of the L-phenylalanine pathway in ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/1475-2859-10-71]
- 53. Systems metabolic engineering of Corynebacterium ... [https://jbioleng.biomedcentral.com/articles/10.1186/s13036-019-0156-5]
- 54. Rapid prototyping of microbial production strains for the ... [https://www.sciencedirect.com/science/article/pii/S109671762030077X]
- 55. Metabolic Engineering of Escherichia coli for De Novo ... [https://www.mdpi.com/2311-5637/11/6/331]
- 56. 2025 Plant Metabolic Engineering Conference GRC [https://www.grc.org/plant-metabolic-engineering-conference/2025/]
- 57. Application of cofactors in the regulation of microbial metabolism ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10126289/]
- 58. Integrated cofactor-centric engineering and energy flux ... [https://www.sciencedirect.com/science/article/abs/pii/S1385894725100223]
- 59. Enzymatic cofactor regeneration systems: A new ... [https://pubmed.ncbi.nlm.nih.gov/36657682/]
- 60. with Phosphite Dehydrogenase... | Newswise Cofactor Engineering [https://www.newswise.com/articles/cofactor-engineering-with-phosphite-dehydrogenase-enables-flexible-regulation-of-lactate-based-copolymer-biosynthesis-in-e-coli]
- 61. Engineering of cofactor regeneration enhances (2S,3S) [https://www.nature.com/articles/srep02643]
- 62. A versatile in situ cofactor enhancing system for meeting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10850783/]
- 63. Evaluation and optimization of quantitative analysis ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267022004615]
- 64. Recent Advances and Challenges in Engineering Pathways... Metabolic [https://www.sciepublish.com/article/pii/472]
- 65. Engineering cofactor metabolism for improved protein and ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-020-01450-w]
- 66. Advances in the optimization of central carbon metabolism in ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-023-02090-6]
- 67. Central metabolic pathway modification to improve L ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6527064/]
- 68. The PEP-pyruvate-oxaloacetate node: variation at the heart of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8100219/]
- 69. Metabolic engineering of Escherichia coli for high-yield ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12175537/]
- 70. Metabolic Pathway Design: A Practical Guide [https://link.springer.com/book/10.1007/978-3-030-29865-4]
- 71. Metabolic Engineering Market Shows Incredible Growth Soon [https://htfmarketinsights.com/report/4386941-metabolic-engineering-market]
- 72. Drug precursors regulated by the EU: 2 new substances [https://www.eqgest.com/en/blog/drug-precursors-regulated-by-the-european-union-2-new-substances-in-delegated-regulation-eu-20251475]
- 73. BioMed X and Novo Nordisk Launch New Collaboration in ... [https://bmedx.com/news-events/press-releases/biomed-x-and-novo-nordisk-launch-new-collaboration-in-oral-peptide-drug-delivery-013510/]
- 74. How Imperial College Lab Advances Metabolic ... [https://www.reshapebiotech.com/case-studies/imperial-college-accelerated-metabolic-engineering]
- 75. Metabolic engineering | Nature Chemical Biology [https://www.nature.com/subjects/metabolic-engineering/nchembio]
- 76. Matching diverse feedstocks to conversion processes for ... [https://www.sciencedirect.com/science/article/pii/S0958166923001271]
- 77. A critical review on various feedstocks as sustainable ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-021-01613-3]
- 78. Laboratory protocols and methods - Research Guides [https://guides.lib.vt.edu/find/lab-methods]
- 79. unlocking the potential of key organic acids for sustainable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11933101/]
- 80. Dynamic control in metabolic engineering: Theories, tools ... [https://www.sciencedirect.com/science/article/pii/S1096717620301440]
- 81. Layered dynamic for improving metabolic regulation ... pathway [https://pubmed.ncbi.nlm.nih.gov/29507236/]
- 82. Machine learning for metabolic pathway optimization: A review [https://pmc.ncbi.nlm.nih.gov/articles/PMC10781721/]
- 83. Chapter 15 Experimental protocols | Lab Handbook [https://ccmorey.github.io/labHandbook/experimental-protocols.html]
- 84. How to Write a Research Protocol: Tips and Tricks - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6172884/]
- 85. A guide to metabolic flux analysis in metabolic engineering [https://www.sciencedirect.com/science/article/abs/pii/S1096717620301683]
- 86. Comparison of metabolic states using genome-scale ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8601616/]
- 87. CFSA: Comparative flux sampling analysis as a guide for ... [https://www.sciencedirect.com/science/article/pii/S2214030124000130]
- 88. All Resources - Site Guide - NCBI - NIH [https://www.ncbi.nlm.nih.gov/guide/all/]
- 89. Metabolic modeling of host-microbe interactions [https://pubmed.ncbi.nlm.nih.gov/41127816/]
- 90. Microbiome Metabolic Modeling as a tool for innovation in ... [https://www.sciencedirect.com/science/article/pii/S2214799325000980]
- 91. List of open-source bioinformatics software [https://en.wikipedia.org/wiki/List_of_open-source_bioinformatics_software]
- 92. Metabolic Modelling: Methods, Applications and Future ... [https://www.mdpi.com/journal/metabolites/special_issues/Met_Mod]
- 93. Metabolic Objectives and Trade-Offs: Inference and Applications [https://pmc.ncbi.nlm.nih.gov/articles/PMC11857637/]
- 94. Balancing growth, reproduction, maintenance, and activity ... [https://www.sciencedirect.com/science/article/pii/S0960982222007771]
- 95. In silico analysis of design of experiment methods ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11087228/]
- 96. Optimizing the strain engineering process for industrial-scale ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10548853/]
- 97. Streamlined identification of strain engineering targets for ... [https://www.nature.com/articles/s41598-023-39661-x]
- 98. Dynamic metabolic engineering: New strategies for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4629492/]
- 99. Relieving metabolic burden to improve robustness and ... [https://www.sciencedirect.com/science/article/pii/S0734975024000958]
- 100. Hierarchical metabolic engineering for rewiring cellular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12514951/]
- 101. Metabolic Engineering for the Production of Natural Products [https://pmc.ncbi.nlm.nih.gov/articles/PMC4077471/]
- 102. Physics-based Modeling in the New Era of Enzyme Engineering [https://pmc.ncbi.nlm.nih.gov/articles/PMC12239909/]
- 103. Improving metabolic engineering design with enzyme ... [https://www.sciencedirect.com/science/article/pii/S1096717625000850]
- 104. Using Computations to Analyze and Reshape Metabolism ... [https://engineering.nyu.edu/events/2025/12/12/using-computations-analyze-and-reshape-metabolism-bioproduction]
- 105. ISMRM 2025 Workshop on Frontiers in Metabolomics & ... [https://www.ismrm.org/workshops/2025/MMI/]
- 106. Effective data visualization strategies in untargeted ... [https://pubs.rsc.org/en/content/articlehtml/2025/np/d4np00039k]
- 107. A Large Language Model–Powered Map of Metabolomics ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11957067/]
- 108. Machine learning prediction of metabolic dysfunction ... [https://www.frontiersin.org/journals/nutrition/articles/10.3389/fnut.2025.1616229/full]
- 109. Machine learning models for predicting metabolic ... [https://pubmed.ncbi.nlm.nih.gov/40155991/]
- 110. A concise guide to choosing suitable gene expression ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10643540/]
- 111. E. coli vs. Yeast: Which Protein Expression System Is Better? [https://synapse.patsnap.com/article/e-coli-vs-yeast-which-protein-expression-system-is-better]
- 112. Advantages & Disadvantages of Protein Expression Systems [https://www.betalifesci.com/blogs/articles/advantages-and-disadvantages-of-various-protein-expression-systems?srsltid=AfmBOopsr6CqSywZNplJbkWfmD3eGcODwgxlv3pQFovQMtrtu8cSyEM0]
- 113. Yeast—a handy multitool in your research: a report from ISSY38 [https://pmc.ncbi.nlm.nih.gov/articles/PMC12570875/]
- 114. Systems metabolic engineering of Escherichia coli for the ... [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2025.1640158/full]
- 115. Switching the yeast metabolism via manipulation of sugar ... [https://www.sciencedirect.com/science/article/abs/pii/S1096717625000230]
- 116. Microbial Metabolic Engineering Technology - Special Issue [https://www.mdpi.com/journal/microorganisms/special_issues/5HT923R9E0]
- 117. Pitfalls in Early Bioprocess Development Using Shake Flask ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11773345/]
- 118. Exploring Principles of Bioreactor Scale-Up [https://www.bioprocessintl.com/bioreactors/lessons-in-bioreactor-scale-up-part-1-mdash-exploring-introductory-principles]
- 119. 5 Things You Need to Do to Scale Up Your Experiments ... [https://www.scientificbio.com/blog/5-things-you-need-to-do-to-scale-up-your-experiments-from-shake-flasks/]
- 120. Challenges in Scaling Up Bioprocesses in Bioprocessing [https://www.idbs.com/knowledge-base/challenges-in-scaling-up-bioprocesses-in-bioprocessing/]
- 121. From shake flasks to stirred fermentors: Scale-up of an ... [https://www.sciencedirect.com/science/article/abs/pii/S1359511306000195]
- 122. How to Scale Up Fermentation from Shake Flask to 5L [https://synapse.patsnap.com/article/how-to-scale-up-fermentation-from-shake-flask-to-5l]
- 123. Challenges in scaling up a perfusion process - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3284882/]
- 124. Guide for authors - Metabolic Engineering - ISSN 1096-7176 [https://www.sciencedirect.com/journal/metabolic-engineering/publish/guide-for-authors]
- 125. The Energy-Economy Nexus of Advanced Air Pollution Control Technologies: Pathways to Sustainable Development [https://www.mdpi.com/1996-1073/18/9/2378]
- 126. Methods and Guidelines for Metabolism Studies [https://www.mdpi.com/1422-0067/26/17/8466]
- 127. Strategies for Comprehensive Multi-Omics Integrative Data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11592251/]
- 128. Integrating Multi-Omics Data: Methods and Applications in ... [https://www.sciencedirect.com/science/article/pii/S2215017X25000657]
- 129. A Guide to Multi-omics Integration Strategies [https://frontlinegenomics.com/a-guide-to-multi-omics-integration-strategies/]