AGORA2: A Comprehensive Guide to Personalized Microbiome Metabolic Modeling for Drug Development and Biomedical Research
The AGORA2 pipeline represents a transformative advance in systems biology, enabling personalized, strain-resolved modeling of gut microbiome metabolism.
AGORA2: A Comprehensive Guide to Personalized Microbiome Metabolic Modeling for Drug Development and Biomedical Research
Abstract
The AGORA2 pipeline represents a transformative advance in systems biology, enabling personalized, strain-resolved modeling of gut microbiome metabolism. This resource of 7,302 manually curated genome-scale metabolic reconstructions provides a mechanistic framework to investigate host-microbiome interactions in health and disease. By integrating multi-omics data, AGORA2 facilitates the prediction of microbial drug metabolism, identification of disease-associated metabolic shifts, and the development of personalized therapeutic interventions. This article explores AGORA2's foundations, methodological applications, optimization strategies, and validation frameworks, offering researchers and drug development professionals a comprehensive guide to leveraging this powerful tool for precision medicine.
The AGORA2 Framework: Building the Foundation for Personalized Microbiome Modeling
The Assembly of Gut Organisms through Reconstruction and Analysis, version 2 (AGORA2) is a pivotal resource in the field of microbiome research, representing a significant scale-up from its predecessor. The original AGORA resource contained 773 manually curated genome-scale metabolic reconstructions of human gut microorganisms [1]. AGORA2 dramatically expands this scope to include 7,302 strains spanning 1,738 species and 25 phyla, enabling unprecedented investigation of host-microbiome metabolic interactions in personalized medicine [1] [2].
This expansion addresses a critical need in precision medicine, as the human gut microbiome influences the efficacy and safety of numerous commonly prescribed drugs [1]. Research has demonstrated that gut microorganisms can metabolize 176 of 271 tested drugs, with this activity varying substantially between individuals [1] [2]. AGORA2 provides the strain- and molecule-resolved computational modeling capability necessary to incorporate these microbial metabolic influences into personalized treatment strategies [1].
Table 1: Key Features of AGORA2 Compared to the Original AGORA Resource
| Feature | AGORA | AGORA2 |
|---|---|---|
| Number of Strains | 773 | 7,302 |
| Number of Species | 605 | 1,738 |
| Number of Phyla | 14 | 25 |
| Drug Metabolism Coverage | Not specified | 98 drugs, 15 enzymes |
| Experimental Data Curation | Limited | 732 papers + textbooks |
The AGORA2 Reconstruction Pipeline: DEMETER
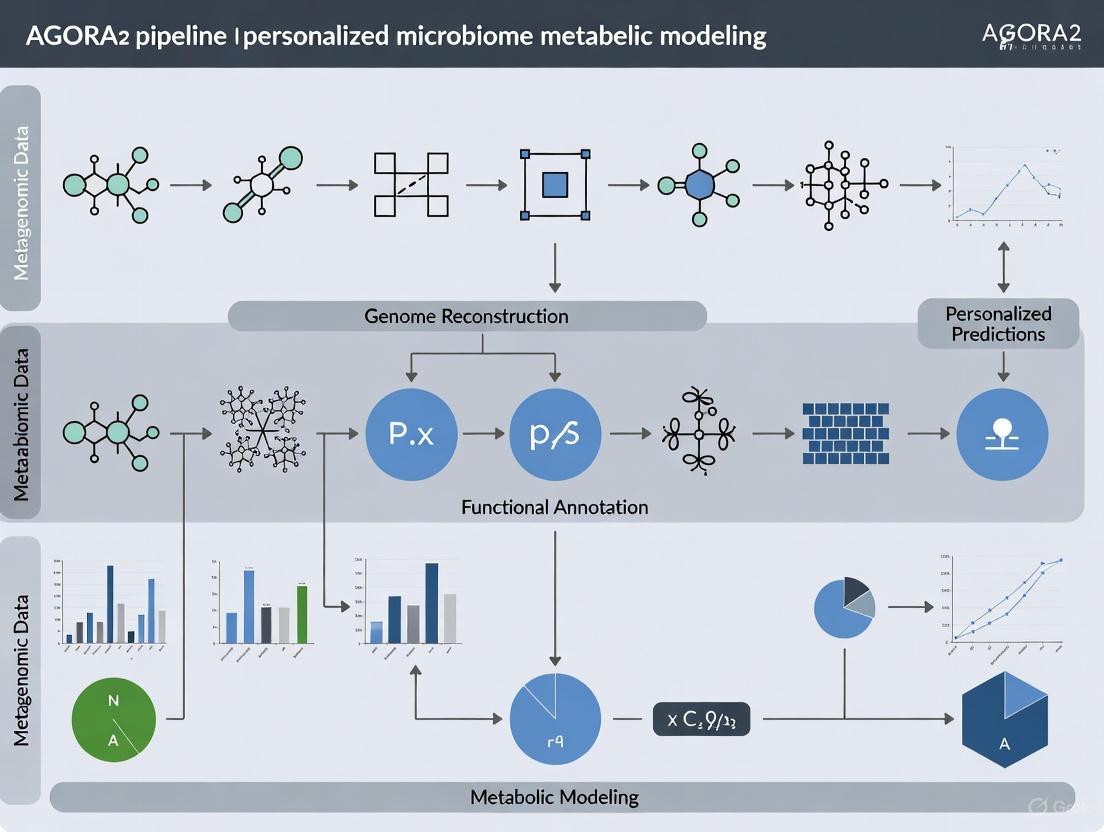
The creation of AGORA2 was facilitated through a substantially revised and expanded data-driven reconstruction refinement pipeline known as DEMETER (Data-drivEn METabolic nEtwork Refinement) [1]. This comprehensive workflow integrates multiple stages to transform raw genomic data into high-quality, predictive metabolic models.
The DEMETER pipeline follows a systematic approach [1]:
- Data Collection and Integration: Gathering genomic and experimental data from diverse sources
- Draft Reconstruction Generation: Initial automated reconstruction via the KBase online platform [1]
- Namespace Translation: Converting reactions and metabolites into the Virtual Metabolic Human (VMH) namespace [1]
- Iterative Refinement: Simultaneous refinement, gap-filling, and debugging through an iterative process [1]
Comprehensive Curation Efforts
A hallmark of the AGORA2 development was the extensive manual curation applied to ensure biological relevance and predictive accuracy. The annotation of 446 gene functions across 35 metabolic subsystems for 74% of genomes (5,438 of 7,302) was manually validated and improved using PubSEED [1] [2]. Additionally, an extensive manual literature search spanning 732 peer-reviewed papers and two microbial reference textbooks provided information for 95% of strains (6,971 of 7,302) [1]. For the remaining 331 strains, either no experimental data were available or all reported biochemical tests were negative [1].
These curation efforts resulted in substantial modifications to the automated drafts, with an average of 685.72 reactions added and 685.72 reactions removed per reconstruction (standard deviation: ±620.83) [1]. The metabolic models derived from these refined reconstructions demonstrated clear improvement in predictive potential compared to models from the original KBase draft reconstructions [1].
Diagram 1: DEMETER Pipeline for AGORA2 Reconstruction
Key Enhancements and Novel Capabilities in AGORA2
Drug Metabolism Capabilities
A groundbreaking enhancement in AGORA2 is the incorporation of strain-resolved drug degradation and biotransformation capabilities. Through comprehensive manual comparative genomic analysis, AGORA2 accounts for [1] [2]:
- 98 drugs with documented microbial metabolism
- 15 enzymes experimentally shown to directly or indirectly modify drug metabolism
- Their subcellular locations
- 12 drug transporter genes
This drug metabolism module includes an average of 188 drug-related reactions, 111 metabolites, and 1,440 drug-related reactions based on genetic evidence, achieving a prediction accuracy of 0.81 when validated against independent experimental data [1] [2].
Taxonomic and Metabolic Diversity
AGORA2 captures the substantial metabolic diversity present across human gut microorganisms. Analysis of the resource reveals that reconstructions cluster by class and family according to their reaction coverage, reflecting their phylogenetic relationships [1]. Several genera in the Bacilli and Gammaproteobacteria classes form distinct subgroups illustrating important metabolic differences between them [1]. These cross-phylum metabolic differences translate to variations in reconstruction sizes, predicted growth rates, and metabolic consumption and secretion potentials [1].
Validation and Performance Assessment
The quality of AGORA2 reconstructions was rigorously assessed against other microbial genome-scale reconstruction resources, including CarveMe, gapseq, MIGRENE (MAGMA), and manually curated reconstructions from the BiGG database [1]. The assessment evaluated the fraction of flux-consistent reactions in each resource, with AGORA2 outperforming all other semi-automated approaches and rivaling manually curated reconstructions [1].
Table 2: Performance Comparison of AGORA2 Against Other Reconstruction Resources
| Resource | Flux Consistency | Accuracy Against Experimental Data | Advantages |
|---|---|---|---|
| AGORA2 | High | 0.72-0.84 | Extensive curation, drug metabolism |
| CarveMe | High (by design) | Not specified | Automatically removes flux inconsistencies |
| gapseq | Lower than AGORA2 | Not specified | High throughput |
| MAGMA | Lower than AGORA2 | Not specified | - |
| BiGG | High | High (limited overlap) | Manual curation |
Validation Against Independent Experimental Datasets
AGORA2 was validated against three independently collected experimental datasets [1]:
- Species-level metabolite uptake and secretion data for 455 species (5,319 strains) from the NJC19 resource [1]
- Species-level metabolite uptake data for 185 species (328 strains) from Madin et al. [1]
- Strain-resolved metabolite uptake and secretion data for 676 AGORA2 strains [1]
AGORA2 achieved an accuracy of 0.72 to 0.84 across these datasets, surpassing other reconstruction resources [1]. This high performance demonstrates the resource's capability to accurately capture known biochemical and physiological traits of the target organisms.
Protocols for AGORA2 Implementation in Personalized Medicine Research
Protocol 1: Building Personalized Community Models for Drug Metabolism Prediction
Purpose: To predict the drug conversion potential of individual gut microbiomes using AGORA2.
Materials:
- AGORA2 resource of 7,302 metabolic reconstructions
- Metagenomic sequencing data from patient samples
- Constraint-based reconstruction and analysis (COBRA) toolbox
- Virtual Metabolic Human (VMH) database
Procedure:
- Metagenomic Mapping: Map metagenomic sequencing data from individual patients to the AGORA2 resource. In a study of 616 patients with colorectal cancer and controls, 97% of detected species could be mapped to AGORA2, compared to only 72% with the original AGORA [1] [2].
- Community Model Construction: Build a personalized microbiome community model for each individual by combining the metabolic reconstructions of detected species.
- Constraint Application: Apply condition-specific constraints, including nutrient availability and metabolic interactions.
- Flux Balance Analysis: Perform flux balance analysis to predict metabolic fluxes, including drug biotransformation capabilities.
- Validation: Compare predictions against metabolomic data where available.
Applications: This protocol was used to predict the overall drug-metabolizing capacity of gut microbiomes in a cohort of 365 patients with colorectal cancer and 251 healthy controls, revealing correlations between microbial drug metabolism potential and clinical parameters including age, sex, and body mass index [1] [2].
Protocol 2: Investigating Early-Life Microbiome Development
Purpose: To evaluate the metabolic capabilities of infant gut microbiomes and the impact of delivery mode.
Materials:
- AGORA2 resource expanded with human milk oligosaccharide (HMO) degradation module
- Metagenomic data from infant and maternal gut samples
- COBRA modeling tools
Procedure:
- Module Expansion: Enhance AGORA2 with specialized metabolic modules for infant gut microbiomes, such as HMO degradation pathways.
- Longitudinal Sampling: Collect metagenomic data at multiple time points (e.g., 5 days, 1 month, 6 months, and 1 year).
- Personalized Modeling: Build metabolic models for each sample time point.
- Comparative Analysis: Compare metabolic capabilities between Cesarian section-delivered (CSD) and vaginally delivered (VD) infants.
- Maternal Comparison: Compare infant gut metabolic capabilities with maternal gut microbiomes.
Applications: This approach revealed that CSD infant gut microbiomes are depleted in their metabolic capabilities at early stages compared to VD infants, with differences in metabolites such as fermentation products, HMO degradation products, and amino acids [3]. Additionally, infant gut microbiomes were found to produce less butyrate but more L-lactate and have enriched potential for B-vitamin synthesis compared to adult gut microbiomes [3].
Diagram 2: APOLLO Resource Construction
Table 3: Essential Research Reagents and Computational Resources for AGORA2 Implementation
| Resource | Type | Function | Availability |
|---|---|---|---|
| AGORA2 Reconstructions | Data Resource | 7,302 strain-resolved metabolic models | https://www.vmh.life/ |
| Virtual Metabolic Human (VMH) | Database | Standardized metabolic namespace | https://www.vmh.life/ |
| COBRA Toolbox | Software | Constraint-based modeling and simulation | Open source |
| DEMETER Pipeline | Computational Method | Semi-automated reconstruction refinement | Described in methodology |
| KBase | Online Platform | Draft reconstruction generation | https://www.kbase.us/ |
| APOLLO | Extended Resource | 247,092 reconstructions from MAGs | https://www.vmh.life/ |
Future Directions: Beyond AGORA2 to APOLLO
The methodology developed for AGORA2 has enabled even larger-scale reconstruction efforts, most notably the APOLLO resource, which encompasses 247,092 genome-scale metabolic reconstructions built from metagenome-assembled genomes (MAGs) [4] [5]. APOLLO spans 19 phyla, contains >60% uncharacterized strains, and accounts for microbes from 34 countries, all age groups, and multiple body sites [4]. This expanded resource enables the construction of sample-specific microbiome community models that can stratify microbiomes by body site, age, and disease state [4].
The AGORA2 resource and its methodological foundations represent a critical advancement in personalized medicine, providing the computational framework necessary to mechanistically understand and predict how individual variations in gut microbiome composition influence drug metabolism, disease progression, and therapeutic outcomes. As these resources continue to expand and integrate with whole-body human metabolic models, they offer unprecedented opportunities for systems-level investigation of host-microbiome interactions in health and disease.
The DEMETER (Data-drivEn METabolic nEtwork Refinement) pipeline represents a foundational framework for the development of high-quality, manually curated genome-scale metabolic reconstructions within the AGORA2 resource. AGORA2 (Assembly of Gut Organisms through Reconstruction and Analysis, version 2) provides a comprehensive resource of 7,302 genome-scale metabolic reconstructions of human microorganisms, dramatically expanding from the first version that contained 773 reconstructions [1]. This massive expansion was enabled by the DEMETER pipeline, which facilitates the systematic refinement of draft metabolic networks through the integration of comparative genomic analyses and manual literature curation [1].
The DEMETER pipeline was specifically designed to overcome limitations of purely automated reconstruction tools by incorporating extensive manual curation efforts based on experimental data from peer-reviewed literature and microbial reference textbooks. This hybrid approach ensures that the resulting metabolic reconstructions capture species-specific metabolic capabilities, including specialized functions such as drug biotransformation and degradation pathways that are often missing from automated annotations [1]. The pipeline's rigorous refinement process has proven essential for generating metabolic models with high predictive accuracy, making AGORA2 a cornerstone resource for personalized microbiome metabolic modeling in biomedical and pharmaceutical research.
Table 1: Key Statistics of the AGORA2 Resource and DEMETER Pipeline
| Component | Metric | Value |
|---|---|---|
| AGORA2 Resource | Total number of strain reconstructions | 7,302 strains |
| Taxonomic coverage | 1,738 species, 25 phyla | |
| Drug transformation coverage | 98 drugs, >5,000 strains | |
| DEMETER Curation | Manual gene function validations | 446 gene functions across 35 subsystems |
| Literature sources | 732 peer-reviewed papers + 2 textbooks | |
| Strains with experimental data | 6,971 (95% of total) |
DEMETER Pipeline: Workflow and Technical Architecture
The DEMETER pipeline operates through a sophisticated, multi-stage workflow that transforms initial draft reconstructions into high-fidelity metabolic models. The technical architecture follows a sequential process of data collection, data integration, draft reconstruction generation, and simultaneous iterative refinement, with continuous verification through automated testing suites [1].
The pipeline begins with the generation of automated draft reconstructions through the KBase (KnowledgeBase) platform, which provides initial genome-scale metabolic networks based on genomic annotations [1]. These draft reconstructions subsequently undergo the DEMETER refinement process, which incorporates both computational and manual curation components. A critical aspect of the pipeline is the translation of reactions and metabolites into the Virtual Metabolic Human (VMH) namespace, ensuring compatibility with existing human metabolic reconstructions and facilitating the study of host-microbiome interactions [1].
The workflow is supported by comprehensive quality control measures, including the generation of quality reports for all reconstructions, which achieved an average quality score of 73% for the AGORA2 resource [1]. The pipeline also incorporates atom-atom mapping for 5,583 enzymatic and transport reactions (65% of total) and retrieves metabolic structures for 1,838 metabolites (51% of total), significantly enhancing the biochemical fidelity of the resulting models [1].
Diagram 1: The DEMETER Pipeline Workflow for Metabolic Reconstruction Refinement. This workflow transforms initial genome sequences into high-quality metabolic reconstructions through sequential stages of data collection, integration, and iterative refinement with manual curation.
Technical Implementation and Functional Considerations
The technical implementation of the DEMETER pipeline incorporates several critical functional considerations to ensure the biological relevance and computational tractability of the resulting models. During the refinement process, reactions are strategically placed in periplasm compartments where appropriate, enhancing the spatial accuracy of transport and exchange reactions [1]. The pipeline also involves careful curation of biomass reactions, which define the composition of essential macromolecules required for cellular growth and serve as key objective functions in constraint-based modeling approaches [1].
The DEMETER pipeline successfully addresses several challenges inherent to metabolic reconstruction, including the removal of futile cycles that can lead to biologically implausible flux distributions and ATP overproduction. Compared to draft reconstructions and other automated tools, models refined through DEMETER demonstrate significantly higher percentages of flux-consistent reactions, ensuring that metabolic networks can support feasible steady-state flux distributions [1]. This improvement is particularly notable given that DEMETER reconstructions maintain larger metabolic content while achieving this higher consistency, reflecting the pipeline's ability to balance comprehensiveness with biochemical plausibility.
Manual Curation Process: Methodology and Implementation
The manual curation component of the DEMETER pipeline represents a critical differentiator that elevates AGORA2 reconstructions beyond purely automated approaches. This process involves systematic validation of gene functions and integration of experimental evidence from biochemical literature, ensuring that metabolic capabilities are accurately represented in the resulting models.
Gene Function Validation and Literature Integration
A cornerstone of the manual curation process is the extensive validation of gene functions using the PubSEED platform, which enabled manual curation of 446 gene functions across 35 metabolic subsystems for 5,438 genomes (74% of total) [1]. This gene-centric validation ensures accurate annotation of enzyme-catalyzed reactions and addresses limitations in automated genome annotation tools, which often miss species-specific metabolic capabilities.
The manual curation process incorporated evidence from 732 peer-reviewed papers and two microbial reference textbooks, providing experimental validation for 6,971 of the 7,302 strains (95%) in AGORA2 [1]. For the remaining 331 strains, either no experimental data were available or all reported biochemical tests were negative. This comprehensive literature review enabled the inclusion of specialized metabolic pathways, particularly drug biotransformation reactions that are often poorly annotated in standard genomic databases.
Table 2: Manual Curation Components in DEMETER Pipeline
| Curation Component | Scope | Tools/Resources | Impact |
|---|---|---|---|
| Gene Function Validation | 446 functions across 35 subsystems | PubSEED platform | Correct enzyme annotations and reaction associations |
| Literature Integration | 732 papers + 2 textbooks | Manual literature search | Experimental validation of metabolic capabilities |
| Drug Metabolism Curation | 98 drugs, 15 enzymes | Comparative genomics + literature | Strain-resolved drug transformation predictions |
| Ontology Mapping | All variables when feasible | OLS, Zooma (EBI) | Enhanced machine readability and interoperability |
Experimental Protocol: Manual Curation of Metabolic Functions
Protocol Title: Manual Curation of Species-Specific Metabolic Capabilities for Genome-Scale Metabolic Reconstructions
Objective: To manually validate and refine the metabolic capabilities of microbial strains based on experimental evidence from biochemical literature and comparative genomics.
Materials and Reagents:
- Genomic sequences and annotated genomes of target microorganisms
- PubSEED platform for manual gene function annotation [1]
- Ontology Lookup Service (OLS) and Zooma for ontology term mapping [6]
- Access to scientific literature databases (e.g., PubMed, Scopus)
- Virtual Metabolic Human (VMH) database and namespace specifications [1]
Methodology:
- Gene Function Annotation Validation:
- Access the target genome in the PubSEED platform
- Systematically review annotations for 446 key gene functions across 35 metabolic subsystems
- Compare automated annotations with experimental evidence from literature
- Correct misannotated genes and add missing functions with experimental support
Literature-Based Pathway Curation:
- Conduct systematic literature searches for each target strain using relevant keywords
- Extract experimental data on substrate utilization, metabolic end products, and growth requirements
- Incorporate positive and negative biochemical evidence into the metabolic reconstruction
- Place reactions in appropriate cellular compartments (e.g., periplasm) based on experimental evidence
Drug Metabolism Pathway Integration:
- Review literature on microbial drug transformation for 98 target compounds
- Manually formulate strain-resolved drug biotransformation and degradation reactions
- Validate predicted transformations against independent experimental data when available
Ontology Mapping and Standardization:
- Map each variable to equivalent ontology codes using OLS and Zooma platforms
- Prioritize domain-specific ontologies that are well-maintained and reliable
- Conduct thorough review to ensure accuracy and correspondence of applied codes
Quality Control:
- Generate quality control reports for all reconstructions
- Verify flux consistency of metabolic networks
- Test biomass production capabilities under defined conditions
- Validate against independently collected experimental datasets
Performance Assessment and Validation Metrics
The performance of the DEMETER pipeline and its associated manual curation processes has been rigorously evaluated through multiple validation frameworks. These assessments demonstrate the significant improvement in predictive accuracy achieved through the pipeline's refinement process compared to automated draft reconstructions.
Quantitative Performance Metrics
The DEMETER-refined reconstructions were evaluated against three independently collected experimental datasets, achieving predictive accuracies ranging from 0.72 to 0.84, surpassing other reconstruction resources [1]. The pipeline also demonstrated exceptional performance in predicting known microbial drug transformations, with an accuracy of 0.81 against independent experimental data [1].
A critical metric for metabolic reconstruction quality is the percentage of flux-consistent reactions, which indicates the biochemical feasibility of the metabolic network. DEMETER reconstructions showed significantly higher percentages of flux-consistent reactions compared to the original KBase drafts, as well as reconstructions generated by other automated tools such as gapseq and MAGMA [1]. This improvement is particularly notable given that the DEMETER reconstructions maintained larger network sizes while achieving this higher consistency.
Table 3: Performance Metrics of DEMETER-Refined Reconstructions
| Validation Metric | DEMETER Performance | Comparison to Draft Reconstructions |
|---|---|---|
| Experimental Data Accuracy | 0.72 - 0.84 | Significantly improved |
| Drug Transformation Prediction | 0.81 accuracy | Not available in drafts |
| Flux Consistent Reactions | Significantly higher | Improved despite larger network size |
| ATP Production Plausibility | Biologically realistic | Drafts showed unrealistic overproduction |
| Taxonomic Clustering | Reflected phylogenetic relationships | Poor clustering in drafts |
Experimental Protocol: Reconstruction Validation and Testing
Protocol Title: Validation and Quality Assessment of Refined Metabolic Reconstructions
Objective: To quantitatively assess the quality and predictive accuracy of metabolic reconstructions refined through the DEMETER pipeline.
Materials and Reagents:
- Flux balance analysis software (e.g., COBRA Toolbox)
- Experimental phenotype data (substrate utilization, growth requirements)
- Drug transformation datasets for validation
- Flux variability analysis algorithms
- Quality control assessment scripts
Methodology:
- Flex Consistency Analysis:
- Perform flux variability analysis on all reactions in the reconstruction
- Calculate the percentage of flux-consistent reactions
- Identify and address thermodynamically infeasible loops
Phenotype Prediction Validation:
- Retrieve species-level metabolite uptake and secretion data from NJC19 resource
- Map species-level positive metabolite uptake data from Madin et al. dataset
- Compare model predictions against experimental data for growth and metabolite utilization
- Calculate accuracy metrics for phenotype predictions
Drug Transformation Validation:
- Test prediction of known microbial drug transformations against independent data
- Validate strain-resolved drug conversion potential using clinical microbiome data
- Assess correlation between predicted drug metabolism and patient factors
Taxonomic Consistency Evaluation:
- Cluster reconstructions by reaction content across taxonomic groups
- Verify that metabolic capabilities reflect phylogenetic relationships
- Assess functional differences between genera and classes
Quality Control:
- Generate unbiased quality control reports for all reconstructions
- Verify biomass production under defined nutritional conditions
- Test production of species-specific metabolites
- Ensure absence of ATP overproduction under standard conditions
Research Reagent Solutions and Resource Toolkit
The implementation of the DEMETER pipeline and utilization of AGORA2 reconstructions requires specific computational resources and data management tools. The following table summarizes essential research reagents and resources for employing these systems in microbiome metabolic modeling research.
Table 4: Essential Research Reagents and Resources for DEMETER and AGORA2 Implementation
| Resource Category | Specific Tools/Resources | Function and Application |
|---|---|---|
| Reconstruction Platforms | KBase, PubSEED, CarveMe, gapseq | Draft reconstruction generation and manual curation |
| Metabolic Modeling | COBRA Toolbox, DEMETER pipeline | Constraint-based modeling and network refinement |
| Data Standardization | Microbiome Research Data Toolkit, REDCap | Standardized metadata reporting and data collection |
| Ontology Services | Ontology Lookup Service (OLS), Zooma | Ontology mapping for improved interoperability |
| Validation Databases | NJC19, Madin et al. datasets, BiGG Models | Experimental data for model validation and testing |
| Community Modeling | MICOM, AGORA2 resource | Personalized microbiome metabolic modeling |
The DEMETER pipeline and its comprehensive manual curation processes represent a transformative approach to genome-scale metabolic reconstruction, addressing fundamental limitations of purely automated methods through the integration of computational refinement and experimental validation. The resulting AGORA2 resource provides strain-resolved, manually curated metabolic models for 7,302 human microorganisms, enabling unprecedented investigation of host-microbiome metabolic interactions in personalized medicine contexts.
The rigorous validation of the DEMETER pipeline demonstrates its superior performance against independently collected experimental data, with particular strength in predicting drug biotransformation capabilities that are essential for pharmaceutical research and precision medicine. The continued refinement of this pipeline and expansion of manually curated metabolic content will further enhance our ability to model and manipulate human microbiome metabolism for therapeutic applications.
The AGORA2 (Assembly of Gut Organisms through Reconstruction and Analysis, version 2) resource represents a foundational knowledge base for personalized, predictive analysis of host-microbiome metabolic interactions [1]. This resource systematically expands upon its predecessor by accounting for 7,302 strains of human microorganisms, spanning 1,738 species and 25 phyla, enabling strain- and molecule-resolved computational modeling of microbial metabolism [1]. A critical advancement in AGORA2 is the manual curation of drug biotransformation and degradation capabilities for 98 pharmaceuticals across more than 5,000 microbial strains, incorporating 15 key enzymes involved in these metabolic processes [1] [7].
This resource bridges biochemistry, pharmacology, genetics, and microbiology, consolidating diverse research fields to illuminate the profound impact of microbial metabolism on drug efficacy and safety [7]. The gut microbiota influences a wide variety of commonly prescribed drugs, with human gut microorganisms demonstrated to metabolize 176 of 271 tested drugs in experimental studies, with activity varying significantly between individuals [1]. AGORA2 provides the computational framework to explore this variability mechanistically, paving the way for precision medicine interventions that incorporate dietary, genetic, and microbial factors [1] [7].
Comprehensive Drug Metabolism Coverage
Quantitative Scope of Drug Biotransformation
The AGORA2 resource incorporates manually formulated, molecule- and strain-resolved drug biotransformation and degradation reactions, enabling personalized, strain-resolved modeling of drug conversion potential in individual gut microbiomes [1].
Table 1: Quantitative Scope of Drug Biotransformation in AGORA2
| Feature | Scope | Details |
|---|---|---|
| Total Microbial Strains | 7,302 strains | 1,738 species, 25 phyla [1] |
| Drugs with Curated Metabolism | 98 compounds | Diverse pharmaceuticals [1] |
| Strains with Drug Metabolism Capabilities | >5,000 strains | Spanning diverse taxa [1] |
| Enzyme Systems Covered | 15 enzymes | Including DPD, enzymes for cardiac glycoside reduction, etc. [1] [7] |
| Prediction Accuracy | 81% (0.81 accuracy) | For known microbial drug transformations [1] |
Key Drug-Microbiome Interactions
Microbial drug metabolism involves biotransformation through enzymatic reactions, leading to metabolites with altered pharmacological properties [7]. These interactions can be categorized by their pharmacodynamic outcomes:
- Activation: Conversion of prodrugs to active compounds
- Modulation: Alteration of drug activity without complete inactivation
- Deactivation: Inactivation of therapeutic compounds
- Toxification: Production of toxic metabolites
- Reactivation: Conversion of metabolites back to active forms [7]
Table 2: Examples of Clinically Relevant Drug-Microbiome Interactions in AGORA2
| Drug/Drug Class | Microbial Reaction | Key Enzymes | Pharmacodynamic Effect | Example Microorganisms |
|---|---|---|---|---|
| 5-Fluorouracil (5-FU) & Capecitabine | Dihydropyrimidine reduction | Dihydropyrimidine dehydrogenase (DPD) encoded by preT/preA genes | Deactivation (reduced cytotoxic effect) | Escherichia coli, Salmonella enterica [7] |
| Cardiac Glycosides (Digoxin) | Reduction to inactive metabolites | Cardiac glycoside reductases | Deactivation | Eggerthella lenta [7] |
| Various Drugs | Azoreduction, deamination, hydrolysis, decarboxylation | Various species-specific enzymes | Activation, deactivation, or toxification | Diverse gut microbiota [1] |
Experimental Protocols for Validation
Protocol 1: Comparative Genomic Analysis for Enzyme Annotation
Objective: To identify and annotate genes encoding 15 drug-metabolizing enzymes across 5,438 bacterial strains.
Methodology:
- Sequence Collection: Retrieve genome sequences for target strains from public repositories (e.g., NCBI, EBI) [1].
- Hidden Markov Model (HMM) Profiling: Use curated HMM profiles for each of the 15 target enzymes to identify putative genes in target genomes [7].
- Manual Curation: Validate automated annotations through manual inspection using the PubSEED platform for 446 gene functions across 35 metabolic subsystems [1].
- Reaction Integration: Incorporate confirmed drug transformation reactions into genome-scale metabolic reconstructions using the DEMETER pipeline [1].
Key Reagents:
- Genomic Databases: NCBI GenBank, EMBL-EBI
- Annotation Platform: PubSEED platform for manual curation [1]
- Analysis Pipeline: Custom DEMETER workflow for data-driven metabolic network refinement [1]
Protocol 2: In Silico Prediction of Personalized Drug Metabolism
Objective: To predict the drug conversion potential of individual gut microbiomes using AGORA2 reconstructions.
Methodology:
- Metagenomic Data Processing:
- Obtain shotgun metagenomic sequencing data from human gut samples.
- Perform quality control, assembly, and binning to reconstruct metagenome-assembled genomes (MAGs) [4].
- Community Model Reconstruction:
- Drug Metabolism Simulation:
- Set constraints to reflect physiological conditions of the gut environment.
- Simulate the biotransformation of target drugs using constraint-based modeling approaches [1].
- Validation Against Experimental Data:
Key Reagents:
- Modeling Resource: AGORA2 reconstructions (7,302 strain models) [1]
- Simulation Software: COBRA Toolbox or similar constraint-based modeling environment
- Validation Datasets: Three independently collected experimental datasets for drug metabolism [1]
Research Reagent Solutions
Table 3: Essential Research Reagents and Resources for AGORA2-Based Drug Metabolism Studies
| Reagent/Resource | Function/Application | Specifications/Examples |
|---|---|---|
| AGORA2 Reconstruction Resource | Genome-scale metabolic models for 7,302 human microbes | Includes drug metabolism for 98 compounds; available through Virtual Metabolic Human (VMH) database [1] |
| DEMETER Pipeline | Data-driven metabolic network refinement | Semiautomated curation workflow; integrates genomic, biochemical, and physiological data [1] |
| PubSEED Annotation Platform | Manual curation of genome annotations | Enables validation of 446 gene functions across 35 metabolic subsystems [1] |
| Constraint-Based Modeling Software | Simulation of metabolic fluxes and drug transformations | COBRA Toolbox, COBRApy; enables prediction of community-level metabolic activities [1] [3] |
| Human Whole-Body Metabolic Reconstructions | Modeling host-microbiome co-metabolism | Compatible with generic and organ-resolved, sex-specific human reconstructions [1] |
Workflow Visualization
AGORA2 Reconstruction and Prediction Workflow
Personalized Drug Metabolism Prediction
The AGORA2 pipeline (Assembly of Gut Organisms through Reconstruction and Analysis, version 2) represents a transformative resource for personalized microbiome metabolic modeling, enabling unprecedented exploration of host-microbiome interactions in health and disease. This resource systematically expands upon previous microbial reconstruction efforts by encompassing 1,738 species across 25 phyla, totaling 7,302 strain-level reconstructions [1]. Such extensive taxonomic representation provides the foundation for investigating the functional metabolic diversity of the human gut microbiome, allowing researchers to move beyond correlative analyses toward mechanistic, predictive modeling of microbial community functions [1] [3].
AGORA2 addresses a critical bottleneck in microbiome research by providing curated, genome-scale metabolic reconstructions that integrate both taxonomic and functional dimensions of microbial diversity. These reconstructions capture the comprehensive metabolic potential of individual microbial strains, enabling the prediction of strain-resolved drug metabolism, nutrient utilization, and metabolite production [1]. The resource has been demonstrated to predict known microbial drug transformations with an accuracy of 0.81 and performs robustly against independently collected experimental datasets with accuracy ranging from 0.72 to 0.84 [1], establishing it as a reliable knowledge base for personalized medicine applications.
Table 1: Key Quantitative Features of the AGORA2 Resource
| Feature | Specification | Significance |
|---|---|---|
| Total Reconstructions | 7,302 strains | Enables strain-resolved modeling of personalized microbiomes |
| Species Coverage | 1,738 species | Captures substantial human gut microbial diversity |
| Phylogenetic Breadth | 25 phyla | Represents broad taxonomic diversity |
| Drug Metabolism Coverage | 98 drugs | Facilitates prediction of personalized drug metabolism |
| Enzyme Coverage | 15 drug-metabolizing enzymes | Provides mechanistic basis for biotransformation predictions |
| Experimental Validation Accuracy | 0.72-0.84 against independent datasets | Ensures biological relevance and predictive power |
| Atom-Atom Mapping | 65% of enzymatic and transport reactions | Enables detailed metabolic tracing studies |
Table 2: Functional Characterization in AGORA2
| Functional Category | Coverage | Application Examples |
|---|---|---|
| Drug Biotransformation | 98 drugs; 5,000+ strains | Predicting interindividual variation in drug efficacy and toxicity |
| Metabolite Production | Short-chain fatty acids, amino acids, vitamins | Linking microbiome composition to host metabolic health |
| Nutrient Utilization | Human milk oligosaccharides, dietary fibers | Understanding diet-microbiome interactions across life stages |
| Community Interactions | Cross-feeding, competition | Modeling ecological dynamics in gut communities |
Protocol: Construction and Curation of AGORA2 Reconstructions
Reconstruction Workflow
The following diagram illustrates the DEMETER pipeline for building curated metabolic reconstructions:
Step-by-Step Methodology
Data Collection and Integration
- Retrieve genome sequences for target microbial strains from public repositories
- Collate experimental data from 732 peer-reviewed papers and microbial reference textbooks covering 6,971 strains (95% of AGORA2) [1]
- Compile biochemical and physiological data from culture-based studies to inform metabolic capabilities
Draft Reconstruction Generation
Manual Curation and Refinement
- Manually validate and improve annotations of 446 gene functions across 35 metabolic subsystems for 5,438 genomes (74%) using PubSEED [1]
- Perform extensive literature-based curation to incorporate species-specific pathways, including drug metabolism capabilities
- Add metabolic structures for 1,838 metabolites (51% of total) and implement atom-atom mapping for 5,583 enzymatic and transport reactions (65%) [1]
Quality Control and Validation
- Execute comprehensive test suite to identify and resolve metabolic gaps and network inconsistencies [1]
- Verify flux consistency of reactions and eliminate futile cycles that generate biologically implausible ATP production
- Assess reconstruction quality using an unbiased scoring system, achieving an average quality score of 73% across all reconstructions [1]
Protocol: Personalized Community Modeling with AGORA2
Workflow for Building Personalized Models
The following diagram outlines the process for constructing personalized microbiome metabolic models:
Step-by-Step Methodology
Input Data Preparation
- Process metagenomic sequencing data from patient samples to determine taxonomic composition
- Quantify relative abundances of microbial taxa present in the AGORA2 resource
- Define environmental constraints based on host diet, physiological conditions, or drug exposures
Community Model Assembly
- Select corresponding AGORA2 reconstructions for detected microbial taxa
- Construct personalized microbiome community models by integrating strain-specific reconstructions in proportion to their relative abundance [3]
- Implement appropriate community modeling frameworks such as the Microbiome Modeling Toolbox [1]
Simulation and Analysis
- Apply constraint-based modeling techniques to predict metabolic fluxes under specified conditions
- Quantify production and consumption of metabolites relevant to host health (e.g., short-chain fatty acids, vitamins, amino acids)
- Predict drug transformation capabilities and potential drug-microbiome interactions [1]
- Perform comparative analyses between patient groups (e.g., healthy vs. disease states) to identify differentially abundant metabolic functions
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Resources for AGORA2-Based Research
| Resource | Type | Function | Availability |
|---|---|---|---|
| AGORA2 Reconstructions | Metabolic Models | Strain-resolved metabolic networks for 7,302 human microbes | Virtual Metabolic Human (VMH) database |
| DEMETER Pipeline | Computational Tool | Data-driven metabolic network refinement workflow | Published protocols [1] |
| VMH Namespace | Standardization | Unified nomenclature for metabolites and reactions | Virtual Metabolic Human platform |
| PubSEED | Annotation Tool | Manual curation and annotation of metabolic functions | Publicly available platform |
| Constraint-Based Modeling | Computational Framework | Predictive simulation of metabolic behavior | COBRA Toolbox |
| Personalized Community Modeling | Computational Framework | Building individual-specific microbiome models | Custom scripts based on published methods [3] |
Application Notes
Investigating Developmental Origins of Health and Disease
The AGORA2 pipeline has been successfully applied to elucidate metabolic differences in infant gut microbiomes based on delivery mode. Personalized metabolic modeling of gut microbiomes from 20 infants at multiple timepoints during the first year of life revealed that Cesarian section delivery (CSD) results in transient depletion of metabolic capabilities compared to vaginally delivered infants [3]. Specifically, CSD microbiomes showed reduced potential for human milk oligosaccharide degradation, bile acid transformation, and synthesis of various fermentation products during early development [3]. This application demonstrates how AGORA2 can generate testable hypotheses about how early-life microbiome perturbations may influence long-term health outcomes.
Predicting Personalized Drug Metabolism
AGORA2 enables prediction of interindividual variation in drug metabolism based on microbiome composition. The resource includes strain-resolved drug degradation and biotransformation capabilities for 98 drugs, accounting for known microbial drug-metabolizing enzymes [1]. When applied to gut microbiomes from 616 patients with colorectal cancer and controls, AGORA2 revealed substantial interpersonal variation in drug conversion potential that correlated with age, sex, body mass index, and disease stage [1]. This application highlights the potential for incorporating microbial metabolism into precision medicine approaches for drug dosing and selection.
Functional Analysis in Inflammatory Bowel Disease
Integration of AGORA2 with multi-omics data from inflammatory bowel disease (IBD) patients has identified key metabolic alterations associated with disease [8]. Modeling approaches have revealed specific taxa associated with variations in amino acids, short-chain fatty acids, and pH in the gut of IBD patients [8]. Furthermore, analysis of functional redundancy in IBD microbiomes using AGORA2-based community models showed that, despite decreased species diversity in IBD, functional redundancy increased for certain metabolites like hydrogen sulphide [9]. This paradoxical finding highlights how functional metrics derived from AGORA2 can provide insights beyond conventional diversity measures.
Troubleshooting Guide
Table 4: Common Challenges and Solutions in AGORA2 Implementation
| Challenge | Potential Cause | Solution |
|---|---|---|
| Gaps in Metabolic Networks | Incomplete genome annotation | Use DEMETER pipeline with experimental data for gap-filling [1] |
| Unrealistically High ATP Production | Futile cycles in metabolic network | Verify flux consistency and apply thermodynamic constraints [1] |
| Inaccurate Growth Predictions | Missing transport reactions or nutrient constraints | Curate uptake capabilities based on experimental literature [1] |
| Computational Intensity | Large community models with many strains | Implement compartmentalization or sampling approaches for complex communities |
Integration with Whole-Body Metabolic Models and Virtual Metabolic Human (VMH) Database
The AGORA2 pipeline represents a foundational resource in personalized microbiome research, providing genome-scale metabolic reconstructions of 7,302 human microorganisms to enable mechanistic modeling of host-microbiome interactions [1]. This pipeline achieves its full translational potential through systematic integration with whole-body metabolic (WBM) models and the Virtual Metabolic Human (VMH) database, creating a unified framework for predicting how microbial metabolism influences human physiology and drug response [1] [10]. This integration enables researchers to move beyond correlation to causation by providing a mechanistic, stoichiometrically accurate representation of the metabolic exchanges between host tissues and the microbiome.
The AGORA2 resource was explicitly designed for compatibility with existing human metabolic reconstructions, including the generic human metabolic reconstruction and organ-resolved, sex-specific whole-body models [1]. These WBM reconstructions capture the metabolism of 26 organs and six blood cell types with over 80,000 biochemical reactions in an anatomically and physiologically consistent manner [10]. The VMH database serves as the central hub connecting these resources, providing a common nomenclature and structured database that links human metabolism with genetics, microbial metabolism, nutrition, and disease [11].
Table 1: Key Components of the Integrated AGORA2 Framework
| Component | Description | Scale/Scope |
|---|---|---|
| AGORA2 | Genome-scale metabolic reconstructions of human gut microorganisms | 7,302 strains, 1,738 species, 25 phyla [1] |
| Whole-Body Metabolic Models | Organ-resolved computational models of human metabolism | 26 organs, 6 blood cell types, >80,000 reactions [10] |
| Virtual Metabolic Human (VMH) | Centralized database connecting metabolic entities | Links to >50 external databases [11] |
Quantitative Capabilities and Performance Metrics
The AGORA2 resource has been rigorously validated against multiple independent experimental datasets, demonstrating superior performance compared to automated reconstruction tools. When assessed against species-level metabolite uptake and secretion data, AGORA2 achieved an accuracy of 0.72 to 0.84, surpassing other reconstruction resources [1]. For predicting known microbial drug transformations, the resource achieved an accuracy of 0.81 [1]. The reconstructions also showed a significantly higher percentage of flux-consistent reactions compared to KBase draft reconstructions, despite having larger metabolic content [1].
The power of this integrated approach is demonstrated in clinical applications. For example, when modeling the drug conversion potential of gut microbiomes from 616 patients with colorectal cancer, AGORA2 revealed substantial variation between individuals that correlated with age, sex, body mass index, and disease stages [1]. In another study of inflammatory bowel disease (IBD), the integrated modeling approach identified 185 different bacterial reactions whose fluxes were associated with inflammation, enriched in nine key metabolic pathways [12].
Table 2: Performance Metrics of AGORA2 and Integrated Modeling
| Validation Metric | Performance | Context |
|---|---|---|
| Experimental Data Accuracy | 0.72 - 0.84 | Against species-level metabolite uptake/secretion data [1] |
| Drug Transformation Prediction | 0.81 accuracy | For known microbial drug transformations [1] |
| Flux Consistency | Significantly higher than draft reconstructions | Despite larger metabolic content [1] |
| Probiotic Engraftment Prediction | >85% accuracy | In synbiotic intervention trial [13] |
Experimental Protocols and Methodologies
Protocol: Building Personalized Whole-Body Models with Integrated Microbiome
Purpose: To construct personalized whole-body metabolic models that incorporate individual-specific gut microbiome composition data for predicting host-microbiome co-metabolism.
Materials:
- AGORA2 resource of microbial metabolic reconstructions [1]
- Whole-body metabolic reconstruction template [10]
- VMH database access [11]
- Host physiological data (age, sex, BMI, diet)
- Microbiome composition data (16S rRNA or metagenomic sequencing)
Procedure:
- Strain-Level Mapping: Map metagenomic sequencing data to microbial reference genomes compatible with AGORA2 reconstructions [12].
- Community Modeling: Reconstruct genome-scale metabolic models for the microbial community using tools such as MicrobiomeGS2 (for cooperation-focused analysis) or BacArena (for competition-focused analysis) [12].
- Host Contextualization: Parameterize the whole-body metabolic model with the individual's physiological, dietary, and metabolomic data [10].
- Integration: Connect the microbiome community model with the whole-body model through defined exchange metabolites in the VMH namespace [1] [11].
- Validation: Compare predicted metabolic outputs (e.g., SCFA production, drug metabolism) with experimental metabolomic data when available [12].
Applications: This protocol was used to model gut microbiomes from 616 colorectal cancer patients and controls, revealing extensive interindividual variation in drug metabolism potential [1].
Protocol: Predicting Microbial Drug Metabolism in Patient Cohorts
Purpose: To assess the strain-resolved drug conversion potential of individual gut microbiomes using AGORA2's manually curated drug metabolism database.
Materials:
- AGORA2 resource with drug metabolism capabilities (98 drugs, 15 enzymes) [1]
- Patient microbiome composition data
- Clinical metadata (age, sex, BMI, disease status)
Procedure:
- Model Personalization: Build personalized microbiome models for each patient using their microbiome composition data and the corresponding AGORA2 reconstructions [1].
- Drug Metabolism Mapping: Identify which strains in the personalized microbiome contain the enzymatic capabilities for specific drug transformations based on AGORA2's manually curated drug degradation and biotransformation reactions [1].
- Flux Prediction: Use constraint-based modeling to predict the potential flux through drug transformation pathways under physiological conditions.
- Correlation Analysis: Associate variations in drug metabolism potential with patient factors such as age, sex, BMI, and disease stage [1].
Applications: This approach demonstrated that the drug conversion potential of gut microbiomes varied substantially between individuals and correlated with clinical factors in colorectal cancer patients [1].
Protocol: Modeling Host-Microbiome Metabolic Interactions in Disease
Purpose: To identify dysregulated host-microbiome metabolic interactions in inflammatory bowel disease using multi-omics data and metabolic modeling.
Materials:
- Longitudinal multi-omics data (microbiome, transcriptome, metabolome)
- AGORA2 resource for microbiome modeling [1]
- Host metabolic reconstructions for tissue and blood [12]
- Linear mixed models for association testing
Procedure:
- Data Collection: Collect dense longitudinal multi-omics data from IBD patients before and after treatment initiation [12].
- Microbiome Modeling: Reconstruct metabolic models of patient gut microbiomes using AGORA2 and map 16S sequencing data to reference genomes [12].
- Host Modeling: Build context-specific metabolic models for host tissues using transcriptomic data from biopsies and blood samples [12].
- Association Analysis: Use linear mixed models to identify metabolic reactions and pathways in both microbiome and host that associate with disease activity [12].
- Cross-talk Analysis: Identify coordinated changes in metabolic activity across host and microbiome data layers, particularly in NAD, amino acid, one-carbon, and phospholipid metabolism [12].
Applications: This protocol revealed concomitant changes in NAD, amino acid, and one-carbon metabolism across host and microbiome in IBD, suggesting novel therapeutic strategies [12].
Signaling Pathways and Metabolic Networks
The integration of AGORA2 with whole-body models has revealed several key host-microbiome metabolic pathways with clinical significance. In inflammatory bowel disease, researchers identified concomitant changes across multiple data layers involving NAD, amino acid, one-carbon, and phospholipid metabolism [12]. On the host level, elevated tryptophan catabolism depleted circulating tryptophan, thereby impairing NAD biosynthesis, while reduced host transamination reactions disrupted nitrogen homeostasis and polyamine/glutathione metabolism [12]. Simultaneously, microbiome metabolic shifts in NAD, amino acid, and polyamine metabolism exacerbated these host metabolic imbalances [12].
In infant gut microbiome development, the integration approach revealed that Cesarian section delivery altered metabolic capabilities, particularly in human milk oligosaccharide degradation, bile acid transformation, and production of fermentation products [14]. These early-life metabolic differences may have long-term implications for health and disease susceptibility through metabolic programming mechanisms [14].
Table 3: Essential Research Reagents and Computational Resources for AGORA2 Integration Studies
| Resource | Type | Function | Access |
|---|---|---|---|
| AGORA2 Resource | Microbial metabolic reconstructions | Provides 7,302 strain-resolved models of human gut microorganisms with drug metabolism capabilities | Available via VMH database [1] |
| Whole-Body Metabolic Reconstructions | Host metabolic models | Sex-specific models of 26 organs and 6 blood cell types for physiological modeling | Available via VMH database [10] |
| Virtual Metabolic Human (VMH) | Database platform | Central hub with common nomenclature connecting microbial and human metabolism | Online access at digitalmetabolictwin.org [11] |
| DEMETER Pipeline | Computational workflow | Data-driven refinement pipeline for reconstruction quality control and gap-filling | Described in AGORA2 publication [1] |
| MicrobiomeGS2 | Modeling tool | Community metabolic modeling with focus on cooperative interactions | Used in IBD studies [12] |
| BacArena | Modeling tool | Agent-based platform for modeling competitive microbial interactions | Used in IBD studies [12] |
| Constraint-Based Reconstruction and Analysis (COBRA) | Modeling framework | Mathematical approach for simulating metabolic fluxes in biological systems | Primary methodology [1] |
Applications and Validation Studies
The integration of AGORA2 with whole-body models has enabled numerous applications across clinical and research domains. In inflammatory bowel disease, modeling revealed a multi-level deregulation of host-microbiome metabolic networks, with reduced within-community metabolic exchange and altered microbiome-host exchange during inflammation [12]. Specifically, researchers identified ten metabolites with altered cross-feeding patterns during inflammation, including increased lactate cross-feeding and reduced exchange of fermentation-related metabolites [12].
In infant development, personalized modeling of gut microbiome metabolism throughout the first year of life revealed that Cesarian section delivery altered metabolic capabilities at the earliest stages, with depletion in fermentation products, human milk oligosaccharide degradation products, and amino acids [14]. These metabolic differences normalized later in the first year of life but may have implications for long-term health outcomes [14].
For therapeutic development, metabolic modeling has been used to identify determinants of synbiotic efficacy in human intervention trials, accurately predicting probiotic engraftment with over 85% accuracy [13]. The models also predicted significant increases in butyrate and propionate production following synbiotic treatment, with predicted changes in propionate production negatively associated with changes in C-reactive protein, a marker of systemic inflammation [13].
The integration framework has also enabled the prediction of dietary interventions that could remodel the microbiome to restore metabolic homeostasis in disease states, suggesting novel therapeutic strategies for complex disorders like IBD [12]. By leveraging the stoichiometric precision of metabolic models, researchers can move beyond correlation to propose and test causal mechanisms underlying host-microbiome interactions in health and disease.
From Theory to Practice: Implementing AGORA2 for Precision Medicine Applications
Personalized microbiome metabolic modeling represents a transformative approach in biomedical research, enabling a mechanistic, systems-level understanding of how gut microbiota influence human health and disease. This paradigm shift from correlation to causation is largely driven by the integration of genome-resolved metagenomics with constraint-based metabolic modeling [15] [16]. The AGORA2 pipeline stands at the forefront of this transition, providing a standardized framework for constructing personalized, strain-resolved community models that predict metabolic functions across individuals [1]. These models have demonstrated remarkable accuracy in predicting clinically relevant metabolites, including short-chain fatty acid production profiles that correlate with cardiometabolic and immunological health markers [17]. This protocol details the comprehensive workflow from raw metagenomic sequencing data to personalized community-scale metabolic models, with particular emphasis on the AGORA2 ecosystem and its applications in precision medicine and therapeutic intervention development.
Background and Significance
The human gut microbiome encodes complex metabolic capabilities that profoundly influence host physiology, including drug metabolism, immune function, and nutrient processing [1] [17]. Traditional 16S rRNA sequencing approaches have limited utility in functional studies due to their inability to resolve species-level taxonomy or predict metabolic capabilities [15]. Genome-resolved metagenomics, which involves reconstructing microbial genomes directly from whole-metagenome sequencing data, has emerged as a pivotal technology for microbiome medicine [15]. When combined with genome-scale metabolic models (GEMs), this approach enables quantitative prediction of microbial community metabolic fluxes and their variation across individuals [16] [17].
The AGORA2 resource (Assembly of Gut Organisms through Reconstruction and Analysis, version 2) provides 7,302 manually curated microbial metabolic reconstructions, representing 1,738 species and 25 phyla, with expanded capabilities for modeling drug biotransformations [1]. This resource, complemented by the APOLLO resource of 247,092 reconstructions from metagenome-assembled genomes (MAGs), enables the construction of personalized microbiome models that account for individual-specific microbial community composition [5]. The resulting models can predict personalized responses to dietary interventions, prebiotics, and probiotics, paving the way for precision microbiome therapeutics [17].
The comprehensive workflow from metagenomic data to personalized community models involves six major stages, each with specific inputs, processes, and outputs as illustrated below:
Experimental Protocols
Metagenomic Data Processing and Genome Assembly
Objective: To reconstruct metagenome-assembled genomes (MAGs) from whole-metagenome sequencing (WMS) data.
Procedure:
- Quality Control: Process raw sequencing reads using FastQC and Trimmomatic to remove adapter sequences and low-quality bases.
- Metagenome Assembly: Assemble quality-filtered reads using either single-sample assembly (metaSPAdes) or co-assembly approaches (MEGAHIT) based on research objectives [15].
- Genome Binning: Group contigs into MAGs using composition-based (tetranucleotide frequency) and abundance-based features across multiple samples.
- Bin Refinement: Apply DAS Tool to obtain an optimal set of non-redundant MAGs from multiple binning approaches.
- Quality Assessment: Evaluate MAG quality using CheckM, retaining medium-quality (≥50% completeness, ≤10% contamination) and high-quality (≥90% completeness, ≤5% contamination) bins [5].
Critical Considerations:
- Single-assembly preserves strain-specific variants but may produce more fragmented contigs [15].
- Co-assembly improves continuity but may blur strain-level differences [15].
- Geographic and population biases in reference databases may affect binning performance; incorporate locally relevant reference genomes when available [15].
Metabolic Reconstruction from Genomic Data
Objective: To convert MAGs into high-quality genome-scale metabolic models (GEMs).
Procedure:
- Draft Reconstruction: Generate draft models using automated tools such as CarveMe or gapseq [16] [18].
- Namespace Standardization: Convert reaction and metabolite identifiers to Virtual Metabolic Human (VMH) namespace using DEMETER pipeline [1] [5].
- Model Refinement: Manually curate models based on comparative genomics and literature data, focusing on:
- Species-specific pathways (e.g., drug metabolism)
- Periplasm compartmentalization where appropriate
- Biomass composition accuracy [1]
- Gap-Filling: Employ algorithms like DNNGIOR to predict missing reactions using phylogenetic and reaction co-occurrence patterns [19].
- Quality Control: Validate models through the DEMETER test suite, checking for:
Validation:
- Compare model predictions against experimental data from resources like NJC19 and Madin et al. [1].
- Test drug transformation predictions against known microbial drug metabolism [1].
Community Model Construction and Simulation
Objective: To integrate individual GEMs into personalized community metabolic models.
Procedure:
- Community Assembly: Construct sample-specific models by combining AGORA2 reconstructions matching the taxonomic profile of the target microbiome [17].
- Environment Specification: Define nutritional environment based on host diet or experimental conditions using VMH database [17].
- Constraint Definition: Apply constraints to represent ecological interactions:
- Flax Balance Analysis: Solve the optimization problem to predict community metabolic fluxes using the MICOM framework [17].
- Result Interpretation: Analyze predicted metabolite exchange, cross-feeding relationships, and community metabolic objectives.
Personalization Approaches:
- Strain-Level Resolution: Incorporate strain-specific GEMs when available [1].
- Abundance Weighting: Weight metabolic contributions by species relative abundance [17].
- Individualized Environments: Customize nutrient constraints based on personalized dietary data [17].
Table 1: Essential Databases for Metabolic Reconstruction and Modeling
| Category | Resource | Description | Application in Workflow |
|---|---|---|---|
| Generalist Databases | KEGG | Biochemical pathways and reactions | General annotation of genes and genomes [18] |
| MetaCyc | Encyclopedia of metabolic reactions | Reference for metabolic pathways [18] | |
| UniProt | Protein function association | Functional annotation [18] | |
| Metabolic Reconstruction Collections | AGORA2 | 7,302 curated microbial GEMs | Reference reconstructions for human microbiome [1] |
| APOLLO | 247,092 MAG-derived reconstructions | Expanded strain diversity [5] | |
| BiGG | High-quality metabolic network reconstructions | Quality reference models [18] | |
| Specialized Databases | CAZy | Carbohydrate-active enzymes | Modeling polysaccharide utilization [18] |
| CARD | Antibiotic resistance | Predicting drug inactivation [18] | |
| TCDB | Transporter classification | Transport reaction annotation [18] | |
| Modeling Platforms | VMH (Virtual Metabolic Human) | Unified namespace and modeling platform | Standardizing reactions and metabolites [1] |
| COBRA Toolbox | MATLAB modeling suite | Constraint-based modeling and analysis [20] |
The Scientist's Toolkit
Table 2: Essential Computational Tools for Metabolic Modeling
| Tool | Function | Application Note |
|---|---|---|
| metaSPAdes | Metagenomic assembly using De Bruijn graphs | Preferred for complex communities; preserves strain variation [15] |
| CarveMe | Automated metabolic reconstruction | Top-down approach; rapid generation (minutes per genome) [16] [18] |
| gapseq | Automated metabolic reconstruction | Curated reaction database; dedicated gap-filling (slower but potentially more accurate) [18] |
| DEMETER | Semi-automated curation pipeline | Converts draft reconstructions to VMH namespace; extensive quality control [1] [5] |
| DNNGIOR | AI-powered gap-filling | Uses neural networks to predict missing reactions; improves accuracy 2-14x [19] |
| COBRA Toolbox | Constraint-based modeling | MATLAB-based suite for simulation and analysis [20] |
| MICOM | Microbial community modeling | Python package for building and simulating community models [17] |
| MicroMap | Metabolic network visualization | Visual exploration of microbiome metabolism; 5064 reactions, 3499 metabolites [20] |
Data Analysis and Interpretation
Validation Against Experimental Data
Objective: To assess model predictive accuracy using experimental data.
Approaches:
- Metabolite Production Validation: Compare predicted short-chain fatty acid (SCFA) production profiles against measured fecal SCFA concentrations [17].
- Growth Capability Validation: Test model predictions of growth on specific carbon sources against phenotypic data [1].
- Drug Transformation Validation: Validate predicted drug metabolism against known microbial biotransformation reactions [1].
Statistical Analysis:
- Calculate accuracy metrics (e.g., AUC, F1-score) against independent experimental datasets [1].
- Assess correlation between predicted metabolite fluxes and clinically measured biomarkers [17].
Personalization and Stratification Analysis
Objective: To identify metabolic differences across host phenotypes.
Approaches:
- Disease Stratification: Compare metabolic capabilities of microbiomes from healthy vs. diseased individuals [17] [21].
- Demographic Analysis: Identify metabolic variations associated with age, sex, BMI, and geography [1] [17].
- Intervention Prediction: Simulate responses to dietary, prebiotic, and probiotic interventions to design personalized therapies [17].
Visualization:
- Use MicroMap to visualize differences in metabolic capabilities between microbial taxa [20].
- Generate heatmaps of relative reaction presence across microbial groups [20].
Troubleshooting and Optimization
Common Challenges and Solutions
Table 3: Troubleshooting Guide for Metabolic Modeling
| Challenge | Potential Cause | Solution |
|---|---|---|
| Low Model Accuracy | Incomplete genome annotation | Apply DNNGIOR for intelligent gap-filling [19] |
| Unrealistic Flux Predictions | Incorrect biomass composition | Manually curate biomass reaction based on literature [1] |
| Poor Generalization | Population-specific biases | Include MAGs from diverse populations using APOLLO resource [5] |
| Low Classification Performance | Inappropriate data transformation | Test multiple transformations; presence-absence often performs well [21] |
| Inconsistent Metabolite Naming | Different database conventions | Standardize to VMH namespace using DEMETER [1] |
Performance Optimization Strategies
Computational Efficiency:
- Use high-performance computing resources for large-scale reconstruction (40x speedup for visualization generation) [20].
- Implement parallel processing for community model simulation.
Model Quality Improvement:
Classification Optimization:
Applications and Future Directions
The integration of metagenomic data with personalized community modeling enables numerous applications in precision medicine, including prediction of individual-specific drug metabolism, design of targeted dietary interventions, and identification of microbial biomarkers for disease [1] [17]. Future developments should focus on addressing geographic biases in microbiome data, improving strain-level resolution, and integrating host metabolism for full host-microbiome modeling [15] [5]. Standardization of metabolite nomenclature and continued expansion of metabolic databases will further enhance model accuracy and interoperability [16]. As these resources mature, personalized microbiome metabolic modeling will become an increasingly powerful tool for understanding and manipulating host-microbiome interactions in health and disease.
The construction of multi-tissue metabolic models that integrate host and microbiome metabolism represents a transformative approach in systems biology. These models provide a computational framework to simulate the complex metabolic interactions between human tissues and the microbial communities that inhabit the gut. Genome-scale metabolic models (GEMs) serve as the foundation for this approach, offering mathematical representations of the metabolic networks of both host and microbial organisms based on their genomic annotations [22]. The AGORA2 pipeline (Assembly of Gut Organisms through Reconstruction and Analysis, version 2) has emerged as a pivotal resource in this field, providing curated, strain-resolved metabolic reconstructions for 7,302 human microorganisms that enable personalized, predictive analysis of host-microbiome interactions [23].
The power of multi-tissue modeling lies in its ability to simulate metabolite flow between host organs and microbial compartments, revealing how gut microbes influence systemic host metabolism and vice versa. This approach moves beyond single-tissue or single-species analyses to capture the full complexity of host-microbiome metabolic crosstalk. By integrating data from multiple biological layers—including metagenomics, transcriptomics, and metabolomics—researchers can build context-specific models that reflect individual physiological states, dietary patterns, and disease conditions [24]. These models have demonstrated significant potential for understanding the mechanistic basis of various diseases, including inflammatory bowel disease [12], Alzheimer's disease [25], and aging-related metabolic decline [24], while also facilitating the development of novel therapeutic interventions such as live biotherapeutic products [26].
Building integrated host-microbiome metabolic models requires access to comprehensive, high-quality metabolic reconstructions for both host tissues and microbial species. Several curated resources have been developed to meet this need, each with distinct strengths and applications.
Table 1: Key Resources for Metabolic Reconstruction and Modeling
| Resource Name | Description | Scale | Primary Application |
|---|---|---|---|
| AGORA2 [23] | Curated genome-scale metabolic reconstructions of human gut microorganisms | 7,302 strains | Personalized modeling of human gut microbiome metabolism |
| APOLLO [4] | Genome-scale reconstructions from diverse human microbiomes | 247,092 genomes | Cross-population and cross-body site metabolic studies |
| Recon3D [22] | Comprehensive human metabolic reconstruction | N/A | Host tissue metabolism representation |
| BiGG [22] | Database of biochemical, genetic, and genomic knowledge | N/A | Standardized metabolic model repository |
| MetaNetX [22] | Platform for genome-scale metabolic networks | N/A | Namespace standardization and model integration |
The AGORA2 resource stands out for its direct relevance to human microbiome studies, incorporating not only comprehensive metabolic capabilities but also strain-resolved drug degradation and biotransformation capabilities for 98 drugs [23]. This resource has been extensively curated based on comparative genomics and literature searches, achieving an accuracy of 0.72-0.84 against independently assembled experimental datasets [23]. The APOLLO resource significantly expands the scope of available models, encompassing strains from 34 countries, all age groups, and multiple body sites, thereby enabling more diverse and personalized metabolic modeling applications [4].
Software Tools for Model Reconstruction and Analysis
The construction and simulation of multi-tissue host-microbiome models relies on specialized software tools that facilitate model reconstruction, integration, and analysis.
Table 2: Essential Software Tools for Metabolic Modeling
| Tool Name | Function | Key Features | Applicability |
|---|---|---|---|
| CarveMe [22] | Automated metabolic model reconstruction | Draft model generation from genome sequences | Microbial model reconstruction |
| gapseq [24] [22] | Metabolic network reconstruction and analysis | Pathway gap filling and validation | Microbial model reconstruction and refinement |
| RAVEN [22] | Metabolic model reconstruction and simulation | Integration of transcriptomic data | Host and microbial model reconstruction |
| ModelSEED [22] | Automated model reconstruction | Rapid generation from genomic data | Microbial model reconstruction |
| Microbiome Modeling Toolbox [25] | Personalized microbiome modeling | Integration of metagenomic data | Building sample-specific community models |
| COBRA Toolbox [22] | Constraint-based reconstruction and analysis | Flux balance analysis and variant methods | Model simulation and analysis |
These tools employ the constraint-based reconstruction and analysis (COBRA) framework, which uses stoichiometric matrices to represent metabolic networks and flux balance analysis (FBA) to predict metabolic fluxes under steady-state assumptions [22] [27]. The modeling approach is based on the fundamental equation S·v = 0, where S represents the stoichiometric matrix and v represents the flux distribution vector [27]. Constraints are applied to limit flux values (Vi,min < Vi < V_i,max), and objective functions are optimized to identify biologically relevant flux distributions [27].
Protocol: Building Integrated Host-Microbiome Metabolic Models
Data Collection and Preparation
Step 1: Host Data Collection
- Collect host genomic, transcriptomic, and metabolomic data from relevant tissues (e.g., colon, liver, brain)
- For human studies, obtain physiological parameters including age, sex, BMI, and clinical metadata
- For tissue-specific modeling, collect transcriptomic data from multiple tissues to capture systemic metabolic interactions [24]
Step 2: Microbiome Data Generation
- Perform shotgun metagenomic sequencing of stool samples with sufficient depth (minimum 2-3 million reads per sample) [25]
- Process sequencing data using tools like Woltka against reference databases such as Web of Life [25]
- Filter species to include only those present in metabolic reconstruction resources (e.g., AGORA2) to ensure model coverage [25]
Step 3: Metabolomic Profiling
- Collect urine and serum samples for metabolomic analysis using NMR or mass spectrometry
- For host-microbiome interaction studies, focus on key microbial metabolites including short-chain fatty acids, bile acids, vitamins, and amino acids [3] [25]
- Normalize metabolomic data (e.g., creatinine normalization for urine samples) to account for technical variations [25]
Model Reconstruction and Integration
Step 4: Host Metabolic Model Reconstruction
- Obtain tissue-specific metabolic models using context-specific reconstruction algorithms
- For multi-tissue modeling, create separate model instances for each tissue (e.g., colon, liver, brain) connected through blood exchange reactions [24]
- Validate model functionality by ensuring production of key biomass components and energy metabolites
Step 5: Microbial Community Model Reconstruction
- Map microbial abundance data to AGORA2 strains using the Microbiome Modeling Toolbox [25]
- Generate pan-species microbial reconstructions consisting of the union of all metabolites and reactions present in corresponding AGORA2 strains of the same species [25]
- Apply abundance cutoffs to remove low-abundance species and reduce model complexity while maintaining representation of key community functions [25]
Step 6: Host-Microbiome Model Integration
- Connect host and microbiome models through a shared gut lumen compartment that enables metabolite exchange
- Define blood circulation systems that connect multiple host tissues and enable systemic metabolic interactions
- Implement namespace standardization using resources like MetaNetX to ensure compatibility between host and microbial model components [22]
Diagram Title: Host-Microbiome Model Integration Workflow
Model Simulation and Validation
Step 7: Constraint Definition and Flux Simulation
- Define nutritional constraints based on dietary intake or culture media composition
- Set tissue-specific metabolic constraints based on transcriptomic data
- Perform flux balance analysis using community-level objective functions or multi-objective optimization approaches [27]
Step 8: Validation with Experimental Data
- Compare predicted metabolite secretion patterns with measured metabolomic profiles
- Validate predicted host-microbiome co-metabolism through isotopic tracing experiments [22]
- Assess model accuracy by comparing predicted microbial functions with experimentally determined capabilities [23]
Step 9: Gap Analysis and Model Refinement
- Identify metabolic gaps in the integrated model through gap filling algorithms
- Refine model components based on literature evidence and experimental data
- Iterate between simulation and validation to improve model predictive performance
Application Notes
Case Study: Aging-Associated Metabolic Decline
The application of multi-tissue host-microbiome modeling to aging research has revealed profound insights into how microbial metabolic activity changes with host age. In a comprehensive study of aging mice, researchers reconstructed integrated metabolic models of the host and 181 gut microorganisms, demonstrating a pronounced reduction in metabolic activity within the aging microbiome [24]. This was accompanied by reduced beneficial interactions between bacterial species and downregulation of essential host pathways in nucleotide metabolism that critically depend on microbial functions [24].
Key Findings:
- Aging microbiomes showed reduced metabolic activity and decreased cross-feeding interactions
- Microbial contribution to host nucleotide metabolism declined with age
- Model predictions suggested microbiome-dependent disruption of intestinal barrier function and cellular replication in aged hosts
- The identified pathways represent potential targets for microbiome-based anti-aging therapies [24]
Protocol Implementation:
- Models integrated transcriptomic data from colon, liver, and brain tissues with metagenomic and metabolomic data from fecal samples
- Microbial metabolic networks were reconstructed using gapseq and validated for functionality [24]
- Host-microbiome associations were identified through correlation analysis between microbiome metabolic functions and host transcript levels
Case Study: Inflammatory Bowel Disease
Multi-tissue metabolic modeling of inflammatory bowel disease (IBD) has uncovered complex disruptions in host-microbiome metabolic networks that drive disease pathology. By analyzing 296 biopsy, 324 blood, and 565 microbiome samples from IBD patients, researchers identified concomitant changes in NAD, amino acid, one-carbon, and phospholipid metabolism across host and microbial compartments [12].
Key Findings:
- Inflammation was associated with reduced within-microbiome metabolic exchanges, particularly for SCFA production
- Host tryptophan catabolism was elevated, depleting circulating tryptophan and impairing NAD biosynthesis
- Reduced host transamination reactions disrupted nitrogen homeostasis and glutathione metabolism
- Microbiome metabolic shifts in NAD and amino acid metabolism exacerbated host metabolic imbalances [12]
Therapeutic Applications:
- Model-predicted dietary interventions were identified that could remodel the microbiome to restore metabolic homeostasis
- The approach revealed novel therapeutic strategies targeting host-microbiome co-metabolism in IBD [12]
Case Study: Alzheimer's Disease
Integrated host-microbiome modeling has provided insights into the potential role of gut microbes in neurodegenerative diseases. Using personalized whole-body metabolic models, researchers discovered that reduced urine formate levels in Alzheimer's disease patients resulted from combined effects of host genetics and altered microbial formate metabolism [25].
Key Findings:
- Urine metabolomics revealed decreased formate and fumarate concentrations in Alzheimer's patients
- Microbial contribution to formate production was altered in Alzheimer's host-microbiome models
- Specific reactions in host amino acid and sugar metabolism were linked to genes associated with Alzheimer's pathology [25]
Methodological Innovation:
- The study combined urine metabolomics from one cohort with whole-genome sequencing and metagenomics from an independent cohort
- Personalized whole-body models enabled quantification of microbial contribution to systemic metabolite levels
- The approach identified formate as a potential early marker of Alzheimer's disease [25]
Diagram Title: Host-Microbiome Metabolic Crosstalk
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Host-Microbiome Metabolic Modeling
| Reagent/Resource | Function | Application Example | Technical Considerations |
|---|---|---|---|
| AGORA2 Resource [23] | Strain-resolved microbial metabolic reconstructions | Personalized modeling of individual gut microbiomes | Covers 7,302 strains; includes drug metabolism capabilities |
| APOLLO Resource [4] | Diverse microbial reconstructions from global populations | Cross-population comparative studies | Includes 247,092 genomes from multiple continents and body sites |
| Recon3D [22] | Comprehensive human metabolic reconstruction | Host tissue metabolism representation | Requires contextualization for specific tissues and conditions |
| Metagenomic Sequencing Kits | Profiling microbial community composition | Species abundance quantification | Minimum 2-3 million reads/sample recommended for sufficient coverage [25] |
| NMR/Mass Spectrometry Platforms | Metabolite quantification and identification | Validation of predicted metabolic fluxes | Requires proper sample normalization and quality controls [25] |
| Gapseq Pipeline [24] | Metabolic network reconstruction and analysis | Generation of microbial metabolic models | Provides functional annotation and pathway gap filling |
| COBRA Toolbox [22] | Constraint-based modeling and simulation | Flux balance analysis of integrated models | Supports various optimization algorithms and constraint methods |
Troubleshooting and Technical Considerations
Common Challenges and Solutions
Challenge 1: Model Integration Incompatibilities
- Problem: Host and microbial models use different metabolite naming conventions, creating integration barriers
- Solution: Implement namespace standardization using MetaNetX or similar resources to harmonize metabolite and reaction identifiers across models [22]
Challenge 2: Thermodynamic Infeasibilities
- Problem: Integrated models contain thermodynamically infeasible cycles that produce energy without input
- Solution: Apply loop law constraints or implement thermodynamic curation pipelines to eliminate energy-generating cycles [22]
Challenge 3: Computational Complexity
- Problem: Large multi-tissue models with hundreds of microbial species become computationally intractable
- Solution: Apply model reduction techniques, remove low-abundance species, or use distributed computing approaches [25]
Challenge 4: Contextualization Accuracy
- Problem: Generic models fail to capture individual-specific metabolic capabilities
- Solution: Integrate multi-omic data (transcriptomics, metabolomics) to create personalized, context-specific models [24] [12]
Quality Control Metrics
- Microbiome Model Coverage: Ensure that mapped species account for >70% of total sequencing reads in metagenomic data [25]
- Host Model Functionality: Verify that tissue-specific models can produce essential biomass components and energy metabolites
- Prediction Validation: Compare model predictions with experimental data for key metabolites (e.g., SCFA levels in gut, serum metabolites)
- Numerical Accuracy: Check for numerical stability in flux solutions and objective function values across simulations
Future Directions and Applications
The field of multi-tissue host-microbiome metabolic modeling is rapidly evolving, with several promising directions for future development. The integration of more sophisticated microbial community modeling approaches, including spatial organization and dynamic interactions, will enhance the biological relevance of model predictions [22]. The expansion of model resources to include fungal and viral components of the microbiome will provide a more comprehensive view of host-microbial interactions [22].
Applications in therapeutic development are particularly promising, with multi-tissue models playing an increasingly important role in the design of live biotherapeutic products (LBPs) [26]. The AGORA2 pipeline enables systematic screening of LBP candidates, assessment of host-microbiome compatibility, and prediction of therapeutic outcomes in a personalized manner [26]. As these models continue to incorporate more diverse human populations and disease states, they will become increasingly valuable for developing precision medicine approaches that account for individual variations in host genetics, microbiome composition, and environmental exposures [4] [23].
The continued refinement of multi-tissue host-microbiome metabolic models represents a crucial step toward understanding the complex metabolic interactions that underlie human health and disease. By providing a computational framework to simulate these interactions, the AGORA2 pipeline and related resources are enabling researchers to move beyond correlation to causation, identifying mechanistic links between microbial metabolism and host physiology that can be targeted for therapeutic intervention.
The AGORA2 (Assembly of Gut Organisms through Reconstruction and Analysis, version 2) pipeline represents a cornerstone for personalized, predictive analysis of host-microbiome metabolic interactions in drug development. This resource provides a mechanistic, systems biology approach to understanding strain-resolved drug metabolism by the human gut microbiome. AGORA2 accounts for 7,302 microbial strains and includes manually curated, strain-resolved drug degradation and biotransformation capabilities for 98 drugs [1]. The reconstructions are generated through a data-driven refinement pipeline (DEMETER), which integrates extensive manual curation based on comparative genomics and literature searches, encompassing 732 peer-reviewed papers and reference textbooks [1]. By linking microbial genomes to metabolic phenotypes, AGORA2 enables researchers to move beyond correlations and generate testable hypotheses about how an individual's gut microbiome will process a therapeutic compound, thereby de-risking drug development and paving the way for precision medicine.
Quantitative Performance Benchmarks
The predictive performance of AGORA2 has been rigorously validated against independent experimental data. The table below summarizes its key quantitative benchmarks.
Table 1: AGORA2 Performance and Scale Metrics
| Metric | Value | Context / Significance |
|---|---|---|
| Number of Modeled Strains | 7,302 | Represents 1,738 species and 25 phyla [1] |
| Number of Drugs with Curated Metabolism | 98 | Covers biotransformation and degradation reactions [1] |
| Prediction Accuracy (Experimental Datasets) | 0.72 – 0.84 | Surpassed other reconstruction resources [1] |
| Prediction Accuracy (Drug Transformations) | 0.81 | Accuracy for predicting known microbial drug transformations [1] |
| Flux Consistent Reactions | High Fraction | Significantly higher than initial drafts and other automated resources (p < 1×10⁻³⁰) [1] |
Experimental Protocols for Validation and Application
This section provides detailed methodologies for key experiments utilizing the AGORA2 pipeline, from in vitro validation to in silico personalized modeling.
Protocol: High-Throughput Culture Collection Screen for Drug Metabolism
This protocol is designed to empirically identify bacterial strains capable of metabolizing a drug of interest, providing validation data for in silico predictions [28].
- Objective: To identify which bacterial isolates from a curated collection metabolize a target drug and characterize the resulting metabolites.
- Background: While in silico tools are powerful, empirical validation in controlled systems is crucial. This method allows for straightforward interpretation by testing individual strains [28].
Materials:
- Research Reagent Solutions:
- Anaerobically Cultured Bacterial Isolates: A representative library of human gut strains (e.g., from the AGORA2 list) [28].
- Reduced Anaerobic Growth Medium: Such as YCFA or M9 with necessary nutrients and a reducing agent (e.g., cysteine-HCl).
- Drug Stock Solution: The compound of interest, dissolved in a suitable, sterile solvent (e.g., DMSO, water).
- Analytical Standards: For the parent drug and any suspected metabolites.
- Quenching Solution: Cold acetonitrile:methanol (1:1, v/v).
- HPLC-MS/MS System: With appropriate columns and mass spectrometry conditions for the drug.
Procedure:
- Inoculum Preparation: Grow each bacterial strain to mid-log phase in anaerobic conditions.
- Incubation Setup: In an anaerobic chamber, aliquot fresh medium into sterile tubes. Add the drug stock solution to a sub-pharmacological concentration (e.g., 10-100 µM). Inoculate with a standardized volume of bacterial culture. Include sterile controls (medium + drug, no bacteria) and uninoculated controls (bacteria, no drug).
- Anaerobic Incubation: Incubate at 37°C for a predetermined period (e.g., 6-48 hours).
- Sample Quenching and Extraction: At the endpoint, add an equal volume of cold quenching solution to the culture. Vortex mix and centrifuge to pellet cells and precipitated protein.
- Analysis: Transfer the supernatant for analysis by HPLC-MS/MS. Monitor for the loss of the parent drug and the appearance of new metabolite peaks.
- Data Analysis: Compare chromatograms from test incubations against sterile and uninoculated controls. Identify metabolites by their retention time and mass fragmentation pattern, confirmed with analytical standards where available.
Protocol: Building a Personalized, Strain-Resolved Community Model
This protocol details the workflow for using the AGORA2 resource to build a predictive metabolic model of an individual's gut microbiome to simulate drug metabolism potential [3].
- Objective: To predict the drug conversion potential of a personalized gut microbiome based on its metagenomic sequencing data.
- Background: AGORA2 reconstructions can be used as building blocks to create personalized community models. These models simulate the metabolic network of an individual's microbiome, allowing for strain-resolved prediction of community-level functions like drug metabolism [1] [3].
Materials:
- Research Reagent Solutions:
- AGORA2 Model Resource: The collection of 7,302 genome-scale metabolic reconstructions [1].
- Metagenomic Sequencing Data: Shotgun metagenomic data from a human fecal sample.
- Metagenomics Analysis Tools: Software for quality control (FastQC), host read removal (Bowtie2), and taxonomic profiling (MetaPhlAn, mOTUs).
- Modeling Software: A constraint-based modeling and analysis (COBRA) toolbox, such as the MATLAB COBRA Toolbox or the Python implementation.
- Personalized Modeling Pipeline: Custom scripts to map taxonomic abundance data to AGORA2 models and construct the community model.
Procedure:
- Metagenomic Data Preprocessing: Perform quality control on raw sequencing reads and remove host-derived reads.
- Taxonomic Profiling: Use a taxonomic profiler to determine the relative abundance of microbial species or strains in the sample.
- Model Selection and Scaling: Map the identified taxa to corresponding AGORA2 reconstructions. Scale the reaction bounds (e.g., uptake and secretion rates) of each strain's model based on its relative abundance in the community.
- Community Model Construction: Create a compartmentalized community model that includes all selected and scaled individual strain models. Define a shared extracellular environment and a common objective function (e.g., community biomass maximization).
- Simulation of Drug Metabolism: Introduce the drug of interest as a potential nutrient source in the extracellular medium. Use constraint-based methods (e.g., Flux Balance Analysis or parsimonious FBA) to simulate community metabolism. Analyze the flux through the relevant drug transformation reactions to predict the rate of conversion.
- Output Analysis: The primary output is a prediction of whether the community can metabolize the drug and which strains are the key contributors to the metabolic flux.
The following workflow diagram illustrates the computational protocol for building a personalized model.
Protocol: Integrating Metabolomic Data for Mechanistic Insight
This protocol leverages the UK Biobank's large-scale metabolomic dataset to complement and validate microbiome model predictions [29] [30].
- Objective: To integrate population-scale metabolomic data with microbiome models to uncover robust microbe-metabolite-drug relationships.
- Background: Metabolomic data provides a readout of the functional state of an ecosystem. Integrating these data with mechanistic microbiome models can help distinguish causal relationships from correlations and identify key metabolic pathways influenced by the microbiome [29] [30].
Materials:
- Research Reagent Solutions:
- Large-Scale Metabolomic Dataset: Such as the UK Biobank dataset comprising ~250 metabolites measured in 500,000 individuals [29].
- Statistical Integration Tools: Software for integrative analysis (e.g., R or Python with packages for Sparse PLS, Procrustes analysis, or MOFA2) [30].
- AGORA2-based Predictions: Simulated metabolite secretion or consumption profiles from personalized community models.
Procedure:
- Data Preprocessing: Normalize and transform both the metabolomic data (e.g., log-transformation) and microbial abundance data (e.g., Centered Log-Ratio transformation) to account for their compositional nature [30].
- Global Association Testing: Use multivariate methods (e.g., Mantel test, Procrustes analysis) to determine if a significant overall association exists between the microbiome composition and the metabolomic profile in the cohort [30].
- Feature Selection: Apply integrative, multivariate feature selection methods like sparse Partial Least Squares (sPLS) to identify the specific microbial species whose abundances are most strongly associated with the levels of particular metabolites [30].
- Triangulation with Model Predictions: Compare the identified microbe-metabolite associations from the statistical analysis with the metabolic capabilities encoded in the AGORA2 models for the same microbes. This convergence strengthens the evidence for a mechanistic link.
- Hypothesis Generation: Formulate testable hypotheses about how drug administration might perturb these core microbe-metabolite relationships, potentially leading to efficacy or safety outcomes.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Resources for AGORA2-Based Research
| Research Reagent / Resource | Function / Application |
|---|---|
| AGORA2 Reconstruction Resource | Provides the foundational, curated genome-scale metabolic models for 7,302 microbial strains to build in silico models [1]. |
| Curated Strain Culture Collection | Enables empirical validation of in silico predictions through high-throughput culturing and drug incubation assays [28]. |
| Quantitative Proteomics (μLC-MS/MS) | Enables ultrasensitive quantification of drug-metabolizing enzymes and transporters (DMETs) in limited samples (e.g., biopsies) [31]. |
| Sparse PLS (sPLS) / MOFA2 | Statistical and multivariate analysis tools for integrating high-dimensional microbiome and metabolome datasets to identify key associations [30]. |
| Constraint-Based Modeling Software (COBRA) | The simulation environment used to run flux balance analysis and other constraint-based methods on AGORA2 models [1] [3]. |
The AGORA2 pipeline, when applied through the detailed protocols outlined in this document, provides a powerful and validated framework for predicting strain-resolved drug metabolism. By integrating high-throughput in vitro screens, personalized in silico modeling, and population-scale multi-omics data integration, researchers can systematically decipher the complex role of the gut microbiome in drug disposition. This mechanistic understanding is critical for de-risking drug development, explaining inter-individual variability in drug response, and ultimately designing personalized therapeutic strategies that account for the patient's unique gut microbiome.
Inflammatory Bowel Disease (IBD), encompassing Crohn's disease (CD) and ulcerative colitis (UC), is a chronic inflammatory condition of the gastrointestinal tract whose pathogenesis is intricately linked to gut microbiome dysbiosis [32] [33]. The gut microbiome in IBD patients demonstrates significant ecological disturbances, characterized by reduced biodiversity and shifts in microbial composition, including depletion of protective bacteria and enrichment of potentially pathogenic species [33]. While these associations are well-established, deciphering the specific mechanistic roles of microbial metabolism in IBD pathogenesis and progression remains a substantial research challenge. The AGORA2 pipeline (Assembly of Gut Organisms through Reconstruction and Analysis, version 2) provides a powerful computational framework to address this challenge through personalized, strain-resolved metabolic modeling of patient microbiomes [1].
AGORA2 represents a comprehensive knowledge base of genome-scale metabolic reconstructions for 7,302 human microbial strains, spanning 1,738 species and 25 phyla [1]. This resource encapsulates current knowledge of human microbial metabolism, including drug biotransformation capabilities for 98 pharmaceuticals, enabling systems-level interrogation of host-microbiome interactions in IBD [1] [34]. By integrating AGORA2 with patient-specific microbial abundance data, researchers can generate personalized microbiome models that predict metabolic fluxes, nutrient competition, cross-feeding relationships, and community-wide metabolic outputs relevant to IBD pathophysiology [35] [36]. This case study outlines application notes and protocols for employing the AGORA2 pipeline to analyze IBD microbiomes, with specific emphasis on identifying metabolic deficiencies and predicting individual variations in microbial community function.
IBD Microbiome Dysbiosis: Key Microbial Alterations
Systematic analyses of IBD microbiomes have consistently identified distinct patterns of dysbiosis that differ from healthy gut ecosystems. The table below summarizes the characteristic microbial alterations observed in IBD patients compared to healthy controls:
Table 1: Characteristic Microbial Alterations in Inflammatory Bowel Disease
| Taxonomic Level | Changes in IBD | Specific Examples | Functional Implications |
|---|---|---|---|
| Alpha Diversity | Significantly reduced | Lower Chao1 & Shannon indices [37] | Decreased functional redundancy & ecosystem stability |
| Phylum Level | Reduced Firmicutes, increased Proteobacteria | Depletion of Faecalibacterium [33] | Reduced SCFA production, increased inflammation |
| Genus Level | Depletion of butyrate producers | Faecalibacterium, Roseburia, Eubacteria [33] [38] | Impaired epithelial barrier function |
| Species Level | Specific pathogen enrichment | Increased Escherichia coli [38] | Potential pro-inflammatory effects |
| Community Structure | Altered co-occurrence networks | Fewer components, lower edge density [37] | Disrupted microbial interactions & ecosystem dynamics |
The functional consequences of these compositional changes are profound. Reductions in butyrate-producing bacteria like Faecalibacterium prausnitzii and Roseburia species diminish the production of short-chain fatty acids (SCFAs), which serve as crucial energy sources for colonic epithelial cells and play important anti-inflammatory roles in maintaining intestinal homeostasis [33]. Simultaneously, expansions of facultative anaerobes like Escherichia coli within the Proteobacteria phylum may introduce or exacerbate inflammatory processes [38]. Network-based analyses further reveal that the gut microbial ecosystems in IBD patients display less robust structures characterized by fewer network components and lower edge density, indicating a systemic disruption of microbial interactions beyond simple taxonomic shifts [37].
AGORA2 Pipeline: Workflow for IBD Microbiome Analysis
The AGORA2 pipeline, implemented through the Microbiome Modeling Toolbox 2.0, provides a standardized workflow for constructing and interrogating personalized microbiome models from microbial abundance data [35]. The following diagram illustrates the comprehensive workflow for analyzing IBD microbiomes:
The workflow begins with the acquisition of microbial abundance data derived from metagenomic sequencing or 16S rRNA profiling of patient stool samples or mucosal biopsies [35]. The Microbiome Modeling Toolbox's mgPipe module processes this data, mapping the observed microbial taxa onto the corresponding AGORA2 reconstructions and normalizing relative abundances [35]. When abundance data is available only at the species or genus level rather than the strain level, the pipeline automatically generates pan-models that represent the metabolic capabilities of all known strains within that taxonomic group [35]. The resulting community models incorporate dietary and environmental constraints to simulate the luminal metabolic environment, enabling flux variability analysis (FVA) to predict the maximal secretion potential for metabolites—a key metric termed net maximal production capacity (NMPC) [39].
Protocol: Constructing and Simulating Personalized IBD Microbiome Models
Materials and Software Requirements
Table 2: Essential Research Reagents and Computational Tools
| Category | Item | Specification | Purpose |
|---|---|---|---|
| Computational Environment | MATLAB | Version 2017b or higher | Primary computational platform |
| COBRA Toolbox | Version 3.0 or higher | Constraint-based modeling infrastructure | |
| Microbiome Modeling Toolbox | Version 2.0 | AGORA2 integration & community modeling | |
| IBM CPLEX | Version 12.8 or compatible solver | Linear programming optimization | |
| Data Resources | AGORA2 Reconstructions | 7,302 strain-resolved models | Genome-scale metabolic knowledge base |
| Patient Microbiome Data | 16S rRNA or metagenomic sequences | Input microbial abundance profiles | |
| Virtual Metabolic Human (VMH) Database | Diet compositions & metabolite information | Constraint definition for simulations | |
| Experimental Validation | Stool Samples | From IBD patients & controls | Model prediction validation |
| Metabolomics Platforms | LC-MS/MS or GC-MS | Measurement of fecal metabolites |
Step-by-Step Computational Protocol
Data Preprocessing and Quality Control
- Obtain microbial abundance data from 16S rRNA gene sequencing or metagenomic profiling of IBD patient cohorts and healthy controls.
- Perform standard bioinformatic processing including quality filtering, denoising, and taxonomic assignment using tools such as QIIME 2 or mothur.
- Export absolute or relative abundance tables with taxonomic identifiers mapped to standard nomenclature (e.g., SILVA, GTDB).
AGORA2 Resource Preparation
- Download the complete AGORA2 resource from the Virtual Metabolic Human database (VMH, https://www.vmh.life).
- Ensure all microbial reconstructions are available in the appropriate format (SBML or MAT) for COBRA Toolbox compatibility.
Personalized Model Construction using mgPipe
- Initialize the MATLAB environment with the COBRA Toolbox and Microbiome Modeling Toolbox 2.0 properly installed.
- Use the
mgPipefunction to map microbial abundance data onto AGORA2 reconstructions: - Specify parameter settings including diet composition ('rich' for unlimited nutrients or defined dietary constraints), solver parameters, and output directory.
- For abundance data at species or genus level, enable the pan-model generation option to create comprehensive metabolic representations.
Flux Variability Analysis and Metabolic Potential Calculation
- For each personalized microbiome model, perform flux variability analysis (FVA) to determine the range of possible metabolic fluxes:
- Compute the net maximal production capacity (NMPC) for metabolites of interest, particularly short-chain fatty acids (butyrate, propionate, acetate), vitamins, and immunomodulatory metabolites.
- Identify microbe-metabolite links by retrieving the microbes contributing most to the uptake and secretion of key metabolites.
Stratification Analysis and Statistical Comparison
- Compare metabolic flux distributions between IBD patients and healthy controls using statistical tests (e.g., Wilcoxon rank-sum test).
- Perform principal component analysis (PCA) on flux distributions to visualize stratification based on metabolic potential.
- Correlate metabolic fluxes with clinical parameters (disease activity, inflammation markers, treatment response) using Spearman or Pearson correlation.
Validation with Experimental Data
- Validate model predictions by comparing with experimentally measured fecal metabolomics data from the same patients.
- Assess accuracy of predicted microbial metabolic capabilities against culture-based assays or isotopically labeled tracer studies.
This protocol has been successfully applied to construct and analyze over 14,000 personalized microbiome models, including 644 models from colorectal cancer patients and controls, demonstrating its scalability to large cohort studies [35].
Application Notes: Insights from AGORA2 Analysis of IBD Microbiomes
Predictive Modeling of Metabolic Deficiencies
AGORA2-based modeling of IBD microbiomes has revealed specific functional deficiencies that extend beyond taxonomic observations. Models consistently predict impaired butyrogenesis in IBD patients, with reduced flux through butyrate production pathways correlated with depletion of key Firmicutes species [33]. This deficiency is particularly pronounced in patients with active disease compared to those in remission. Additionally, models predict altered bile acid metabolism and vitamin biosynthesis pathways, potentially contributing to the inflammatory milieu and nutritional deficiencies observed in IBD patients [38].
Interindividual Variability in Drug Metabolism Potential
A critical application of AGORA2 in IBD research involves predicting patient-specific variations in microbial drug metabolism [1] [34]. The resource includes manually curated drug transformation reactions for 98 commonly prescribed pharmaceuticals, enabling researchers to predict:
- Which patients may experience altered drug efficacy due to microbial biotransformation
- Potential microbial activation of prodrugs or inactivation of active compounds
- Correlations between drug metabolism potential and clinical parameters such as age, sex, BMI, and disease stage
For instance, AGORA2 can predict the variable conversion of the anti-inflammatory drug sulfasalazine into its active components across different patient microbiomes, potentially explaining differential treatment responses [1].
Identification of Therapeutic Targets
AGORA2-driven analysis facilitates the identification of potential microbiome-targeted therapeutic interventions for IBD. By modeling the metabolic dependencies of enriched taxa in IBD dysbiosis, researchers can identify nutritional interventions or probiotic strategies that might suppress pro-inflammatory species while promoting beneficial taxa. For example, models might predict that specific dietary fibers could selectively enhance the growth of depleted butyrate-producers, thereby restoring metabolic homeostasis [36].
Integration with Multi-Omics Data and Future Directions
The AGORA2 pipeline gains additional power when integrated with other omics technologies. Combining metabolic modeling with metatranscriptomic and metabolomic data from resources like the IBD Multi'omics Database (IBDMDB) enables more accurate constraint of models and validation of predictions [38]. Future developments should focus on:
- Incorporating host metabolic models to simulate host-microbe co-metabolism
- Expanding the drug metabolism repertoire to include newer biologic therapies
- Developing dynamic rather than steady-state models to capture temporal fluctuations in IBD activity
- Integrating microbial and host immune signaling pathways to bridge metabolic and inflammatory processes
The AGORA2 pipeline represents a transformative approach for moving beyond correlation to mechanistic understanding of microbiome contributions to IBD pathogenesis, ultimately advancing toward personalized microbiome-directed therapies.
Parkinson's disease (PD) is the second most common age-related neurodegenerative disease, with recent estimates suggesting a doubling of PD patients every ~30 years [40]. There is growing recognition of the gut-brain axis as an integral bi-directional communication system that may facilitate the spread of α-synuclein pathology [41]. While altered gut microbiome composition in PD is well-established, the underlying metabolic mechanisms linking microbial disruptions to disease-related metabolic changes remain incompletely characterized [42].
The AGORA2 pipeline (Assembly of Gut Organisms through Reconstruction and Analysis, version 2) provides a powerful computational framework for addressing this challenge. AGORA2 comprises 7,302 genome-scale metabolic reconstructions of human microorganisms, enabling strain- and molecule-resolved modeling of host-microbiome interactions [1]. This resource, combined with constraint-based reconstruction and analysis (COBRA) methods, allows for mechanistic investigation of metabolic interplay between the gut microbiome and host metabolism in PD [36] [43].
Methods
AGORA2-Based Metabolic Modeling Workflow for PD
The following protocol outlines the process for linking gut microbial compositional shifts to metabolic disruptions in Parkinson's disease using the AGORA2 pipeline.
Table 1: Key Input Data Requirements for PD Metabolic Modeling
| Data Type | Specifications | Example Sources | Purpose |
|---|---|---|---|
| Gut Metagenomics | 435 PD patients, 219 healthy controls [42] | Whole-genome shotgun sequencing of fecal samples | Personalize models with strain-resolution |
| Blood Metabolomics | 116 metabolites with replicated PD associations [42] | Untargeted LC-MS/MS on plasma/serum | Validate metabolic predictions |
| Clinical Phenotyping | PD subtypes (with/without RBD), disease duration, medication [44] [41] | MDS-UPDRS, RBD screening questionnaire | Stratify patients by clinical presentation |
| Dietary Information | Nutritional profiles, medication records [41] | Food frequency questionnaires, medication logs | Constrain model inputs |
Protocol Steps:
Reconstruction Personalization
- Obtain strain-level microbial abundance profiles from metagenomic data using tools such as HUMAnN2 or METAnnotatioN [43].
- Map identified strains to corresponding AGORA2 genome-scale metabolic reconstructions [1].
- Construct personalized microbiome models for each sample by combining individual microbial models based on abundance.
Metabolic Contextualization
- Apply diet-derived constraints to represent nutrient availability in the gut environment [43].
- Incorporate host-derived metabolic constraints relevant to PD, including dopamine precursors and medication compounds [41].
- Use the Virtual Metabolic Human (VMH) database to ensure consistent biochemical nomenclature across host and microbial metabolites [1].
Flux Simulation and Analysis
- Apply constraint-based modeling techniques, particularly flux balance analysis, to simulate metabolic network behavior under PD and healthy control conditions [42] [43].
- Identify differential production and consumption capabilities for metabolites with established PD associations.
- Trace metabolic disruptions to specific microbial species and pathways through gene knockout simulations and pathway enrichment analysis.
Visualizing the AGORA2 Workflow for PD Investigation
The following diagram illustrates the logical workflow and data integration process for applying the AGORA2 pipeline to Parkinson's disease research:
Results & Key Findings
PD-Associated Metabolic Disruptions Identified Through AGORA2 Modeling
Application of the AGORA2 pipeline to personalized modeling of PD gut microbiomes revealed specific metabolic disruptions in host-microbiome co-metabolism.
Table 2: Key Metabolic Disruptions in PD Predicted by AGORA2 Modeling
| Metabolite | Predicted Change in PD | Associated Microbial Species | Potential Pathophysiological Relevance |
|---|---|---|---|
| L-leucine & Leucylleucine | Reduced production capacity | Roseburia intestinalis (reduced production)Methanobrevibacter smithii (increased consumption) | Branched-chain amino acid metabolism; muscle function |
| Butyrate | Reduced production capacity | Faecalibacterium prausnitzii (reduced abundance) | Gut barrier integrity; anti-inflammatory effects |
| Myristic Acid | Reduced production capacity | Faecalibacterium prausnitzii (reduced abundance) | Saturated fatty acid metabolism; membrane integrity |
| Pantothenate | Reduced production capacity | Faecalibosterium prausnitzii (reduced abundance) | Vitamin B5 synthesis; coenzyme A precursor |
| Nicotinic Acid | Reduced production capacity | Ruthenibacterium lactatiformans (increased consumption) | NAD+ precursor; energy metabolism |
PD Subtype-Specific Metabolic Patterns
Metabolomic profiling of PD patients stratified by REM sleep behavior disorder (RBD) status reveals distinct metabolic signatures that align with proposed body-first (with RBD) and brain-first (without RBD) subtypes [44].
Table 3: Metabolic Differences Between PD Subtypes
| PD Subtype | Enriched Metabolites | Potential Microbial Origins | Proposed Pathway |
|---|---|---|---|
| PD with RBD ("Body-First") | Secondary bile acids (lithocholate sulfate, glycolithocholate), p-cresol sulfate, phenylacetylglutamine | Clostridioides difficile (p-cresol producer) | Gut-derived metabolites entering circulation |
| PD without RBD ("Brain-First") | Glucose, cortisol; decreased caffeine | Host-derived metabolic changes | Neuroendocrine and energy metabolism disruption |
Visualizing Metabolic Pathways in PD Subtypes
The diagram below illustrates the distinct metabolic pathways associated with different Parkinson's disease subtypes, highlighting the gut-brain axis involvement in body-first PD:
The Scientist's Toolkit
Table 4: Essential Research Reagent Solutions for PD Microbiome Metabolism Studies
| Research Tool | Function/Application | Specifications |
|---|---|---|
| AGORA2 Resource | Genome-scale metabolic reconstructions of human gut microbes | 7,302 strain-resolved models; includes 98 drug biotransformation pathways [1] |
| COBRA Toolbox | MATLAB suite for constraint-based modeling and simulation | Includes methods for pairwise and community modeling of microbiota [43] |
| Virtual Metabolic Human (VMH) | Database for human and gut microbial metabolism | Standardized biochemical nomenclature; links microbial and host metabolites [1] |
| HUMAnN2 | Metagenomic functional profiling pipeline | Quantifies microbial pathway abundances from shotgun sequencing data [43] |
| Personalized Community Models | Condition-specific modeling of individual microbiomes | Integrates metagenomic data with AGORA2 reconstructions; predicts metabolic output [42] |
Discussion
This case study demonstrates how the AGORA2 pipeline enables mechanistic interpretation of microbial metabolic disruptions in Parkinson's disease. The predictions generated through constraint-based modeling provide testable hypotheses about how specific microbial species contribute to PD pathophysiology through their metabolic activities [42].
The identification of reduced production capacities for key metabolites including L-leucine, butyrate, and pantothenate in PD patients suggests multiple potential intervention points. These predictions align with independent observations of persistent underrepresentation of short-chain fatty acid-producing bacteria in longitudinal PD studies [41]. Furthermore, the distinct metabolic signatures observed in PD patients with and without RBD support the existence of different etiological subtypes that may require personalized therapeutic approaches [44].
Future applications of the AGORA2 pipeline in PD research could include predicting individual responses to dietary interventions, identifying microbial targets for probiotic therapies, and understanding drug-microbiome interactions that may affect medication efficacy [3] [41]. The integration of multi-omics datasets with mechanistic modeling approaches represents a promising path toward personalized medicine approaches for Parkinson's disease targeting the gut-brain axis.
The infant gut microbiome is a dynamic ecosystem whose initial colonization and development are critical for long-term health, influencing nutrient absorption, immune education, and neurodevelopment [45] [46]. Its establishment is shaped by a complex interplay of early-life factors, with delivery mode being one of the most significant determinants [45] [3]. Disruptions in this developmental sequence, such as those caused by Cesarean section (C-section) delivery, have been associated with altered metabolic capabilities and an increased risk of non-communicable diseases later in life [3]. The AGORA2 pipeline, a curated resource of genome-scale metabolic reconstructions for over 7,300 human microorganisms, provides a powerful framework for moving beyond taxonomic composition to achieve strain- and molecule-resolved modeling of microbiome metabolic functions [1] [3]. These Application Notes detail protocols for analyzing the infant gut microbiome, with a specific focus on the impacts of delivery mode, and guide the integration of resulting data into the AGORA2 pipeline for personalized, mechanistic modeling of metabolic potential.
Key Impacts of Delivery Mode on the Infant Gut Microbiome
Delivery mode is a major factor shaping the initial neonatal gut microbiome. Vaginally delivered infants acquire microbes from maternal vaginal and intestinal sources, whereas C-section infants are initially colonized by bacteria more commonly associated with the hospital environment and maternal skin [3]. These initial differences in microbial seeding have cascading effects on the microbiome's composition and function.
Table 1: Comparative Analysis of Infant Gut Microbiome by Delivery Mode
| Aspect | Vaginally Delivered (VD) Infants | Cesarean-Delivered (CSD) Infants | Citation |
|---|---|---|---|
| Initial Microbial Source | Maternal vaginal and intestinal microbes | Maternal skin and hospital environment | [3] |
| Detectable Microbes in Meconium | ~69% of samples (Spontaneous Vaginal) | 17% of samples (Elective C-section) | [45] |
| Early Colonizing Genera | Escherichia, Bifidobacterium, Bacteroides | Staphylococcus epidermis, Bacteroides fragilis | [3] [47] |
| Metabolic Potential (Early Life) | Higher potential for HMO degradation, bile acid transformation | Depleted metabolic capabilities | [3] |
| Vitamin Synthesis | Comparable B-vitamin synthesis potential to VD infants later in life | [3] | |
| Long-term Diversity | Effect detectable up to at least 2 years of age | Reduced diversity in short-term; differences diminish over time | [45] [3] [47] |
Beyond delivery mode, other factors significantly influence the trajectory of the infant gut microbiome. Infant sex has been associated with differences in community composition, with male neonates exhibiting lower alpha diversity and elevated levels of Enterobacteriales compared to females [45]. Birth order also plays a role; neonates born to primiparous mothers have a lower abundance of the keystone genus Bifidobacterium [45]. The subsequent introduction of solid foods during weaning represents another major developmental milestone, driving a shift from a microbiome dominated by Bifidobacterium to one enriched with Bacteroides, Clostridium, Roseburia, and Faecalibacterium prausnitzii, marking the transition toward an adult-like microbiome [48] [49].
Experimental Protocols for Infant Gut Microbiome Characterization
A robust, multi-omics approach is essential for comprehensively characterizing the infant gut microbiome and generating high-quality data for AGORA2 modeling.
Protocol: Fecal Sample Collection and DNA Isolation from Infants
This protocol is optimized for the unique challenges of infant fecal samples, which are often low in biomass [50].
Research Reagent Solutions:
- Stool DNA Stabilizer: Preserves microbial DNA at point of collection to prevent shifts in community structure.
- Lysis Buffer for Complex Gram-Positive Bacteria: Contains enzymes (e.g., lysozyme) to break down tough cell walls of bacteria like Bifidobacterium and Clostridium.
- Inhibitor Removal Technology Columns: Specifically designed to remove PCR inhibitors common in stool, such as humic acids and bile salts, which is critical for downstream sequencing success.
Procedure:
- Collection: Collect first-pass meconium or infant stool using a sterile spatula. For extremely preterm infants, swabs may be used. Immediately place the sample in a tube containing a commercial stool DNA stabilizer and store at -80°C [50].
- Homogenization: Thaw samples on ice and homogenize 180-220 mg of stool in the provided stabilizer solution using a vortex mixer with adapter.
- Enzymatic Lysis: Add 20 µL of proteinase K and 200 µL of a specialized lysis buffer to the homogenate. Incubate at 70°C for 10-30 minutes to fully lyse cells, including tough Gram-positive bacteria.
- Inhibitor Removal: Bind DNA to a silica-based membrane in the presence of a high-concentration salt solution. Wash with ethanol-based buffers.
- Elution: Elute pure, inhibitor-free DNA in a low-salt elution buffer or nuclease-free water. Quantify DNA using a fluorometric method and store at -20°C.
Protocol: 16S rRNA Gene Sequencing and Metagenomic Profiling
This two-tiered sequencing approach provides both cost-effective community profiling and deep functional insights.
Research Reagent Solutions:
- 16S rRNA Gene Primers (V3-V4 region): For amplifying the hypervariable region to determine taxonomic composition.
- Shotgun Metagenomic Library Prep Kit: For fragmenting and preparing total DNA for untargeted sequencing.
- High-Fidelity DNA Polymerase: Essential for accurate amplification with low error rates in 16S sequencing.
- Dual-IndeX Barcodes and Adapters: Enable multiplexing of hundreds of samples in a single sequencing run.
Procedure for 16S rRNA Gene Sequencing [50] [51]:
- Amplification: Amplify the hypervariable V3-V4 region of the 16S rRNA gene using region-specific primers (e.g., 341F/806R) and a high-fidelity polymerase.
- Library Preparation: Clean amplified products and attach dual-index barcodes via a limited-cycle PCR to create sequencing libraries.
- Sequencing: Pool equimolar amounts of all libraries and sequence on an Illumina MiSeq or similar platform using 2x250 bp paired-end chemistry.
Procedure for Shotgun Metagenomic Sequencing [51]:
- Library Preparation: Fragment total DNA (including from the DNA isolation protocol above) to an average size of 400-500 bp using a commercial library prep kit.
- Size Selection and Adapter Ligation: Select the appropriate fragment size, and ligate sequencing adapters containing unique dual indexes.
- Sequencing: Pool libraries and sequence on an Illumina NovaSeq or similar platform to a minimum depth of 10 million paired-end reads per sample to ensure adequate coverage for strain-level resolution.
Protocol: Personalized Metabolic Modeling with AGORA2
This protocol translates raw sequencing data into predictive, personalized metabolic models.
Procedure:
- Data Preprocessing and Metagenomic Assembly: Process raw shotgun metagenomic reads by removing adapters and low-quality bases. Assemble the cleaned reads into contigs using a dedicated metagenomic assembler (e.g., MEGAHIT, metaSPAdes).
- Gene Prediction and Abundance Profiling: Predict open reading frames (ORFs) on the assembled contigs. Map high-quality reads back to the gene catalog to calculate the relative abundance of each microbial strain in the community.
- AGORA2 Model Integration: Map the identified and quantified microbial strains to the corresponding genome-scale metabolic reconstructions in the AGORA2 resource [1] [3].
- Personalized Community Modeling: Create a personalized microbiome model for each infant sample by combining the AGORA2 reconstructions of detected microbes, constrained by their relative abundances. Use constraint-based reconstruction and analysis (COBRA) methods to simulate metabolic outputs such as SCFA production, HMO degradation potential, and vitamin synthesis [3].
The following workflow diagram illustrates the complete pipeline from sample to model simulation:
Data Analysis and Integration with AGORA2
The power of AGORA2 lies in its ability to contextualize multi-omics data within a mechanistic, metabolic framework. After generating microbiome profiles, the following steps are critical:
- Stratification by Variables: Group infant microbiome data by key variables such as delivery mode (VD vs. CSD), infant sex, birth order, and feeding status. This allows for comparative analysis of metabolic potentials between cohorts [45] [3].
- Metabolic Gap-Filling: For CSD infants, modeling may reveal gaps in metabolic pathways, such as those for processing human milk oligosaccharides (HMOs). AGORA2 can identify which specific bacterial strains are missing and could potentially be supplemented [3].
- Cross-Feeding Analysis: Use the community modeling capabilities of AGORA2 to simulate metabolic interactions between microbes. For example, model how the byproducts of Bifidobacterium's HMO metabolism can support the growth of butyrate-producing bacteria like Faecalibacterium [3].
Table 2: AGORA2-Predicted Metabolic Differences in Early Infant Gut Microbiomes
| Metabolic Feature | Vaginally Delivered Infants | Cesarean-Delivered Infants | Biological Significance |
|---|---|---|---|
| HMO Degradation | Higher potential | Depleted potential | Affects energy harvest and gut barrier integrity |
| B-Vitamin Synthesis | Enriched | Becomes comparable later | Crucial for host metabolism and epigenetics |
| Short-Chain Fatty Acids | Higher butyrate potential | Lower butyrate, higher L-lactate | Butyrate is key for immune regulation and colon health |
| Bile Acid Transformation | Higher potential | Depleted potential | Influences host lipid metabolism and signaling |
The protocols outlined herein provide a comprehensive roadmap for researchers to characterize the infant gut microbiome and quantify the functional impact of delivery mode and other early-life factors. By integrating high-quality multi-omics data with the AGORA2 pipeline, scientists can transition from observing taxonomic correlations to generating testable, mechanistic hypotheses about microbiome metabolism. This personalized modeling approach holds significant promise for advancing our understanding of how early microbial colonization influences long-term health and for developing targeted nutritional or therapeutic interventions to correct microbiome-related deficits in at-risk infant populations.
Optimizing AGORA2 Performance: Addressing Computational and Integration Challenges
The advancement of personalized microbiome metabolic modeling, particularly through pipelines like AGORA2, is critically dependent on the seamless integration of high-quality, standardized metabolic models. Genome-scale metabolic models (GEMs) are mathematically-structured knowledge bases that encapsulate the biochemical, genetic, and genomic information of an organism's metabolism [52]. The AGORA2 (Assembly of Gut Organisms through Reconstruction and Analysis, version 2) resource exemplifies this approach, comprising 7,302 strain-resolved reconstructions of human gut microorganisms to enable personalized, predictive analysis of host-microbiome interactions [1]. However, the interoperability of such resources is severely hampered by a fundamental challenge: the inconsistent use of metabolite nomenclature across different biochemical databases.
The BiGG Models database and the Virtual Metabolic Human (VMH) knowledge base represent two cornerstone resources for metabolic modeling research. BiGG Models serves as a centralized repository for over 75 high-quality, manually-curated genome-scale metabolic models, employing standardized identifiers for metabolites, reactions, and genes to facilitate consistent modeling efforts [52]. Conversely, the VMH database provides an interdisciplinary platform connecting human metabolism with gut microbiome, disease, nutrition, and metabolic maps, capturing 5,180 unique metabolites and 17,730 unique reactions [53]. While VMH extensively cross-references BiGG identifiers (covering 90.2% of its metabolites) [53], the mapping between these namespaces is not bijective or without complications, creating significant hurdles for researchers integrating models from both sources into unified modeling frameworks such as AGORA2.
The problem of namespace inconsistency is pervasive in metabolic modeling. Studies have revealed that identifier inconsistency between biochemical databases can be as high as 83.1% [54]. This inconsistency manifests as both identifier multiplicity (where a single identifier links to multiple names) and name ambiguity (where the same name refers to multiple distinct chemical entities) [54]. For researchers working with the AGORA2 pipeline, which uses the VMH namespace [1], integrating models from BiGG requires careful navigation of these nomenclature differences to avoid fundamental errors in model construction and simulation.
Comparative Analysis of BiGG and VMH Namespaces
Database Scope and Primary Focus
Table 1: Fundamental Characteristics of BiGG and VMH Databases
| Characteristic | BiGG Models | Virtual Metabolic Human (VMH) |
|---|---|---|
| Primary Scope | Genome-scale metabolic models (GEMs) | Human and gut microbial metabolism in disease context |
| Core Content | 75+ manually curated GEMs; standardized reactions, metabolites, genes | 5,180 metabolites; 17,730 reactions; 3,695 human genes; 255 diseases |
| Key Feature | Standardized identifiers for modeling; SBML export with FBC | Integration of nutrition, disease, microbial metabolism; therapeutic diet design |
| Visualization | Escher-based pathway maps | Seven comprehensive human metabolic maps |
| API Access | Comprehensive REST API | Well-documented REST API |
| Model Applications | Constraint-based reconstruction and analysis (COBRA) | Personalized host-microbiome modeling; systems medicine |
The BiGG Models knowledgebase was specifically designed to address the challenge of standardization in metabolic modeling by providing "reaction and metabolite identifiers have been standardized across models to conform to community standards and enable rapid comparison across models" [52]. This focus on computational modeling is reflected in its export formats (SBML Level 3 with FBC) and integration with modeling tools like the COBRA Toolbox.
In contrast, the VMH database takes a more holistic, interdisciplinary approach, "connecting human metabolism with genetics, human-associated microbial metabolism, nutrition, and diseases" [53]. This broader scope makes it particularly valuable for personalized medicine applications, as it incorporates elements beyond core metabolism, including "8790 food items" and "255 Mendelian diseases" that can be integrated into condition-specific models [53].
Identifier Characteristics and Mapping Challenges
Table 2: Identifier Properties and Cross-Reference Challenges
| Property | BiGG Models | VMH | Mapping Implications |
|---|---|---|---|
| Identifier Multiplicity | 1.0141 ± 0.126 average IDs per name [54] | Not explicitly quantified | Low multiplicity in BiGG reduces ambiguity |
| Name Ambiguity | 1.31% of names are ambiguous [54] | Not explicitly quantified | Minimal ambiguous names in BiGG |
| Cross-Reference Coverage | Reference standard for models | 90.2% of VMH metabolites linked to BiGG [53] | High coverage facilitates mapping |
| External Database Links | KEGG, MetaCyc, Reactome, HMDB, Model SEED [52] | 57 external resources including BiGG, ChEBI, HMDB, PubChem [53] | Multiple connection points enable validation |
| Primary Namespace Use in AGORA2 | Not the primary namespace | Used as primary namespace [1] | AGORA2 models require mapping for BiGG content integration |
The fundamental challenge in mapping between BiGG and VMH arises from what the community recognizes as "identifier multiplicity" and "name ambiguity." Identifier multiplicity occurs when a single identifier is linked to multiple names, while name ambiguity describes the situation where the same name refers to different chemical entities [54]. Research has shown that these inconsistencies can be particularly problematic when combining models from different sources, as "the same metabolite can be added many times with different names and, consequently, considered as different chemical entities which can, in the worst case, invalidate the model" [54].
While BiGG maintains relatively strict control over its namespace (with only 1.31% of names being ambiguous) [54], the process of mapping to VMH introduces complexities due to differences in biochemical representation and compartmentalization. Furthermore, the AGORA2 pipeline, which builds upon the VMH namespace, demonstrates the scalability of this resource for large-scale modeling efforts, encompassing 7,302 microbial strains [1]. This creates a practical imperative for researchers to develop robust mapping methodologies between these namespaces.
Protocols for Metabolite Identifier Mapping and Validation
Experimental Protocol: Cross-Database Metabolite Mapping
Purpose: To establish accurate mapping between BiGG and VMH metabolite identifiers to enable integration of metabolic models from both databases into the AGORA2 pipeline for personalized microbiome modeling.
Materials and Reagents:
- BiGG Models database (accessible via http://bigg.ucsd.edu) [52]
- Virtual Metabolic Human database (accessible via https://vmh.life) [53]
- AGORA2 genome-scale reconstructions (available through VMH) [1]
- MetaNetX database (https://www.metanetx.org/) for cross-reference validation [53] [54]
- Python environment with required packages (cobrapy, requests, pandas)
Procedure:
Identifier Extraction:
- Download target metabolic models from BiGG Models in SBML format using the BiGG API [52].
- Extract all metabolite identifiers from the BiGG model using a parser such as cobrapy.
- For each metabolite identifier, retrieve the corresponding chemical name, formula, and charge from the BiGG database.
Initial Mapping via VMH Cross-References:
- Access the VMH database programmatically via its REST API [53].
- Query the VMH for each BiGG metabolite identifier using the cross-reference table.
- Record all VMH metabolite identifiers that reference the target BiGG identifier.
- For metabolites without direct matches, proceed to secondary mapping.
Secondary Mapping via InChI Keys and CheBI Identifiers:
- For metabolites without direct BiGG-VMH mapping, extract InChI keys or CheBI identifiers from BiGG when available.
- Query VMH using these standardized chemical identifiers to find corresponding metabolites.
- Validate chemical structure consistency using structural data from PubChem or ChEBI.
Manual Verification and Conflict Resolution:
- For each mapped metabolite pair, verify consistency in:
- Molecular formula and charge
- Compartmentalization (if specified)
- Biochemical role in metabolic pathways
- Resolve conflicts by consulting original literature or biochemical databases.
- Document all ambiguous mappings for further refinement.
- For each mapped metabolite pair, verify consistency in:
Model Integration and Functional Testing:
- Incorporate successfully mapped metabolites into the target AGORA2-based model.
- Test model functionality by comparing growth predictions or metabolic capabilities before and after integration.
- Validate integrated model against experimental data when available.
Troubleshooting:
- For metabolites with multiple potential mappings, prioritize based on compartment specificity and pathway context.
- If mapping performance is poor for specific metabolite classes (e.g., lipids), consider class-specific mapping approaches.
- When name ambiguity is detected, utilize structural identifiers (InChI) for definitive resolution.
Workflow Visualization
BiGG to VMH Metabolite Mapping Workflow
Integration with AGORA2 Pipeline
Protocol: AGORA2 Model Enhancement with BiGG Content
Purpose: To enhance AGORA2-based personalized microbiome models with specialized metabolic pathways from BiGG Models while maintaining namespace consistency and model functionality.
Materials and Reagents:
- AGORA2 microbial reconstructions [1]
- Curated BiGG-VMH metabolite mapping table (from Protocol 3.1)
- COBRA Toolbox or equivalent modeling environment [52] [53]
- Personalized metagenomic data from patient samples
- Computing infrastructure for constraint-based modeling
Procedure:
Model Preprocessing:
- Select target AGORA2 models corresponding to microbial taxa present in the personalized microbiome profile.
- Identify metabolic gaps or pathways of interest that could be supplemented from BiGG Models.
- Select appropriate BiGG models containing the target pathways or reactions.
Namespace Harmonization:
- Apply the precomputed BiGG-to-VMH metabolite mapping table to the selected BiGG model.
- Convert all metabolite identifiers in the BiGG model to their VMH equivalents.
- Verify reaction stoichiometry consistency after identifier conversion.
- Check for and resolve any duplicated metabolites or reactions.
Pathway Integration:
- Extract target reactions and associated genes from the namespace-harmonized BiGG model.
- Add these components to the target AGORA2 model while maintaining GPR associations.
- Verify mass and charge balance for all added reactions.
Contextualization for Personalized Modeling:
- Constrain the integrated model using patient-specific metagenomic abundance data.
- Apply nutritional constraints based on patient diet or intervention.
- Validate model functionality by testing known metabolic capabilities.
Simulation and Analysis:
- Perform flux balance analysis to predict metabolic phenotypes.
- Simulate drug metabolism or intervention responses using the enhanced model.
- Compare predictions with experimental data or clinical observations when available.
Validation Metrics:
- Percentage of successfully integrated reactions without stoichiometric inconsistencies
- Maintenance of model growth capabilities on standard media
- Improved prediction accuracy for metabolic phenotypes
- Enhanced drug metabolism prediction concordance with experimental data
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for Metabolic Namespace Mapping
| Resource | Type | Function in Namespace Mapping | Access |
|---|---|---|---|
| BiGG Models API [52] | Web API | Programmatic access to BiGG metabolites, reactions, and models | http://bigg.ucsd.edu/api/v2 |
| VMH REST API [53] | Web API | Access to VMH database content and cross-references | https://vmh.life/api/docs |
| MetaNetX [54] | Database Platform | Cross-reference mapping between biochemical namespaces | https://www.metanetx.org |
| COBRA Toolbox [52] [53] | Software Package | Metabolic model simulation and manipulation | https://opencobra.github.io |
| GEMsembler [55] | Python Package | Consensus model assembly and structural comparison | https://github.com/SystemsBiologyInst/GEMsembler |
| Escher [52] | Visualization Tool | Pathway visualization for mapped metabolic networks | https://escher.github.io |
The integration of BiGG and VMH metabolite nomenclature represents a critical challenge in advancing personalized microbiome research using the AGORA2 pipeline. While significant hurdles exist due to namespace inconsistencies, systematic mapping approaches and validation protocols can enable researchers to leverage the unique strengths of both databases. The continued development of standardized mapping resources and computational tools will be essential for achieving seamless interoperability between these foundational resources, ultimately accelerating progress in personalized metabolic modeling and therapeutic development.
The advent of high-throughput meta-omics technologies has generated unprecedented volumes of data on microbial composition and function, creating a critical need for computational frameworks capable of integrating this information into mechanistic models. Constraint-Based Reconstruction and Analysis (COBRA) has emerged as a powerful computational approach for modeling metabolic networks in microbial communities [56]. This methodology enables the contextualization of meta-omics data and allows for mechanistic prediction of metabolic fluxes, making it particularly valuable for studying microbiomes [56]. The COBRA approach relies on genome-scale metabolic models (GEMs) that are built in a bottom-up manner and curated through manual efforts based on genomic, biochemical, and physiological knowledge [3]. These reconstructions represent comprehensive knowledge bases of an organism's metabolic capabilities.
For human microbiome research, the AGORA2 resource represents a significant advancement, containing 7,302 curated genome-scale reconstructions of human microorganisms [1]. This resource enables strain- and molecule-resolved prediction of metabolic differences between individuals through dedicated modeling pipelines [3]. AGORA2 accounts for 1,738 species and 25 phyla, substantially expanding the coverage of previous resources and incorporating manually formulated drug biotransformation and degradation reactions covering over 5,000 strains and 98 drugs [1]. The DEMETER (Data-drivEn METabolic nEtwork Refinement) pipeline used to develop AGORA2 employs a rigorous workflow of data collection, integration, draft reconstruction generation, and simultaneous iterative refinement, gap-filling, and debugging [1]. This extensive curation process has resulted in models that demonstrate high predictive accuracy against independently collected experimental datasets, with accuracy scores ranging from 0.72 to 0.84 [1].
Key Computational Challenges in Large-Scale Modeling
Technical and Conceptual Limitations
Large-scale community modeling faces several significant computational constraints that impact both model construction and simulation. The sheer complexity of microbial communities, which can contain hundreds to thousands of species spanning all domains of life, presents a fundamental challenge [57]. Each species engages in diverse ecological interactions including cross-feeding, competition, signaling, and predation, creating dynamic, non-linear relationships that are difficult to capture computationally [57]. This complexity is compounded by the multi-scale nature of microbiome systems, where molecular-level interactions between genes, proteins, and metabolites give rise to emergent community-level behaviors [57].
The quality and completeness of genome annotations represents another critical constraint. Inaccurate or incomplete annotations introduce substantial uncertainty in the predictive potential of genome-scale reconstructions [1]. This challenge is particularly acute for novel microbial species, with recent research identifying approximately 77% (3,796) of species in the human microbiome as previously unknown [58]. Furthermore, an estimated 75% of genes associated with the human microbiome lack functional annotation, creating significant "functional dark matter" that limits model completeness [58]. The DEMETER pipeline addresses these limitations through manual validation and improvement of 446 gene functions across 35 metabolic subsystems for 74% of genomes and extensive literature searches spanning 732 peer-reviewed papers for 95% of strains [1].
Data Integration and Computational Demand
Integrating diverse data types across multiple scales presents additional computational challenges. Multi-omics data integration requires sophisticated computational methods to reconcile metagenomic, metatranscriptomic, metaproteomic, and metabolomic data into coherent constraint-based models [58]. Each data type possesses different characteristics, resolutions, and noise profiles that must be accounted for during integration. The computational intensity of simulating large communities is another significant constraint. As model size increases, the computational resources required for simulation grow exponentially, creating practical limitations on community complexity that can be feasibly modeled [56] [57].
Table 1: Key Computational Constraints in Large-Scale Community Modeling
| Constraint Category | Specific Challenges | Potential Mitigation Strategies |
|---|---|---|
| Biological Complexity | High species diversity (100s-1000s of taxa) Non-linear ecological interactions Multi-scale system behaviors | Model reduction techniques Modular community modeling Hierarchical approaches |
| Data Limitations | Incomplete genome annotations Functional dark matter (75% genes unannotated) Limited experimental validation data | Manual curation pipelines Comparative genomics Literature mining (732+ papers for AGORA2) |
| Computational Demands | Exponential growth in resource requirements Multi-omics data integration Scalability of simulation algorithms | High-performance computing Efficient optimization algorithms Model decomposition techniques |
Protocol for Building Personalized Community Models
Data Acquisition and Preprocessing
The construction of personalized community models begins with comprehensive data collection from multiple sources. For microbiome studies, this typically includes metagenomic sequencing data to determine taxonomic composition, metabolomic data to identify available nutrients and metabolic end products, and potentially transcriptomic or proteomic data to constrain model activity [58] [3]. The quality control of input data is critical, requiring careful processing to remove contaminants, correct for batch effects, and normalize across samples. For metagenomic data, this involves preprocessing steps such as quality filtering, adapter removal, and host DNA depletion [3].
Taxonomic profiling transforms raw sequencing data into abundance estimates for microbial taxa. For the AGORA2 framework, these taxonomic profiles are mapped to the 7,302 reference strains in the resource [1]. The model initialization process involves creating a personalized community model for each sample by combining the metabolic reconstructions of detected microorganisms weighted by their relative abundance [3]. This results in a sample-specific metabolic network that can be simulated using constraint-based approaches.
Model Simulation and Validation
Once personalized community models are constructed, constraint-based simulation techniques are applied to predict metabolic fluxes. Flux Balance Analysis (FBA) is commonly used to optimize an objective function, typically community biomass production or production of specific metabolites [3]. The simulations require appropriate environmental constraints that define available nutrients and other growth conditions. For gut microbiome models, these constraints often include dietary components, host-derived metabolites, and physical parameters such as pH and oxygen availability [3].
Model validation is essential to ensure predictive accuracy. AGORA2 has been validated against three independently collected experimental datasets, demonstrating accuracy between 0.72-0.84 [1]. Validation can include comparison of predicted growth rates with experimental measurements, assessment of metabolite consumption and secretion patterns, and evaluation of known metabolic capabilities [1]. For personalized models, validation may involve comparing predicted metabolite levels with experimentally measured metabolomic data or assessing the model's ability to recapitulate known host phenotypes [3].
Table 2: Essential Research Reagents and Computational Resources
| Resource Category | Specific Tools/Databases | Primary Function |
|---|---|---|
| Genome-Scale Reconstruction Resources | AGORA2 (7,302 strains) [1] Virtual Metabolic Human (VMH) database [58] BiGG Models [1] | Reference metabolic reconstructions Metabolic knowledge base Standardized reaction database |
| Modeling Software & Platforms | COBRA Toolbox [57] CarveMe [1] gapseq [1] KBase [1] | Constraint-based modeling Automated reconstruction Metabolic pathway prediction Draft reconstruction generation |
| Data Analysis Tools | HUMAnN [58] MAMBO [58] DEMETER pipeline [1] | Metabolic pathway profiling Metabolomic analysis of metagenomes Data-driven reconstruction refinement |
Application Example: Modeling Infant Gut Microbiome Development
Experimental Design and Model Setup
A recent study demonstrated the application of large-scale community modeling to investigate the metabolic capabilities of the infant gut microbiome during the first year of life [3]. The researchers analyzed metagenomic sequencing data from 20 infants at four time points (5 days, 1 month, 6 months, and 1 year) as well as maternal gut microbiome samples [3]. The personalized modeling workflow began with extending the AGORA2 resource with a human milk oligosaccharide (HMO) degradation module to better represent infant gut microbial metabolism [3].
For each sample, a personalized metabolic model was constructed by mapping the metagenomic abundance data to the appropriate AGORA2 reconstructions [3]. The models were constrained based on available nutrients, with particular attention to breast milk components for infant samples. The simulation objectives included predicting production of key metabolites such as short-chain fatty acids, amino acids, and vitamins, as well as assessing overall metabolic potential differences between delivery modes [3].
Key Findings and Technical Insights
The modeling approach revealed that gut microbiomes of infants delivered by Cesarian section were depleted in metabolic capabilities at early time points compared with vaginally delivered infants [3]. Specifically, CSD microbiomes showed reduced potential for HMO degradation and bile acid transformation [3]. The models also predicted that infant gut microbiomes produce less butyrate but more L-lactate than maternal gut microbiomes and are enriched in B-vitamin biosynthesis potential [3].
From a technical perspective, this study demonstrated the value of temporal modeling for capturing microbiome developmental trajectories [3]. The workflow successfully integrated longitudinal metagenomic data with mechanistic metabolic models, revealing how metabolic capabilities evolve during the first year of life. The models also provided insights into community-level metabolic interactions, including cross-feeding relationships and collective metabolic potential that emerges from the combination of microbial taxa present in each individual [3].
Workflow Diagram for Community Metabolic Modeling
Community Metabolic Modeling Workflow
Advanced Methodologies and Optimization Strategies
Computational Efficiency Techniques
Addressing the computational constraints of large-scale community modeling requires sophisticated optimization strategies. Model reduction approaches can decrease complexity while preserving predictive accuracy by removing redundant reactions, combining similar metabolic pathways, or focusing on core metabolic processes [57]. Parallel computing techniques enable distribution of simulations across multiple computing nodes, significantly reducing computation time for large-scale analyses [57]. This is particularly valuable when simulating hundreds of personalized community models or performing extensive parameter sensitivity analyses.
Approximation algorithms provide another strategy for managing computational complexity. Rather than seeking exact solutions to optimization problems, these algorithms find near-optimal solutions with substantially reduced computational requirements [57]. For dynamic simulations, adaptive time-stepping methods can improve efficiency by adjusting temporal resolution based on system dynamics, using finer resolution during periods of rapid change and coarser resolution during relatively stable periods [57].
Data Integration and Multi-Scale Modeling
Effective multi-omics data integration remains a challenge in large-scale community modeling. Bayesian approaches can probabilistically combine data from different sources while accounting for measurement uncertainty and technical variability [58]. Multi-scale modeling frameworks that link metabolic models with higher-level community dynamics or host interactions provide a more comprehensive representation of microbiome systems but introduce additional computational complexity [36] [57].
The DEMETER pipeline developed for AGORA2 exemplifies a sophisticated approach to data integration, incorporating manual curation based on comparative genomics, extensive literature review, and experimental validation [1]. This pipeline employs a test suite for continuous verification during reconstruction refinement and generates quality control reports for all reconstructions, achieving an average quality score of 73% for AGORA2 models [1]. Such rigorous quality control processes are essential for ensuring model reliability despite the computational constraints inherent in large-scale modeling.
Future Directions and Concluding Remarks
The field of large-scale community metabolic modeling continues to evolve rapidly, with several promising directions for addressing current computational constraints. Machine learning integration offers potential for developing surrogate models that can approximate COBRA simulations with substantially reduced computational requirements [57]. Improved reconstruction algorithms that better leverage comparative genomics and automated literature mining could accelerate model building while maintaining quality [1]. The ongoing expansion of curated metabolic resources like AGORA2 will further enhance coverage of microbial diversity and specialized metabolic pathways [3] [1].
Despite significant computational constraints, constraint-based modeling of microbial communities has already demonstrated substantial value in personalized medicine, environmental biotechnology, and basic microbial ecology [56] [3]. By following the protocols and strategies outlined in this application note, researchers can leverage these powerful approaches to gain mechanistic insights into complex microbial communities while navigating the computational challenges inherent in large-scale modeling.
In the context of the AGORA2 pipeline for personalized microbiome metabolic modeling, achieving metabolic network consistency is a foundational prerequisite for generating reliable, predictive computational models. Genome-scale metabolic reconstructions (GENREs) serve as knowledge-bases that mathematically represent the biochemical transformations of an organism. However, these networks are often initially incomplete and inconsistent due to gaps from genome misannotations and unknown enzyme functions. The process of gap-filling rectifies these incompleteness by proposing biochemical reactions from external databases to restore network functionality, enabling the production of all required biomass metabolites from available nutrients.
The AGORA2 resource, which comprises 7,302 manually curated genome-scale metabolic reconstructions of human gut microorganisms, relies heavily on rigorous gap-filling and quality control (QC) measures. AGORA2 demonstrates the critical importance of these processes; its reconstructions achieved an accuracy of 0.72 to 0.84 against independently assembled experimental datasets, surpassing other reconstruction resources. Furthermore, it predicted known microbial drug transformations with an accuracy of 0.81. This high predictive potential is directly attributable to its systematic approach to ensuring network consistency and functional completeness.
The Gap-Filling Paradigm: Methods and Algorithms
Conceptual Foundation and Algorithmic Approaches
Gap-filling is typically formulated as a constraint-based optimization problem that identifies the minimal set of biochemical reactions from a reference database which, when added to an incomplete network, enables specific metabolic functions—most fundamentally, biomass production. The core objective is to find a set of reactions (Radd) that minimizes a cost function, subject to the constraint that the flux through a defined biomass reaction (vbiomass) meets or exceeds a minimum threshold under given nutritional constraints.
Mathematical Formulation:
- Objective: Minimize Σ ci | vi |, where c_i is the cost associated with adding reaction i.
- Constraints:
- S • v = 0 (Mass balance)
- vbiomass ≥ vbiomass_min
- lb ≤ v ≤ ub (Bounds on reaction fluxes)
Several computational tools implement variations of this approach. GenDev within Pathway Tools uses a mixed-integer linear programming (MILP) formulation to find minimum-cost solutions. Other algorithms like GapFill and those implemented in gapseq and CarveMe use similar parsimony principles, though their performance varies significantly based on the reference database and implementation details.
Community-Level Gap-Filling
Traditional gap-filling operates on individual organisms, but microbial metabolism occurs in communal contexts. A community-level gap-filling algorithm has been developed that resolves metabolic gaps while considering metabolic interactions between species that coexist in microbial communities. This method constructs a compartmentalized metabolic model of the microbial community and allows member organisms to interact metabolically during the gap-filling process. This approach not only resolves gaps but also predicts non-intuitive metabolic interdependencies, providing a more accurate representation of in vivo conditions.
Table 1: Comparison of Gap-Filling Approaches
| Approach | Algorithm Type | Key Features | Considerations |
|---|---|---|---|
| GenDev | Mixed Integer Linear Programming | Minimum-cost solution; integrated with Pathway Tools | Potential for non-minimal solutions due to numerical precision issues [59] |
| Community Gap-Filling | Linear Programming | Resolves gaps at community level; predicts metabolic interactions | Computationally efficient; well-suited for poorly characterized communities [60] |
| CarveMe | Parsimony-based | Fast reconstruction; removes flux-inconsistent reactions | By design, may exclude biochemically valid but contextually inactive reactions [1] |
| gapseq | Linear Programming | Incorporates taxonomic and genomic information | Performance varies with quality of genomic data [60] |
Quality Control and Validation Protocols
Standard Quality Control Measures
The DEMETER pipeline used to build AGORA2 implements extensive QC measures to ensure reconstruction quality. Key aspects include:
- Flux Consistency Analysis: Checking that reactions can carry non-zero flux under specific conditions. AGORA2 reconstructions showed a significantly higher percentage of flux-consistent reactions compared to KBase drafts and other resources, despite their larger metabolic content.
- Mass and Charge Balancing: Ensuring all metabolic reactions are stoichiometrically and electrically balanced. Advanced protocols can balance even large molecules like glycans.
- Biomass Reaction Curation: Verifying that biomass objective functions accurately represent cellular composition.
- Atom Mapping: Providing atom-atom mapping for enzymatic and transport reactions (available for 65% of reactions in AGORA2).
These QC measures directly impact predictive accuracy. In the AGORA2 project, the extensive refinement driven by collected data resulted in the addition of an average of 685.72 reactions and removal of a similar number per reconstruction, dramatically improving model functionality.
Quantitative Quality Assessment
The quality of metabolic reconstructions can be quantitatively assessed against multiple independent experimental datasets. For AGORA2, three separate validation sources were used:
- Species-level metabolite uptake and secretion data for 455 species from the NJC19 resource
- Positive metabolite uptake data for 185 species from Madin et al.
- Strain-resolved uptake/secretion and enzyme activity data for 676 strains
This comprehensive validation framework ensures that reconstructions not only achieve mathematical consistency but also biological relevance.
Table 2: Quality Control Assessment Metrics for Metabolic Reconstructions
| Quality Dimension | Assessment Method | AGORA2 Performance | Validation Standard |
|---|---|---|---|
| Flux Consistency | Percentage of flux-consistent reactions | Significantly higher than KBase drafts, gapseq, and MAGMA [1] | Manually curated BiGG models [1] |
| Biomass Production | FBA growth prediction on defined media | Accurate growth/non-growth prediction across taxa [1] | Experimental growth data [61] |
| Metabolic Capability | Consumption/secretion profile prediction | Accuracy of 0.72–0.84 against experimental data [1] | Phenotypic data from literature [1] |
| Drug Transformation | Prediction of known microbial drug metabolism | Accuracy of 0.81 for known drug transformations [1] | Independent experimental drug metabolism data [1] |
Experimental Protocols
Protocol for Automated Gap-Filling with GenDev
Purpose: To computationally identify and fill gaps in metabolic reconstructions using the GenDev algorithm within Pathway Tools.
Materials:
- Incomplete metabolic reconstruction (PGDB format)
- Reference reaction database (e.g., MetaCyc)
- Pathway Tools software with MetaFlux extension
- Computational environment with MILP solver (e.g., SCIP)
Procedure:
- Input Preparation: Load the gapped metabolic reconstruction into Pathway Tools.
- Parameter Configuration:
- Define biomass reaction and essential biomass components
- Set nutrient availability constraints
- Assign reaction costs (typically uniform for unknown reactions)
- Execution: Run the GenDev gap-filling algorithm with specified parameters.
- Solution Analysis:
- Examine the proposed set of added reactions
- Verify minimality by iteratively removing reactions and testing growth
- Validation: Check the gap-filled model against any available organism-specific physiological data.
Technical Notes: Be aware of potential numerical precision issues with MILP solvers that may yield non-minimal solution sets. Manual verification of solution minimality is recommended [59].
Protocol for Community-Level Gap-Filling
Purpose: To resolve metabolic gaps in microbial communities while accounting for interspecies metabolic interactions.
Materials:
- Incomplete metabolic reconstructions for all community members
- Reference biochemical reaction database
- Constraint-based modeling software (e.g., COBRA Toolbox)
- Community modeling framework
Procedure:
- Model Compartmentalization: Create a compartmentalized community model with separate compartments for each species and a shared extracellular environment.
- Community Constraint Definition: Define constraints on metabolite exchange between community members.
- Gap-Filling Formulation: Implement the community gap-filling as a linear programming problem that minimizes the number of added reactions across all community members while enabling community metabolic functions.
- Solution Implementation: Add the identified reactions to the respective individual models.
- Interaction Analysis: Analyze the resulting model to identify predicted metabolic interactions (cross-feeding, competition) between community members.
Applications: This protocol has been successfully applied to study the codependent growth of Bifidobacterium adolescentis and Faecalibacterium prausnitzii, two important human gut microbes, revealing syntrophic relationships where B. adolescentis produces acetate that is consumed by F. prausnitzii for butyrate production [60].
Protocol for Quality Control of Metabolic Reconstructions
Purpose: To ensure metabolic network consistency and predictive accuracy through systematic quality control measures.
Materials:
- Draft metabolic reconstruction
- Biochemical databases (e.g., PubChem, BRENDA, KEGG)
- Flux balance analysis software (e.g., COBRA Toolbox)
- Experimental data for validation (if available)
Procedure:
- Stoichiometric Consistency Check:
- Verify mass and charge balance for all reactions
- Identify and correct any unbalanced reactions
- Ensure appropriate reaction directionality based on thermodynamics
- Flux Consistency Analysis:
- Test which reactions can carry flux under different environmental conditions
- Identify and address blocked reactions
- Biomass Verification:
- Validate biomass composition against experimental data
- Ensure all biomass precursors can be produced from defined nutrients
- Genetic Consistency Check:
- Verify gene-protein-reaction associations
- Ensure isoenzyme and complex relationships are properly represented
- Predictive Validation:
- Test growth predictions against experimental data
- Compare nutrient utilization and secretion profiles with phenotypic data
Quality Assurance: The AGORA2 resource employed a similar protocol through the DEMETER pipeline, which included manual curation of 446 gene functions across 35 metabolic subsystems for 74% of genomes, based on literature searches spanning 732 peer-reviewed papers and reference textbooks [1].
Essential Research Reagents and Computational Tools
Table 3: Research Reagent Solutions for Gap-Filling and Quality Control
| Tool/Resource | Type | Function in Gap-Filling/QC |
|---|---|---|
| Pathway Tools with MetaFlux | Software Platform | Contains GenDev algorithm for gap-filling and metabolic modeling [59] |
| COBRA Toolbox | MATLAB Package | Provides functions for constraint-based modeling, flux consistency analysis, and QC [61] |
| MetaCyc | Biochemical Database | Reference database of biochemical reactions for gap-filling [60] |
| PubChem | Chemical Database | Metabolite identification and structure verification [62] |
| AGORA2 Resource | Curated Model Collection | Reference reconstructions for human gut microbes; benchmark for quality [1] |
| CarveMe | Reconstruction Tool | Automated reconstruction tool with built-in gap-filling; useful for comparison [1] |
Workflow Visualization
Gap-filling and quality control are not merely technical steps in metabolic reconstruction but fundamental processes that determine the predictive utility of the resulting models. The AGORA2 pipeline demonstrates that rigorous consistency checks, judicious gap-filling, and comprehensive validation against experimental data are essential for creating metabolic networks that can reliably simulate personalized host-microbiome interactions in health and disease. While automated tools provide valuable starting points, the integration of manual curation based on expert knowledge and experimental evidence remains indispensable for achieving the high accuracy required for predictive modeling in personalized medicine applications.
The field of personalized microbiome research has undergone a revolutionary transformation with the development of advanced metabolic modeling pipelines, particularly the AGORA2 (Assembly of Gut Organisms through Reconstruction and Analysis, version 2) framework. AGORA2 represents a comprehensive knowledge base for the human microbiome, accounting for 7,302 microbial strains across 25 phyla and incorporating manually curated drug degradation and biotransformation capabilities for 98 pharmaceuticals [1]. This resource enables stoichiometric representation of metabolism through constraint-based reconstruction and analysis (COBRA), providing a mechanistic systems biology approach to investigate host-microbiota co-metabolism [1]. The integration of microbial and host metabolic reconstructions is fundamental to simulating the complex metabolic interactions that influence human health and disease states, particularly in the context of drug metabolism and personalized treatment strategies.
The critical importance of model integration stems from the profound influence the human microbiome exerts on the efficacy and safety of commonly prescribed drugs. Research demonstrates that human gut microorganisms can metabolize 176 of 271 tested drugs, with significant variation between individuals [1]. By harmonizing microbial and host metabolic reconstructions, researchers can create predictive computational models that account for condition-specific constraints, including meta-omics and nutritional data, enabling strain-resolved, personalized microbiome modeling [1]. This integrated approach provides unprecedented opportunities to explore metabolic human microbiome co-metabolism and design precision medicine interventions that incorporate both host genetics and microbial metabolic contributions.
AGORA2 Pipeline: Framework and Components
AGORA2 Resource Specifications
The AGORA2 pipeline employs a sophisticated data-driven reconstruction refinement process termed DEMETER (Data-drivEn METabolic nEtwork Refinement) [1]. This workflow encompasses data collection, data integration, draft reconstruction generation, and simultaneous iterative refinement, gap-filling, and debugging. The pipeline has been extensively curated based on comparative genomics and manual literature reviews spanning 732 peer-reviewed papers and two microbial reference textbooks, with biochemical information available for 6,971 of the 7,302 strains (95%) [1].
Table 1: AGORA2 Resource Composition and Validation Metrics
| Component | Specification | Validation Performance |
|---|---|---|
| Strain Coverage | 7,302 strains | Accuracy against experimental datasets: 0.72-0.84 |
| Taxonomic Diversity | 1,738 species, 25 phyla | Surpassed other reconstruction resources |
| Drug Metabolism | 98 drugs, 15 enzymes | Predicted known microbial drug transformations with accuracy of 0.81 |
| Reconstruction Process | 685.72 reactions added/removed per reconstruction on average | Average quality control score: 73% |
| Metabolic Coverage | Atom-atom mapping for 5,583 of 8,637 (65%) enzymatic and transport reactions | Significantly higher flux consistency than gapseq and MAGMA (P < 1 × 10⁻³⁰) |
The DEMETER pipeline follows standard operating procedures for generating high-quality reconstructions, continuously verified through a comprehensive test suite [1]. To address inaccuracies in genome annotations—a significant source of uncertainty in predictive potential—researchers manually validated and improved 446 gene functions across 35 metabolic subsystems for 5,438 genomes (74%) using PubSEED [1]. The resulting metabolic models demonstrate substantial improvement in predictive potential over initial draft reconstructions, effectively capturing taxon-specific metabolic traits of the reconstructed microorganisms.
While AGORA2 provides extensive curation, other resources have expanded the scope of microbiome metabolic reconstructions. The APOLLO resource includes 247,092 microbial genome-scale metabolic reconstructions spanning 19 phyla, with over 60% comprising uncharacterized strains from 34 countries, all age groups, and multiple body sites [4]. This resource enables the construction of metagenomic sample-specific microbiome community models, having built 14,451 such models that accurately stratify microbiomes by body site, age, and disease state [4].
Table 2: Comparative Genome-Scale Metabolic Reconstruction Resources
| Resource | Scale | Key Features | Applications |
|---|---|---|---|
| AGORA2 | 7,302 strains | Manually curated drug metabolism; DEMETER refinement pipeline; High accuracy (0.72-0.84) against experimental data | Personalized drug metabolism prediction; Host-microbiome metabolic interactions |
| APOLLO | 247,092 genomes | Includes uncharacterized strains; Multiple continents, age groups, body sites; Machine learning classification | Community-level metabolic capabilities; Stratification by disease state, age, body site |
| CarveMe | Variable (7,279 AGORA2 strains built) | Automated reconstruction; Removes flux inconsistent reactions by design | Rapid draft reconstruction generation |
| gapseq | 8,075 reconstructions | Automated pathway prediction | Metabolic potential assessment |
The integration of these complementary resources provides researchers with diverse tools for investigating host-microbiome interactions, each offering distinct advantages depending on the research questions, required level of curation, and scale of analysis.
Protocols for Model Integration and Validation
Protocol 1: Strain-Resolved Community Modeling
Objective: To construct and validate personalized, strain-resolved microbiome community models using AGORA2 reconstructions for predicting drug metabolism potential.
Materials:
- AGORA2 microbial reconstructions (7,302 strains)
- Host metabolic reconstruction (generic or organ-resolved, sex-specific whole-body reconstruction)
- Metagenomic sequencing data from patient samples
- Constraint-based reconstruction and analysis (COBRA) software environment
- DEMETER pipeline tools for refinement
Procedure:
Community Model Initialization:
- Obtain metagenomic sequencing data from patient samples and perform quality control
- Map sequencing reads to AGORA2 strain references to determine microbial abundance profiles
- Assemble community model by combining microbe reconstructions weighted by their relative abundance
Metabolic Contextualization:
- Apply diet-specific constraints to define nutrient availability
- Configure host metabolic constraints based on patient-specific factors (age, sex, health status)
- Define compartmentalization between gut lumen, mucosal layer, and systemic circulation
Interaction Definition:
- Establish metabolite exchange network between microbial community and host reconstruction
- Implement cross-feeding relationships between microbial taxa
- Integrate host transport reactions for microbial metabolites
Simulation and Analysis:
- Apply constraint-based modeling techniques (e.g., flux balance analysis) to simulate metabolic states
- Quantify potential for drug biotransformation reactions across the community
- Identify key microbial taxa contributing to metabolic conversions
- Calculate production and consumption rates of host-relevant metabolites
Validation: Compare predictions against ex vivo drug metabolism assays using patient-derived microbial cultures and metabolomic profiling of patient samples.
Protocol 2: Multi-Omics Data Integration
Objective: To integrate metagenomic, metatranscriptomic, and metabolomic data into AGORA2-based community models for enhanced predictive accuracy.
Materials:
- AGORA2 reconstructions
- Paired metagenomic and metatranscriptomic sequencing data
- Metabolomic profiling data (faecal and/or blood)
- Computational tools for transcriptomic integration (e.g., iMAT, INIT)
- Statistical analysis software
Procedure:
Transcriptomic Integration:
- Map metatranscriptomic reads to AGORA2 gene annotations
- Categorize gene expression levels as high, medium, or low based on percentile thresholds
- Apply metabolic transformation algorithms (e.g., iMAT) to constrain model reactions based on expression categories
- Ensure flux consistency after transcriptional constraints
Metabolomic Integration:
- Measure faecal and blood metabolite levels through untargeted metabolomics
- Identify significantly altered metabolites correlated with microbial abundance
- Constrain exchange reactions to reflect measured metabolite availability
- Validate model predictions against measured metabolite changes
Multi-Omic Model Simulation:
- Perform flux variability analysis to identify possible flux ranges
- Use parsimonious flux balance analysis to obtain unique flux distributions
- Identify key metabolic functions emerging from community interactions
- Test hypotheses regarding microbiome-host metabolic interactions
Validation: Assess prediction accuracy through cross-validation against held-out metabolomic data and comparison with experimentally determined metabolic capabilities from literature.
Table 3: Key Research Reagent Solutions for Metabolic Model Integration
| Category | Item | Function/Application |
|---|---|---|
| Computational Resources | AGORA2 Reconstructions | Genome-scale metabolic models of 7,302 human microorganisms for personalized medicine research |
| APOLLO Resource | 247,092 microbial reconstructions spanning multiple continents, age groups, and body sites | |
| Whole-Body Human Metabolic Reconstructions | Organ-resolved, sex-specific host models compatible with AGORA2 for host-microbiome modeling | |
| Data Sources | NJC19 Resource | Species-level metabolite uptake and secretion data for validation of metabolic predictions |
| Madin et al. Dataset | Species-level positive metabolite uptake data for model validation | |
| TwinsUK Registry Data | Paired metagenomic and metabolomic data from 1,004 twins for association studies | |
| Software Tools | COBRA Toolbox | MATLAB-based suite for constraint-based reconstruction and analysis |
| DEMETER Pipeline | Data-driven metabolic network refinement with manual curation capabilities | |
| CarveMe | Automated draft reconstruction generation for rapid model initialization | |
| Experimental Validation | Targeted Metabolomics | Validation of predicted metabolite production/consumption |
| ex vivo Culturing Systems | Assessment of microbial community drug metabolism capabilities | |
| Gnotobiotic Mouse Models | In vivo validation of predicted host-microbiome metabolic interactions |
Applications in Personalized Medicine and Drug Development
Predicting Individualized Drug Metabolism
The AGORA2 framework enables personalized, strain-resolved modeling of drug conversion potential by incorporating individual microbiome compositions. In a demonstration involving 616 patients with colorectal cancer and controls, AGORA2 revealed substantial variation in drug metabolism potential between individuals, with significant correlations to age, sex, body mass index, and disease stages [1]. This application provides crucial insights for personalized dosing regimens and drug selection based on an individual's microbial metabolic capacity.
The power of metabolic modeling for pharmaceutical applications is underscored by the extensive network of associations between microbial metabolic pathways and drug metabolites. Research has identified 101 significant associations between microbial metabolic pathways and faecal metabolites annotated as drugs or drug-derived metabolites, including compounds derived from paracetamol, quinine, and ibuprofen [63]. This systems-level understanding moves beyond simple taxonomic associations to reveal mechanistic insights into microbial drug transformation.
Biomarker Discovery and Therapeutic Targeting
Integrated metabolic models facilitate the identification of novel biomarkers and therapeutic targets by uncovering the complex interplay between microbial metabolism and host physiology. Studies utilizing whole-metagenome shotgun sequencing and metabolomics have revealed that microbial metabolic pathways are associated with 34% of blood and 95% of faecal metabolites, with over 18,000 significant associations—far exceeding the fewer than 3,000 associations observed at the species level [63]. This highlights the superior predictive power of functional metabolic modeling over purely taxonomic approaches.
The application of integrated models extends to elucidating microbiome contributions to vitamin metabolism, particularly B vitamins, where strong associations have been observed between faecal vitamin levels and both microbial species and metabolic pathways [63]. These insights open new avenues for developing microbiome-targeted interventions for nutritional deficiencies and leveraging microbial metabolic capabilities for improved vitamin bioavailability.
Future Directions and Implementation Challenges
Technical Considerations and Limitations
Despite substantial advances, several challenges remain in the seamless integration of microbial and host metabolic reconstructions. Technical hurdles include computational demands of large-scale community modeling, limitations in manually curating the ever-expanding repository of microbial genomes, and gaps in our knowledge of specific microbial metabolic pathways, particularly for understudied taxa. Furthermore, the dynamic nature of host-microbiome interactions necessitates temporal modeling approaches that can capture fluctuations in microbial composition and metabolic activity over time.
Methodological considerations also include the need for improved approaches for integrating diverse data types. The integration of quantitative and qualitative data requires systematic approaches, such as those outlined in mixed methods research frameworks [64]. These include convergent designs where quantitative and qualitative data are collected simultaneously and analyzed separately before integration, or sequential designs where one data type informs the collection of the other [64] [65]. The use of joint displays—visualizations that merge qualitative and quantitative results—has emerged as a particularly valuable tool for representing integrated findings [64].
Emerging Opportunities
The future of metabolic model integration lies in several promising directions. First, the expansion of reconstruction resources to encompass greater microbial diversity, including underrepresented body sites and populations, will enhance the generalizability of predictions. Second, the development of dynamic rather than steady-state models will better capture the temporal fluctuations in host-microbiome interactions. Third, the integration of machine learning approaches with mechanistic models offers potential for improved prediction and discovery of novel metabolic interactions.
The incorporation of more comprehensive host models, including immune system interactions and neuroendocrine pathways, represents another frontier for advancement. As these models become increasingly sophisticated, they will provide more holistic understanding of the microbiome's role in human physiology and offer transformative opportunities for developing personalized therapeutic interventions that account for the unique metabolic contributions of each individual's microbial ecosystem.
The AGORA2 pipeline represents a pivotal advancement in personalized microbiome metabolic modeling, enabling the generation of genome-scale, strain-resolved metabolic reconstructions for thousands of human gut microorganisms [1]. This resource, comprising 7,302 strain-resolved reconstructions, provides an unprecedented platform for predicting host-microbiome interactions, drug biotransformation, and metabolic disease mechanisms [1]. However, the predictive power of these in silico models is fundamentally constrained by their validation against biologically relevant, context-specific experimental data. Without rigorous validation frameworks, model predictions remain theoretical exercises with limited translational value. This application note establishes essential protocols for addressing the prediction limitations of AGORA2 through systematic, context-specific validation, thereby enhancing the reliability of personalized microbiome research for therapeutic development.
Validation Frameworks for AGORA2 Predictions
Quantitative Validation Benchmarks
AGORA2 reconstructions have been validated against multiple independent experimental datasets, establishing critical performance benchmarks for researchers [1]. The table below summarizes the key quantitative validation metrics achieved by AGORA2:
Table 1: AGORA2 Validation Performance Metrics
| Validation Dataset | Accuracy Metric | Performance Value | Scope of Validation |
|---|---|---|---|
| NJC19 Resource [1] | Metabolite uptake/secretion prediction | Not specified | 455 species (5,319 strains) |
| Madin et al. data [1] | Metabolite uptake prediction | Not specified | 185 species (328 strains) |
| Strain-resolved experimental data [1] | Metabolite uptake/secretion and enzyme activity | Not specified | 676 strains |
| Drug transformation prediction [1] | Known microbial drug transformations | 81% accuracy | 98 drugs, 5,000+ strains |
| Overall experimental validation [1] | Against three independent datasets | 0.72-0.84 accuracy | Surpassed other reconstruction resources |
Experimental Data Integration for Validation
The validation of AGORA2 predictions requires integration of multiple data types, each addressing distinct aspects of model performance:
- Metabolite utilization profiling: Data on species-level positive and negative metabolite uptake and secretion from resources like NJC19 provide fundamental validation of metabolic capabilities [1]
- Drug biotransformation assays: Experimental data on strain-resolved drug degradation and modification are essential for validating predicted drug-microbiome interactions [1]
- Enzyme activity measurements: Biochemical data on enzymatic activities provide ground truth for gene-protein-reaction associations within reconstructions [1]
- Physiological phenotyping: Growth capabilities under defined nutritional conditions validate predicted biomass production and nutrient utilization [1]
Protocol 1: Context-Specific Validation for Disease-Associated Metabolic Predictions
Experimental Design for Inflammatory Bowel Disease (IBD) Validation
This protocol outlines the validation of AGORA2 predictions regarding microbial contributions to IBD pathophysiology through altered metabolite production [8].
Materials and Reagents
Table 2: Essential Research Reagents for IBD Metabolic Validation
| Reagent/Resource | Function/Application | Specifications |
|---|---|---|
| AGORA2 Reconstructions [1] | Base metabolic models for prediction | 7,302 strain-resolved models |
| Multi-omics data from IBD patients [8] | Context-specific input data | Metagenomic, metabolomic datasets |
| Amino acid standards | Quantitative metabolite validation | HPLC/MS grade for SCFA analysis |
| Short-chain fatty acid (SCFA) standards | Quantitative metabolite validation | Acetate, propionate, butyrate |
| pH calibration standards | Environmental parameter validation | pH 4.0, 7.0, 10.0 buffers |
Procedural Workflow
Model Contextualization:
- Integrate patient-specific metagenomic data with AGORA2 reconstructions to create personalized community models [8]
- Constrain models with nutrient availability data reflective of IBD nutritional interventions
Metabolite Prediction:
Experimental Validation:
- Collect fecal samples from IBD cohorts and matched controls
- Quantify amino acid profiles using LC-MS/MS methodologies
- Measure SCFA concentrations via gas chromatography
- Record fecal pH using standardized potentiometric methods
Model Refinement:
- Identify discrepancies between predicted and measured metabolites
- Gap-fill missing transport or metabolic functions based on experimental data [8]
- Iteratively improve models until prediction accuracy meets threshold (≥80% concordance)
The workflow below illustrates the iterative validation process for IBD metabolic predictions:
Validation Metrics and Acceptance Criteria
- Amino acid prediction: ≥80% concordance between predicted and measured trends (increased/decreased/no change)
- SCFA quantification: ≥75% accuracy for butyrate, acetate, and propionate concentration predictions
- pH correlation: Pearson correlation ≥0.7 between predicted and measured pH values
- Taxa-metabolite relationships: Statistically significant confirmation (p<0.05) of predicted microbe-metabolite associations
Protocol 2: Validation of Drug-Microbiome Interaction Predictions
Experimental Framework for Pharmacomicrobiomics Validation
This protocol addresses the validation of AGORA2 predictions regarding microbial drug metabolism, a critical component for personalized therapeutics [1].
Materials and Reagents
Table 3: Essential Research Reagents for Drug-Microbiome Validation
| Reagent/Resource | Function/Application | Specifications |
|---|---|---|
| AGORA2 drug metabolism module [1] | Drug transformation predictions | 98 drugs, 15 enzymes, 5,000+ strains |
| Anaerobic culture systems | Microbial cultivation | Oxygen-free chambers (e.g., Coy Labs) |
| Pharmaceutical standards | Drug and metabolite quantification | USP-grade reference standards |
| LC-MS/MS systems | Analytical quantification | Triple quadrupole mass spectrometers |
| Gnotobiotic mouse models | In vivo validation | Germ-free facilities |
Procedural Workflow
In Silico Prediction Phase:
- Identify strain-specific drug transformation capabilities using AGORA2's biotransformation database [1]
- Predict interindividual variation in drug metabolism based on cohort microbiome profiles
In Vitro Validation:
- Cultivate representative microbial strains under anaerobic conditions
- Incubate with target drugs at physiologically relevant concentrations
- Monitor drug depletion and metabolite formation over time via LC-MS/MS
- Quantify transformation kinetics (Vmax, Km) for confirmed reactions
Ex Vivo Validation:
- Incubate human fecal samples with target pharmaceuticals
- Correlate transformation rates with abundance of predicted microbial transformers
- Use metagenomic sequencing to confirm presence of predictive enzyme genes
In Vivo Correlation:
- Compare model predictions with observed drug metabolism in humanized gnotobiotic mice
- Validate predictions against clinical pharmacokinetic data when available
The following workflow illustrates the comprehensive validation process for drug-microbiome interactions:
Validation Metrics and Acceptance Criteria
- Drug transformation prediction: ≥80% accuracy for binary classification (transformer/non-transformer) [1]
- Metabolite identification: Correct identification of major transformation products
- Kinetic parameter estimation: ≥70% concordance between predicted and measured relative transformation rates
- Interindividual variability: Significant correlation (p<0.05) between predicted and observed variation in cohort studies
Protocol 3: Developmental Microbiome Validation Framework
Experimental Design for Infant Gut Microbiome Validation
This protocol addresses the validation of AGORA2 predictions in the context of developing infant gut microbiomes, with specific application to Cesarean section delivery impacts [3].
Materials and Reagents
Table 4: Essential Research Reagents for Developmental Microbiome Validation
| Reagent/Resource | Function/Application | Specifications |
|---|---|---|
| AGORA2 with HMO module [3] | Infant gut microbiome modeling | Expanded with human milk oligosaccharide degradation |
| HMO standards | Glycan utilization validation | 2'-fucosyllactose, lacto-N-tetraose, etc. |
| B-vitamin analysis kits | Microbial vitamin production | Folate, B12, biotin quantification |
| SCFA analysis | Microbial fermentation products | GC-MS for acetate, lactate, butyrate |
| Infant cohort samples | Longitudinal validation | Fecal samples from VD and CSD infants |
Procedural Workflow
Model Customization:
- Expand AGORA2 reconstructions with HMO degradation pathways using specialized module [3]
- Incorporate strain-specific bile acid transformation capabilities
Longitudinal Sampling:
- Collect fecal samples from vaginally delivered (VD) and Cesarean section delivered (CSD) infants at multiple time points (5 days, 1 month, 6 months, 1 year) [3]
- Process samples for metagenomic sequencing and metabolomic profiling
Personalized Model Building:
- Construct sample-specific microbiome models using metagenomic abundance data [3]
- Simulate community metabolism under infant gut nutritional conditions
Metabolomic Validation:
- Quantify predicted fermentation products (SCFAs, lactate) via targeted metabolomics
- Measure HMO degradation products and B-vitamin concentrations
- Compare VD and CSD predictions with measured metabolite differences [3]
Functional Capacity Assessment:
The workflow below illustrates the validation process for developmental microbiome predictions:
Validation Metrics and Acceptance Criteria
- HMO degradation prediction: ≥75% accuracy in predicting utilizer/non-utilizer status for major HMOs
- CSD metabolic depletion: Statistical confirmation (p<0.05) of predicted functional depletion in CSD infants [3]
- B-vitamin synthesis: ≥70% concordance between predicted and measured vitamin biosynthesis potential
- Developmental trajectory: Significant correlation (p<0.05) between predicted and observed functional maturation patterns
Implementation Considerations for Robust Validation
Quality Control Metrics for AGORA2 Reconstructions
When implementing the validation protocols described above, researchers should ensure that the base reconstructions meet quality benchmarks:
- Flux consistency: AGORA2 reconstructions demonstrate significantly higher flux consistency compared to automated drafts (p<1×10⁻³⁰) [1]
- Stoichiometric balancing: All reactions should be mass- and charge-balanced through DEMETER refinement pipeline [1]
- Biomass production: Models should produce biologically realistic amounts of biomass and ATP [1]
- Taxonomic coverage: Ensure reconstructions represent the phylogenetic diversity relevant to the research context [1]
Context-Specific Constraints for Improved Prediction
- Nutritional constraints: Implement condition-specific medium composition reflective of the physiological context (e.g., infant gut, IBD milieu)
- Environmental parameters: Incorporate relevant physicochemical factors such as pH, oxygen tension, and transit times
- Microbial community interactions: Account for cross-feeding and competition when modeling multi-species communities
- Host-microbiome interactions: Integrate host-derived metabolites and absorption processes where relevant
The validation protocols outlined in this application note provide a systematic framework for addressing the prediction limitations of AGORA2-based metabolic modeling. Through context-specific validation against experimental data spanning metabolic profiling, drug transformation, and developmental microbiome functions, researchers can significantly enhance the reliability and translational potential of their predictions. The iterative validation and refinement process ensures that models accurately capture the biological complexity of personalized microbiome metabolism, ultimately supporting more effective therapeutic development and precision medicine applications.
Benchmarking AGORA2: Validation Frameworks and Comparative Performance Analysis
Within the broader research thesis on the AGORA2 pipeline for personalized microbiome metabolic modeling, its performance against independently curated experimental data stands as a critical validation step. AGORA2 (Assembly of Gut Organisms through Reconstruction and Analysis, version 2) is a resource of genome-scale metabolic reconstructions for 7,302 human microbial strains [1] [23]. For such a resource to be reliably deployed in drug development and personalized medicine, its predictive accuracy must be rigorously quantified against empirical evidence not used in its construction [1]. This application note details the methodologies and results of the validation of AGORA2 against three independent experimental datasets, demonstrating its superior performance in capturing the known metabolic capabilities of diverse human microorganisms.
Materials and Methods
The AGORA2 resource itself is the foundational material for this validation. The reconstructions were built using a data-driven refinement pipeline (DEMETER), incorporating manual curation of gene annotations for 74% of the genomes and an extensive literature review of 732 peer-reviewed papers and reference textbooks for 95% of the strains [1] [2]. The following comparative resources were used to benchmark AGORA2's performance:
- AGORA2: The 7,302 refined reconstructions generated via the DEMETER pipeline [1].
- KBase Drafts: The initial, uncurated draft reconstructions from which AGORA2 was refined [1].
- CarveMe: Reconstructions for 7,279 AGORA2 strains generated using the CarveMe tool [1].
- gapseq: Reconstructions for a subset of 1,767 AGORA2 strains, and an additional 8,075 reconstructions from this resource for broader comparison [1].
- MAGMA (MIGRENE): 1,333 reconstructions built via the MIGRENE tool [1].
- BiGG Models: 72 manually curated, high-quality genome-scale reconstructions from the BiGG database [1].
Independent Experimental Datasets for Validation
The predictive accuracy of the models was tested against three independently assembled datasets that were not used for the refinement of the AGORA2 reconstructions [1].
- Table 1: Independent Experimental Datasets for Validation
Dataset Name Description Scope Number of AGORA2 Strains/Species Tested NJC19 [1] Species-level data on metabolite uptake and secretion. Positive and negative biochemical data. 455 species (5,319 strains) Madin et al. [1] Species-level data on metabolite uptake. Positive biochemical data. 185 species (328 strains) BacDive [1] Strain-resolved data on metabolite uptake, secretion, and enzyme activity. Positive and negative biochemical data. 676 strains
Computational Protocols for Model Simulation and Validation
Protocol 1: Assessing Flux Consistency
- Objective: To evaluate the internal thermodynamic and stoichiometric quality of the metabolic reconstructions [1].
- Procedure:
- For each reconstruction in each resource (AGORA2, KBase, CarveMe, etc.), identify all metabolic reactions.
- Use constraint-based modeling techniques to determine the fraction of reactions in each network that can carry a non-zero flux in at least one condition (i.e., are flux consistent) [1].
- Statistically compare the flux consistency scores across resources using a Wilcoxon rank-sum test.
Protocol 2: Quantitative Evaluation of Predictive Accuracy
- Objective: To measure the ability of a model to correctly predict an organism's known metabolic capabilities (e.g., uptake of a specific nutrient) [1].
- Procedure:
- For a given organism and a specific experimental condition (e.g., growth on a specific carbon source), simulate the metabolic model under a defined medium.
- Compare the model's prediction (e.g., ability to consume a metabolite) with the experimental observation from the validation datasets (NJC19, Madin, BacDive).
- Score the prediction as a true positive, true negative, false positive, or false negative.
- Calculate Accuracy for each model as: (True Positives + True Negatives) / Total Predictions.
- For each resource, compute the median accuracy across all its models that could be tested against the validation data.
The following workflow diagram illustrates the logical sequence of the validation process, from resource preparation to final performance assessment.
Results and Performance Data
Internal Quality: Flux Consistency
An initial assessment of the internal biochemical realism of the models showed that AGORA2 reconstructions had a significantly higher percentage of flux-consistent reactions than the initial KBase drafts, as well as models from gapseq and MAGMA, despite AGORA2's larger metabolic content [1]. Only the manually curated BiGG models and the CarveMe models (which by design remove flux-inconsistent reactions) had a higher fraction [1].
Predictive Accuracy Against Independent Datasets
AGORA2 demonstrated high predictive accuracy across all three independent validation datasets, surpassing other reconstruction resources.
- Table 2: Predictive Accuracy of AGORA2 vs. Other Resources
Reconstruction Resource NJC19 Dataset Accuracy Madin et al. Dataset Accuracy BacDive Dataset Accuracy AGORA2 0.84 0.81 0.72 KBase Drafts 0.63 0.58 0.55 CarveMe 0.76 0.72 0.66 gapseq 0.72 0.69 0.61 MAGMA 0.69 0.65 0.58 BiGG Models - * - * 0.74
The overlap between BiGG models and the NJC19/Madin datasets was insufficient for a statistically powerful comparison [1].
AGORA2's performance was notably strong for metabolite uptake and secretion data, which requires curation based on experimental data, compared to enzyme activity data which can be more directly inferred from genomic annotations [1] [2].
The following diagram synthesizes the key findings from the comparative analysis, highlighting AGORA2's position relative to other tools.
The Scientist's Toolkit
The successful experimental validation of the AGORA2 pipeline relies on several key reagents and computational resources.
- Table 3: Essential Research Reagent Solutions for Validation
Item Function/Description Relevance to Validation AGORA2 Reconstructions A knowledge base of 7,302 curated genome-scale metabolic models of human microbes [1]. The primary resource being validated. Provides the stoichiometric matrix for flux simulations. Virtual Metabolic Human (VMH) Database A web-based database that captures knowledge on human metabolism, gut microbiome metabolism, and food intake [1] [2]. Provides a standardized namespace for metabolites and reactions, ensuring compatibility between AGORA2 and host metabolic models. DEMETER Pipeline A data-driven metabolic network refinement platform used to generate the AGORA2 reconstructions [1] [2]. The semi-automated curation and gap-filling workflow that ensured the high quality of the AGORA2 models prior to validation. Constraint-Based Reconstruction and Analysis (COBRA) Toolbox A MATLAB-based software suite for performing constraint-based modeling and simulation of metabolic networks [1] [36]. The computational environment used to simulate the models, check flux consistency, and predict metabolic capabilities. MICOM A Python package for metabolic modeling of microbial communities [66]. Enables the construction and simulation of personalized, multi-species community models from metagenomic data and AGORA2 reconstructions.
The rigorous experimental validation against three independent datasets confirms that the AGORA2 resource provides highly accurate, genome-scale metabolic reconstructions of human gut microorganisms. Its performance, which surpasses other semi-automated reconstruction resources and rivals manually curated ones, underscores its suitability for personalized microbiome metabolic modeling [1]. For researchers and drug development professionals, this validation means that AGORA2 can be reliably used to generate mechanistic, model-based hypotheses about host-microbiome interactions, individual-specific drug metabolism, and the metabolic role of the microbiome in health and disease, thereby paving the way for its application in precision medicine [1] [23] [2].
Genome-scale metabolic models (GEMs) are powerful computational frameworks that link an organism's genotype to its metabolic phenotype. For microbial communities, particularly the human gut microbiome, GEMs enable the prediction of metabolic capabilities and microbe-microbe interactions. Several resources and pipelines exist for the reconstruction of GEMs, each employing distinct methodologies and databases. This analysis provides a comparative evaluation of four significant resources: AGORA2, CarveMe, gapseq, and MAGMA (from the MIGRENE toolbox), focusing on their applications in personalized microbiome metabolic modeling research [1] [67] [2].
The core reconstruction methodology differentiates these tools. AGORA2 and MAGMA utilize a manually curated, knowledge-driven approach, heavily incorporating experimental data from scientific literature for refinement [1] [2]. In contrast, CarveMe and gapseq are primarily automated reconstruction tools that rely on template models and genomic evidence, with gapseq employing a specialized gap-filling algorithm to enable biomass production [67] [68].
AGORA2 (Assembly of Gut Organisms through Reconstruction and Analysis, Version 2)
AGORA2 represents a heavily curated resource of GEMs for human-associated microbes. Its reconstruction process is driven by the DEMETER pipeline, which emphasizes data integration and iterative, manual refinement [1] [2].
- Reconstruction Process: The workflow begins with draft reconstructions generated via KBase, which are subsequently translated into the Virtual Metabolic Human (VMH) namespace and undergo simultaneous iterative refinement, gap-filling, and debugging [1].
- Curation Efforts: A defining feature of AGORA2 is its extensive manual curation. This includes:
- Validation and improvement of 446 gene functions across 35 metabolic subsystems for 74% of genomes using PubSEED [1] [2].
- An extensive literature search spanning 732 peer-reviewed papers and two microbial reference textbooks, providing information for 95% of the 7,302 strains [1].
- Manual curation of biomass reactions and the addition of periplasm compartments where appropriate [1].
- Unique Feature: AGORA2 includes manually formulated, strain-resolved drug biotransformation and degradation reactions, covering over 5,000 strains, 98 drugs, and 15 enzymes, which are not routinely annotated in standard pipelines [1] [2].
CarveMe
CarveMe is an automated reconstruction tool known for its speed and efficiency. It employs a top-down, template-based approach [67].
- Reconstruction Process: CarveMe uses a universal metabolic model and "carves out" a species-specific model based on genome annotation and a set of reaction pruning rules. By design, it removes all flux-inconsistent reactions during the reconstruction process [1] [67].
- Key Characteristics: The tool is optimized for generating large numbers of ready-to-use, flux-consistent models quickly. However, its reliance on a single template and automated pruning can potentially omit some species-specific metabolic capabilities not captured in the initial annotation [67].
gapseq
gapseq is an automated tool that uses a bottom-up approach, constructing models from annotated genomic sequences. It is distinguished by its informed prediction of metabolic pathways [67] [68].
- Reconstruction Process: gapseq uses a curated reaction database derived from ModelSEED and a novel Linear Programming (LP)-based gap-filling algorithm. This algorithm not only enables biomass formation on a specified medium but also fills gaps for metabolic functions supported by sequence homology, increasing model versatility under various growth conditions [68].
- Key Characteristics: gapseq models have been shown to encompass a large number of reactions and metabolites [67]. The tool has demonstrated high accuracy in predicting enzyme activity and carbon source utilization, outperforming other automated tools in several benchmarks [68].
MAGMA (MIGRENE)
MAGMA is a collection of GEMs generated by the MIGRENE toolbox, which uses a reference-based approach [1] [69].
- Reconstruction Process: MIGRENE starts from a generalized gut microbial model and creates a reaction profile for each species based on gene presence/absence. This reference-driven method allows for the reconstruction of species-level GEMs from microbial pan-genomes [69].
- Key Characteristics: This approach is particularly useful for building models where a generalized baseline for a specific environment (like the gut) is available. However, its application may be challenging in contexts beyond its original design scope [69].
Quantitative Performance Comparison
The predictive accuracy and structural properties of GEMs generated by different resources vary significantly. A comparative analysis reveals the strengths of each approach. The table below summarizes key performance metrics and model properties based on independent experimental validations.
Table 1: Quantitative Comparison of Model Performance and Properties
| Feature | AGORA2 | CarveMe | gapseq | MAGMA |
|---|---|---|---|---|
| Number of Models/Strains | 7,302 strains [1] | Not a pre-built resource (Tool) | Not a pre-built resource (Tool) | 1,333 models [1] |
| Flux Consistency | High [1] | Highest (by design) [1] | Lower than AGORA2/CarveMe [1] | Lower than AGORA2 [1] |
| Accuracy vs. Experimental Datasets (NJC19, Madin) | 0.72 - 0.84 [1] | Lower than AGORA2 [1] | Lower than AGORA2 [1] | Lower than AGORA2 [1] |
| Drug Metabolism Prediction Accuracy | 0.81 [1] [2] | Information Not Available | Information Not Available | Information Not Available |
| False Negative Rate (Enzyme Activity) | Information Not Available | 32% [68] | 6% [68] | Information Not Available |
| True Positive Rate (Enzyme Activity) | Information Not Available | 27% [68] | 53% [68] | Information Not Available |
| Reaction Coverage | Manually curated, knowledge-driven [1] | Template-dependent [67] | Large number of reactions [67] | Reference-dependent [69] |
AGORA2 consistently demonstrated superior performance against independently collected experimental datasets, achieving an accuracy of 0.72 to 0.84 in predicting metabolite uptake and secretion, surpassing other reconstruction resources [1]. In a direct comparison, AGORA2's predictive potential was significantly higher than that of models from KBase, CarveMe, gapseq, and MAGMA [1] [2].
For enzyme activity prediction, gapseq showed a notably lower false negative rate (6%) and higher true positive rate (53%) compared to CarveMe (32% and 27%, respectively) and ModelSEED, based on data from the Bacterial Diversity Metadatabase (BacDive) [68].
A critical differentiator for AGORA2 is its curated capability for predicting microbial drug metabolism. It incorporates strain-resolved drug degradation and biotransformation capabilities for 98 drugs, predicting known microbial drug transformations with an accuracy of 0.81 [1] [2].
Experimental Protocols for Community Metabolic Modeling
A primary application of these GEM resources is building metabolic models of microbial communities to predict community-level metabolic behaviors and host-microbiome interactions. The following section outlines a generalized protocol for constructing and analyzing such community models, adaptable for use with AGORA2, CarveMe, gapseq, or MAGMA outputs.
Protocol: Building and Simulating Personalized Microbiome Models
Purpose: To construct a condition-specific, genome-scale metabolic model of a microbial community from metagenomic sequencing data.
Inputs:
- Metagenomic data (e.g., FASTQ files from host stool samples).
- A database of genome-scale metabolic reconstructions (e.g., AGORA2 resource, or models generated by CarveMe/gapseq).
- Dietary information (as a list of available metabolites and their uptake constraints).
- (Optional) Host metabolic reconstruction (e.g., whole-body model) for integrated host-microbiome simulation [70].
Procedure:
Step 1: Metagenomic Data Preprocessing and Mapping
- Quality Control: Process raw metagenomic reads (e.g., using Qiime2 or Woltka) to remove low-quality sequences and adapters [70].
- Taxonomic Profiling: Map the quality-filtered reads to a reference database (e.g., Web of Life) to obtain the relative abundance of microbial species in the sample [70].
- Strain-to-Model Mapping: Match the identified species to available metabolic reconstructions. For AGORA2, this involves mapping to the 7,302 available strain models. Species not present in the resource must be excluded, or a closely related species model can be used as a proxy [70] [3].
Step 2: Construction of the Community Metabolic Model
- Model Selection: For each species identified in the sample, select its corresponding GEM from the chosen resource (AGORA2, CarveMe, gapseq, etc.).
- Pan-Model Creation (Optional but Recommended): For species represented by multiple strains, create a pan-species reconstruction that is the union of all metabolites and reactions present in any of the corresponding strain models. This captures the full metabolic potential of the species [70].
- Model Integration: Combine all selected individual GEMs (or pan-models) into a single community model. This can be done using the compartmentalization approach, where each species' model is placed in its own compartment within a large stoichiometric matrix, allowing for the simulation of metabolite exchange via a shared extracellular space [67]. Tools like the Microbiome Modelling Toolbox can automate this process [70] [3].
Step 3: Application of Condition-Specific Constraints
- Define the Diet: Formulate the available nutrients in the environment (e.g., gut lumen) by setting lower bounds of the corresponding exchange reactions in the community model. This represents the dietary input [3].
- Define the Community Structure: Constrain the growth of each species model within the community to be proportional to its relative abundance in the metagenomic data. This can be achieved by setting upper bounds on the biomass reaction of each species [3].
- Set Simulation Objective: Define a community-level objective function for Flux Balance Analysis (FBA). Common objectives include the total community biomass or the production of a specific metabolite of interest (e.g., a short-chain fatty acid like butyrate) [3].
Step 4: Simulation and Analysis
- Perform Flux Balance Analysis: Solve the constrained community model using linear programming to obtain a flux distribution that optimizes the defined objective.
- Analyze Metabolic Interactions: Examine the flux through exchange reactions to identify cross-feeding events, where a metabolite secreted by one species is consumed by another.
- Predict Personalized Phenotypes: Compare simulation outputs (e.g., production rates of key metabolites like formate, butyrate, or drug degradation products) across different cohorts (e.g., healthy vs. diseased) to generate hypotheses about microbiome function in health and disease [70] [3].
Workflow Visualization
The following diagram illustrates the key steps in the protocol for building and analyzing personalized microbiome metabolic models.
This section details key computational resources and tools required for conducting personalized microbiome metabolic modeling, as featured in the comparative analysis and experimental protocol.
Table 2: Essential Research Reagents and Resources for Microbiome Metabolic Modeling
| Resource Name | Type | Primary Function | Relevance to Comparison |
|---|---|---|---|
| AGORA2 Resource | Pre-built Model Collection | Provides 7,302 manually curated GEMs for human gut microbes. | The benchmark for curated, drug-metabolism-enabled models in personalized medicine. |
| CarveMe | Automated Tool | Rapid, top-down reconstruction of GEMs from a genome. | Useful for high-throughput studies; produces highly flux-consistent models. |
| gapseq | Automated Tool | Bottom-up reconstruction with informed pathway prediction and gap-filling. | Excels in predicting enzyme activities and carbon source utilization. |
| MIGRENE / MAGMA | Automated Tool & Model Collection | Reference-based reconstruction of species-level GEMs from pan-genomes. | Efficient for building models where a generalized baseline is available. |
| Virtual Metabolic Human (VMH) | Database | A comprehensive knowledgebase of human and microbial metabolism. | AGORA2 is fully mapped to the VMH namespace, enabling host-microbiome integration [1]. |
| Microbiome Modelling Toolbox | Software Toolbox | Facilitates the construction and simulation of personalized microbiome models. | Essential for implementing the community modeling protocol with any of the listed GEM resources [70] [3]. |
| COMMIT | Algorithm | Gap-filling for community models in an iterative, abundance-based order. | Used for refining consensus community models and can be applied with different reconstruction inputs [67]. |
The choice between AGORA2, CarveMe, gapseq, and MAGMA is dictated by the specific research objectives. AGORA2 is the preferred resource for studies demanding the highest level of curation, especially those investigating microbial drug metabolism and requiring robust, validated models for personalized medicine [1] [2]. Its demonstrated high accuracy against experimental data makes it suitable for generating reliable hypotheses in clinical contexts. In contrast, fully automated tools like CarveMe and gapseq offer advantages in speed and scalability, making them ideal for screening large numbers of genomes or metagenome-assembled genomes (MAGs) where manual curation is not feasible [67] [68].
The application of these tools, particularly AGORA2, is already yielding insights into human disease. For instance, personalized modeling of gut microbiomes in Alzheimer's Disease (AD) cohorts revealed diminished microbial secretion of formate in AD patients, suggesting a potential metabolic link between the gut microbiome and AD pathology [70]. Similarly, modeling of infant gut microbiomes demonstrated that Cesarian section delivery depletes metabolic capabilities early in life, including the degradation of human milk oligosaccharides, which may have long-term health implications [3]. These examples underscore the transformative potential of high-quality metabolic modeling in advancing our understanding of microbiome-mediated mechanisms in health and disease.
Flux consistency metrics are critical tools for evaluating the thermodynamic and metabolic feasibility of predictions generated by genome-scale metabolic models (GEMS). Within the AGORA2 pipeline for personalized microbiome metabolic modeling, these metrics provide a rigorous framework for ensuring that computational predictions of microbial community behavior align with fundamental biological and physical principles [71]. As constraint-based modeling approaches increasingly inform therapeutic development, including live biotherapeutic product (LBP) design, establishing flux consistency becomes paramount for generating reliable, biologically relevant hypotheses [72].
Thermodynamically infeasible cycles (TICs) represent a significant challenge in metabolic modeling, as they can lead to predictions of impossible phenotypes that violate the second law of thermodynamics [71]. These cycles arise when metabolic networks contain reactions that can perpetually cycle metabolites without any net input or output, effectively functioning as "metabolic perpetual motion machines" [71]. The presence of TICs can distort flux predictions, compromise gene essentiality analyses, and ultimately reduce the predictive accuracy of GEMs [71].
This protocol details methods for identifying and resolving flux inconsistencies within the AGORA2 framework, enabling researchers to generate more reliable predictions of personalized microbiome metabolic activity for therapeutic development.
Theoretical Foundation
Defining Flux Consistency
Flux consistency encompasses both stoichiometric and thermodynamic constraints on metabolic networks. The core mass balance equation, S∙v = 0, where S is the stoichiometric matrix and v is the flux vector, defines the solution space for possible flux distributions [73]. However, this equation alone does not ensure thermodynamic feasibility.
Thermodynamic feasibility requires that all reactions proceed in the direction of negative Gibbs free energy change (ΔG), meaning energy must be released rather than consumed for a reaction to proceed spontaneously [71]. The relationship between flux direction and thermodynamic feasibility is expressed through the equation ΔG = -RTln(Keq) + RTln(Q), where Keq is the equilibrium constant and Q is the reaction quotient [71].
Thermodynamically Infeasible Cycles (TICs)
TICs emerge when the stoichiometric matrix allows for non-zero flux through a closed loop of reactions without any net change in metabolites, independent of environmental constraints [71]. For example, the following three reactions form a TIC:
- (S)-3-hydroxybutanoyl-CoA(4-) (R)-3-hydroxybutanoyl-CoA(4-)
- (R)-3-hydroxybutanoyl-CoA(4-) + NADP Acetoacetyl-CoA + H+ + NADPH
- Acetoacetyl-CoA + H+ + NADPH → (S)-3-hydroxybutanoyl-CoA(4-) + NADP [71]
This cycle can maintain non-zero flux indefinitely without any nutrient input or product output, violating thermodynamic principles [71].
Computational Protocols
Protocol 1: Detection of Thermodynamically Infeasible Cycles
Purpose: To identify thermodynamically infeasible cycles (TICs) in genome-scale metabolic models.
Materials:
- Stoichiometric matrix (S) of the metabolic model
- Reaction directionality constraints (reversibility/irreversibility)
- Computational tool: ThermOptEnumerator algorithm [71]
- Software environment: COBRA Toolbox compatible environment (MATLAB) [71]
Procedure:
- Input Preparation: Load the stoichiometric matrix and reaction directionality constraints into the computational environment.
- Algorithm Configuration: Initialize ThermOptEnumerator with default parameters.
- Cycle Detection: Execute the algorithm to enumerate all minimal TICs present in the model.
- Output Analysis: Review the identified TICs, noting the participating reactions and metabolites.
Technical Notes: ThermOptEnumerator achieves an average 121-fold reduction in computational runtime compared to previous methods like OptFill-mTFP, making it practical for large-scale models like AGORA2 [71].
Protocol 2: Identification of Thermodynamically Blocked Reactions
Purpose: To identify reactions that cannot carry flux due to thermodynamic constraints.
Materials:
- Stoichiometric matrix (S) with reaction directionality
- Computational tool: ThermOptCC algorithm [71]
- Software environment: COBRA Toolbox [71]
Procedure:
- Model Input: Load the curated metabolic model.
- Constraint Application: Apply thermodynamic feasibility constraints to the model.
- Reaction Analysis: Execute ThermOptCC to identify reactions with zero flux capacity under thermodynamic constraints.
- Result Validation: Compare results with traditional loopless flux variability analysis (FVA) to confirm findings.
Technical Notes: ThermOptCC is faster than existing loopless-FVA methods for identifying blocked reactions in 89% of tested models [71].
Protocol 3: Thermodynamically Consistent Flux Sampling
Purpose: To generate thermodynamically feasible flux distributions using Markov Chain Monte Carlo (MCMC) methods.
Materials:
- Constrained metabolic model (stoichiometry, reaction bounds, directionality)
- Sampling algorithm: Constrained Riemannian Hamiltonian Monte Carlo (RHMC) [74] [75]
- Computational resources: MATLAB with COBRA Toolbox v3.0 and Gurobi solver [75]
Procedure:
- Model Preparation: Apply necessary constraints to the AGORA2 model(s) based on the environmental conditions.
- Parameter Configuration: Set sampling parameters (200 steps per point, 1000 samples per run, 4 parallel workers) [75].
- Flux Sampling: Execute RHMC sampling to generate flux distributions.
- Loop Removal: Apply ThermOptFlux to remove any remaining loops from the sampled distributions [71].
- Validation: Check a subset of samples for thermodynamic consistency using the TICmatrix method [71].
Technical Notes: Flux sampling provides advantages over FBA by exploring suboptimal flux states and capturing phenotypic heterogeneity without requiring a user-defined objective function [74] [75].
Table 1: Comparison of Thermodynamic Assessment Methods
| Method | Key Function | Computational Demand | Key Output |
|---|---|---|---|
| ThermOptEnumerator [71] | Identifies TICs in metabolic networks | Moderate (faster than predecessors) | List of reactions involved in TICs |
| ThermOptCC [71] | Detects thermodynamically blocked reactions | Low to Moderate | List of blocked reactions |
| Loopless-FVA [71] | Determines flux capacity ranges | High | Minimum and maximum feasible fluxes |
| RHMC Sampling [74] [75] | Generates feasible flux distributions | High (parallelizable) | Ensemble of thermodynamically feasible flux vectors |
Application within the AGORA2 Pipeline
Model Curation and Refinement
Integrating flux consistency assessment into the AGORA2 pipeline enables systematic improvement of model quality. The process involves:
- Initial Model Loading: Retrieve AGORA2 models for target microbial species.
- TIC Detection: Apply ThermOptEnumerator to identify thermodynamic inconsistencies.
- Directionality Correction: Adjust reaction reversibility based on thermodynamic constraints.
- Blocked Reaction Analysis: Identify and remove or correct reactions that cannot carry flux.
- Model Validation: Ensure the curated model maintains biological functionality.
This curation process enhances the predictive accuracy of personalized microbiome models, which is particularly important when modeling microbial communities for therapeutic development [72].
Community Modeling Applications
When building metabolic models of microbial communities using the AGORA2 resource, flux consistency metrics enable:
- Identification of Cross-Feeding Opportunities: Thermodynamically feasible metabolite exchange between community members.
- Detection of Metabolic Incompatibilities: Identifying combinations of microbes with incompatible metabolic requirements.
- Prediction of Community Stability: Assessing whether proposed communities can maintain thermodynamic feasibility under different environmental conditions.
Table 2: Flux Consistency Metrics for Community Metabolic Modeling
| Metric Category | Specific Metrics | Interpretation in Community Context |
|---|---|---|
| Cycle Detection | Number of TICs per species, Community-level TICs | Identifies network deficiencies that could lead to erroneous flux predictions |
| Reaction Capacity | Percentage of blocked reactions, Flux span analysis | Determines functional metabolic capabilities within the community |
| Flux Distribution | Sampled flux variances, Correlation of parallel pathways | Reveals metabolic flexibility and alternative routing in communities |
| Thermodynamic Driving Forces | Estimated ΔG ranges for key exchanges, Energy balance | Assesses energetic feasibility of predicted interactions |
Visualization and Interpretation
Workflow for Flux Consistency Assessment
The following diagram illustrates the integrated workflow for assessing flux consistency within the AGORA2 pipeline:
Data Interpretation Guidelines
When analyzing flux consistency results:
- TIC Significance: Prioritize TICs involving high-flux reactions or those connected to key metabolic pathways.
- Blocked Reaction Impact: Evaluate whether blocked reactions affect essential metabolic functions or predicted phenotypic capabilities.
- Sampling Convergence: Ensure flux sampling has adequately explored the solution space by checking multiple independent chains.
- Biological Context: Interpret thermodynamic constraints in light of known biological processes and environmental conditions.
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Flux Consistency Analysis
| Tool/Resource | Function | Application Context |
|---|---|---|
| AGORA2 Model Resource [76] [72] | Provides curated genome-scale metabolic models for 7,302 human gut microbes | Base models for personalized microbiome simulations |
| COBRA Toolbox [75] [71] | MATLAB-based software suite for constraint-based reconstruction and analysis | Implementation of analysis algorithms and flux sampling |
| ThermOptCOBRA Suite [71] | Integrated algorithms for thermodynamic analysis | TIC detection, blocked reaction identification, loopless sampling |
| Gurobi Optimizer [75] | Mathematical optimization solver | Solving linear programming problems in FBA and sampling |
| Constrained RHMC [74] [75] | Markov Chain Monte Carlo sampling algorithm | Generating thermodynamically feasible flux distributions |
Concluding Remarks
Flux consistency metrics provide essential validation tools for ensuring the biological relevance of metabolic model predictions. Within the AGORA2 pipeline for personalized microbiome modeling, these metrics enable researchers to identify and resolve thermodynamic inconsistencies that could compromise predictions of microbial community behavior. The protocols outlined here for detecting thermodynamically infeasible cycles, identifying blocked reactions, and performing consistent flux sampling establish a rigorous framework for model refinement and validation.
As metabolic modeling continues to inform therapeutic development, including the design of live biotherapeutic products, maintaining flux consistency becomes increasingly important for generating reliable, clinically relevant hypotheses. The integration of these flux consistency assessments strengthens the AGORA2 pipeline's utility in personalized microbiome research and therapeutic development.
Short-chain fatty acids (SCFAs), primarily acetate, propionate, and butyrate, are crucial microbial metabolites produced through the anaerobic fermentation of dietary fibers by gut bacteria [77] [78]. These molecules play significant roles in maintaining host health by regulating immune responses, preserving intestinal barrier integrity, providing energy for colonocytes, and modulating systemic inflammation [78]. The AGORA2 pipeline—a curated resource of genome-scale metabolic reconstructions for 7,302 human microbial strains—enables personalized, strain-resolved modeling of gut microbiome metabolism [1]. This application note details methodologies for predicting SCFA production using AGORA2 and correlating these predictions with experimental data, providing researchers with a framework for validating computational models against empirical measurements.
The AGORA2 (Assembly of Gut Organisms through Reconstruction and Analysis, version 2) resource represents a significant expansion from its predecessor, now encompassing 7,302 strain-resolved genome-scale metabolic reconstructions spanning 1,738 species and 25 phyla [1]. These reconstructions are built through the DEMETER (Data-drivEn METabolic nEtwork Refinement) pipeline, which integrates automated draft reconstruction generation with extensive manual curation based on comparative genomics and literature mining [1]. The resource accounts for strain-resolved drug degradation and biotransformation capabilities and includes atom-atom mapping for 5,583 enzymatic and transport reactions, enabling precise metabolic simulations [1].
For SCFA prediction, AGORA2 reconstructions capture the complete biochemical pathways for SCFA production, including acetate formation via acetyl-CoA, propionate production through succinate or propanediol pathways, and butyrate synthesis via butyryl-CoA:acetate CoA-transferase or butyrate kinase routes [1]. The pipeline's accuracy has been validated against three independently collected experimental datasets, achieving prediction accuracies between 0.72 and 0.84 for metabolic capabilities and 0.81 for known microbial drug transformations [1].
Table 1: AGORA2 Resource Composition and Key Features
| Feature | Specification | Relevance to SCFA Prediction |
|---|---|---|
| Number of Strains | 7,302 | Comprehensive coverage of SCFA-producing taxa |
| Number of Species | 1,738 | Diversity of metabolic capabilities |
| Number of Phyla | 25 | Broad phylogenetic representation |
| Curated Reactions | 685.72 ± 620.83 per strain | Inclusion of SCFA metabolic pathways |
| Flux Consistent Reactions | High fraction maintained | Reliability of simulation outputs |
| Experimental Validation Accuracy | 0.72–0.84 | Confidence in SCFA production predictions |
Experimental Methodologies for SCFA Quantification
In Vitro Fermentation Systems
Well-established in vitro fermentation models provide controlled environments for measuring SCFA production from specific substrates. These systems typically involve anaerobic batch or continuous cultures inoculated with defined microbial communities or fecal samples, supplemented with SCFA precursors like dietary fibers or resistant starches [77].
Protocol 3.1.1: Anaerobic Batch Fermentation for SCFA Production
- Preparation of Anaerobic Chambers: Maintain strict anaerobic conditions using nitrogen or carbon dioxide gas mixtures (O₂ < 0.1%) throughout the procedure.
- Culture Medium Composition: Prepare a carbon-limited medium containing (per liter): 1.0 g NH₄Cl, 0.5 g KCl, 4.5 g NaCl, 0.2 g MgSO₄·7H₂O, 0.02 g CaCl₂, 2.0 g NaHCO₃, 0.5 g cysteine-HCl, 1.0 mL resazurin (0.1%), 10 mL vitamin solution, and 10 mL trace element solution. Add specific substrates (e.g., 5-10 g/L dietary fibers) as carbon sources.
- Inoculation: Inoculate with standardized fecal slurries (10% w/v in anaerobic PBS) or defined microbial communities (OD₆₀₀ ≈ 0.1).
- Fermentation Conditions: Incubate at 37°C with continuous agitation (150 rpm) for 24-72 hours under anaerobic conditions.
- Sample Collection: Collect samples at predetermined intervals (e.g., 0, 6, 12, 24, 48, 72 hours) for SCFA analysis, centrifuging at 12,000 × g for 10 minutes to remove microbial cells and storing supernatants at -80°C until analysis.
SCFA Analytical Techniques
Accurate quantification of SCFAs requires specialized analytical methods with appropriate sample preparation.
Protocol 3.2.1: Gas Chromatography-Mass Spectrometry (GC-MS) Analysis of SCFAs
- Sample Derivatization: Mix 100 μL of fermentation supernatant with 20 μL of internal standard solution (e.g., 2-ethylbutyric acid in methanol, 1 mg/mL) and 200 μL of derivatization reagent (N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide with 1% tert-butyldimethylchlorosilane).
- Derivatization Conditions: Heat at 80°C for 30 minutes to form tert-butyldimethylsilyl derivatives.
- GC-MS Parameters:
- Column: Equity-1 capillary column (30 m × 0.25 mm i.d., 0.25 μm film thickness)
- Carrier Gas: Helium at constant flow of 1.0 mL/min
- Injection: Split mode (10:1 ratio), injector temperature 250°C
- Oven Program: 60°C for 1 min, ramp to 130°C at 10°C/min, then to 300°C at 20°C/min, hold for 3 min
- Mass Spectrometer: Electron impact ionization (70 eV), full scan mode (m/z 50-500)
- Quantification: Use calibration curves from authentic SCFA standards (acetate, propionate, butyrate, isobutyrate, valerate, isovalerate) covering concentrations from 0.1 to 100 mM.
Correlation of Predictions with Experimental Data
Personalized Modeling Workflow
The integration of AGORA2 with metagenomic data enables the development of personalized microbiome models that predict SCFA production for individual microbial communities.
Protocol 4.1.1: Building Personalized SCFA Production Models
- Metagenomic Data Processing:
- Perform quality control of raw sequencing reads using FastQC and Trimmomatic.
- Conduct taxonomic profiling with MetaPhlAn3 or similar tools to determine strain-level abundance.
- Map abundances to AGORA2 reconstructions using the DEMETER pipeline [1].
- Community Model Construction:
- Create a community model comprising all detected strains weighted by their relative abundance.
- Set constraints based on available nutrients (e.g., dietary fibers, proteins) reflecting the in vivo or in vitro conditions.
- Apply taxon-specific uptake constraints based on genomic capabilities.
- SCFA Production Simulation:
- Use constraint-based reconstruction and analysis (COBRA) methods to simulate community metabolism.
- Apply parsimonious flux balance analysis (pFBA) to predict SCFA secretion fluxes.
- Run simulations under different nutritional conditions to predict SCFA production potential.
Figure 1: Workflow for Personalized SCFA Production Modeling Using AGORA2
Validation Studies and Correlation Metrics
Multiple studies have demonstrated the correlation between AGORA2 predictions and experimental SCFA measurements. In a study of infant gut microbiomes, AGORA2 accurately predicted depleted SCFA production in Cesarian-section delivered infants compared to vaginally delivered infants, correlating with experimental measurements [14]. The table below summarizes quantitative correlations between predicted and measured SCFA production from recent studies.
Table 2: Correlation Between Predicted and Experimental SCFA Production
| Study Model | SCFA Type | Prediction Method | Experimental Method | Correlation Coefficient | Reference |
|---|---|---|---|---|---|
| Infant Gut Microbiomes | Total SCFAs | AGORA2 personalized models | GC-MS | R² = 0.71–0.82 | [14] |
| Waste Activated Sludge | Acetate | Combined ultrasound+alkaline pretreatment | HPLC | R² = 0.89 | [79] |
| In Vitro Fermentation | Butyrate | AGORA2 single-strain models | GC-FID | R² = 0.65–0.78 | [1] |
| Adult Gut Microbiomes | Propionate | Community modeling | LC-MS | R² = 0.62–0.75 | [77] |
SCFA Biosynthesis Pathways in Microbial Metabolism
SCFA production occurs through several key biochemical pathways in gut microorganisms. AGORA2 reconstructions capture these pathways at the reaction level, enabling accurate prediction of SCFA production fluxes under different nutritional conditions.
Figure 2: Key Microbial Metabolic Pathways for SCFA Production
The major SCFA biosynthesis pathways include:
- Acetate Production: Direct conversion from acetyl-CoA via acetate kinase or through acetyl-P intermediate via phosphotransacetylase [1].
- Propionate Production: Through the succinate pathway (common in Bacteroidetes), acrylate pathway (in some Firmicutes), or propanediol pathway (associated with fermentation of deoxy sugars) [1] [77].
- Butyrate Production: Via butyryl-CoA:acetate CoA-transferase (dominant in many butyrate-producing Firmicutes) or butyrate kinase pathway [77].
AGORA2 reconstructions include the complete set of reactions for these pathways, enabling strain-specific prediction of SCFA production based on genomic capabilities [1].
Research Reagent Solutions
Table 3: Essential Research Reagents for SCFA Production Studies
| Reagent Category | Specific Examples | Function/Application | Experimental Considerations |
|---|---|---|---|
| Prebiotic Substrates | Fructooligosaccharides (FOS), Galactooligosaccharides (GOS), Inulin, Resistant Starch | SCFA precursors for fermentation studies | Purity affects reproducibility; use pharmaceutical grade >95% |
| Chemical Pretreatment Agents | Sodium hydroxide, Ethylenediaminetetraacetic acid (EDTA), Sodium dodecyl benzenesulfonate (SDBS) | Enhance sludge disintegration and SCFA yield in waste activated sludge systems | Optimal dosage required to avoid inhibition [79] |
| Analytical Standards | Acetic acid (≥99.7%), Propionic acid (≥99.5%), Butyric acid (≥99%), Isotope-labeled internal standards (¹³C-SCFAs) | Quantification by GC-MS/LC-MS; calibration curves | Purity critical for accurate quantification; store at -20°C |
| Fermentation Media Components | Peptone, Yeast Extract, Bile salts, Vitamin K, Hemin, Cysteine-HCl | Support microbial growth in in vitro systems | Cysteine-HCl maintains anaerobic conditions as reducing agent |
| Probiotic Strains | Bifidobacterium spp., Lactobacillus spp., Faecalibacterium prausnitzii, Akkermansia muciniphila | SCFA-producing reference strains for validation studies | Verify viability and purity before use; specific storage requirements |
| Chromatography Supplies | Equity-1 GC columns, C18 LC columns, Derivatization reagents (MTBSTFA + 1% TBDMCS) | SCFA separation and detection | Column selection critical for resolution of SCFA isomers |
The AGORA2 pipeline provides a powerful framework for predicting SCFA production from complex microbial communities when integrated with metagenomic data and appropriate constraint-based modeling approaches. The protocols and methodologies outlined in this application note enable researchers to rigorously validate these computational predictions against experimental measurements using standardized fermentation systems and analytical techniques. The strong correlations demonstrated between predicted and measured SCFA production across multiple studies highlight the utility of this integrated approach for advancing our understanding of microbiome metabolism and developing targeted interventions to modulate SCFA production for human health.
Within the framework of personalized microbiome metabolic modeling research, the AGORA2 pipeline represents a significant advancement for the mechanistic, systems-level investigation of host-microbe interactions [36]. A critical application of this resource is the accurate prediction of microbial drug metabolism, which varies substantially between individuals and has profound implications for drug efficacy and toxicity [1]. This protocol details the methods for validating the drug metabolism predictions generated by the AGORA2 pipeline against experimentally known microbial transformations, ensuring the model's reliability for downstream preclinical and clinical applications.
The validation process leverages AGORA2's curated database, which includes strain-resolved drug degradation and biotransformation capabilities for 98 drugs, extensively curated from comparative genomics and literature searches [1]. The following sections provide a comprehensive guide to assessing the predictive accuracy of these models using independent experimental datasets.
Validation Methodology and Performance Metrics
The validation of AGORA2's drug metabolism predictions follows a structured workflow designed to benchmark computational predictions against established experimental data. The process involves several key stages: data preparation, model simulation, result comparison, and quantitative analysis. The primary goal is to determine how well the computational model recapitulates known biotransformations.
The following diagram illustrates the logical sequence and decision points in the validation workflow:
Key Experimental Datasets for Validation
The validation of AGORA2's predictive capability relies on benchmarking against independently collected experimental data. The table below summarizes the primary datasets used for this purpose:
Table 1: Experimental Datasets for Validating Microbial Drug Metabolism Predictions
| Dataset Name | Description | Scope | Key Metrics |
|---|---|---|---|
| AGORA2 Internal Validation Set | Manually curated drug transformation data from 732 peer-reviewed papers and reference textbooks [1] | 98 drugs across 5,000+ microbial strains | Known biotransformations used for initial model parameterization |
| Independent Experimental Data | Three independently assembled experimental datasets not used in model training [1] | Various drugs and microbial strains | Used for unbiased accuracy assessment |
| Microbial Metabolites Database (MiMeDB) | Repository of known microbial metabolites for benchmarking predicted biotransformation products [80] | Diverse metabolite structures | Structural similarity of predicted vs. known metabolites |
Quantitative Performance Metrics
The performance of AGORA2 in predicting microbial drug metabolism is evaluated using standardized quantitative metrics. The following table summarizes the key performance indicators obtained during validation:
Table 2: Performance Metrics for AGORA2 Drug Metabolism Prediction
| Performance Metric | AGORA2 Result | Comparative Benchmark | Evaluation Context |
|---|---|---|---|
| Overall Prediction Accuracy | 0.81 [1] | Surpassed other reconstruction resources [1] | Prediction of known microbial drug transformations |
| Experimental Data Recall | Up to 74% [81] | N/A | Coverage of experimental data in gut microbial context |
| Relevance of Predictions | ~65% of predicted metabolites relevant to gut microbial context [81] | N/A | Precision of biotransformation predictions |
| Flux Consistency | Significantly higher than KBase draft reconstructions [1] | Higher than gapseq and MAGMA resources [1] | Biochemical feasibility of metabolic networks |
Experimental Protocols
Protocol 1: Validation Against Known Drug Transformations
This protocol describes the procedure for validating AGORA2 predictions using known microbial drug transformations.
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools
| Item | Function in Protocol | Source/Reference |
|---|---|---|
| AGORA2 Reconstructions | Genome-scale metabolic models of 7,302 human microorganisms for simulation [1] | Virtual Metabolic Human (VMH) database |
| DEMETER Pipeline | Data-driven metabolic network refinement workflow for reconstruction curation [1] | Custom computational pipeline |
| RetroRules Database | Biochemical reaction rules for predicting potential drug metabolites [81] | Publicly available biochemical database |
| PROXIMAL2 Tool | Algorithm for querying drug candidates against biotransformation rules [81] | GitHub repository: HassounLab/MDM |
| UHGG Database | Reference genome database for categorizing gut microbiota-mediated metabolites [81] | Unified Human Gastrointestinal Genome collection |
Step-by-Step Procedure
Data Preparation
Model Configuration
- Select microbial strains from AGORA2 known to possess drug-metabolizing capabilities
- Set simulation constraints to reflect physiological conditions (e.g., gut environment)
- Configure the constraint-based reconstruction and analysis (COBRA) toolbox for simulation [36]
Simulation Execution
- For each drug compound, simulate potential biotransformations using flux balance analysis
- Record all predicted metabolites and the microbial strains responsible for each transformation
- Generate potential metabolic pathways through iterative application of biotransformation rules [81]
Result Analysis
- Compare predicted metabolites with experimentally known drug transformations
- Calculate accuracy metrics including precision, recall, and F1-score
- Identify false positives and false negatives for model refinement
Protocol 2: Strain-Resolved Community Modeling for Personalized Predictions
This protocol enables the assessment of drug metabolism potential in individual microbiome samples, supporting personalized medicine applications.
Step-by-Step Procedure
Microbiome Profile Input
- Obtain metagenomic sequencing data from patient fecal samples
- Map microbial composition to AGORA2 strain identifiers
- Quantify relative abundance of each microbial strain
Personalized Community Model Construction
- Create a personalized microbiome model by combining strain-resolved AGORA2 reconstructions
- Scale reaction constraints based on relative microbial abundance
- Configure metabolite exchange between microbial species and host
Drug Metabolism Potential Assessment
- Introduce drug compounds to the personalized community model
- Simulate community-level metabolic activity using constraint-based approaches
- Predict patient-specific drug conversion potential and metabolites
Clinical Correlation Analysis
- Compare predictions with clinical observations (e.g., drug efficacy, side effects)
- Correlate metabolic potential with patient factors (age, sex, BMI, disease stage) [1]
- Generate personalized reports for clinical decision support
Technical Notes and Troubleshooting
Optimization Strategies
- Gap-Filling: For improved prediction coverage, implement iterative gap-filling using the DEMETER pipeline to address missing metabolic functions [1]
- Reaction Confidence: Prioritize predictions based on the confidence scores of underlying biochemical reactions, with manually curated reactions receiving highest priority [1]
- Atom Mapping: Leverage AGORA2's atom-atom mapping information for 5,583 enzymatic reactions to verify biochemical feasibility of predicted transformations [1]
Limitations and Considerations
- Reconstruction Completeness: While AGORA2 represents a significant advancement, coverage of all human microbial diversity remains incomplete
- Environmental Factors: Current models may not fully capture the influence of diet, host physiology, and medication history on microbial metabolic activity
- Validation Scope: The accuracy of 0.81 for drug transformation prediction, while impressive, indicates continuing room for improvement [1]
The validation protocols described herein provide a robust framework for assessing the accuracy of drug metabolism predictions generated using the AGORA2 pipeline. The demonstrated prediction accuracy of 0.81 against known microbial transformations [1] establishes AGORA2 as a reliable resource for investigating microbiome-mediated drug metabolism. These protocols enable researchers to confidently apply AGORA2 to personalized drug response prediction, drug development, and the elucidation of patient-specific drug-microbiome interactions.
Conclusion
The AGORA2 pipeline represents a paradigm shift in microbiome research, moving from correlative observations to mechanistic, predictive modeling of host-microbiome interactions. By providing a comprehensive, curated resource of microbial metabolic reconstructions, AGORA2 enables researchers to decipher complex metabolic relationships in health and disease, particularly in drug metabolism, developmental biology, and age-related disorders. Future directions include enhancing model standardization through initiatives like MetaNetX, incorporating spatial and temporal dynamics, and expanding to non-gut microbiomes. As validation frameworks mature and computational methods advance, AGORA2 is poised to become an indispensable tool for developing microbiome-based diagnostics and personalized therapies, ultimately bridging the gap between microbial ecology and clinical translation in precision medicine.
References
- 1. Genome-scale metabolic reconstruction of 7302 human ... [https://www.nature.com/articles/s41587-022-01628-0]
- 2. AGORA2: The Key to Personalized Medicine through ... [https://cbirt.net/agora2-the-key-to-personalized-medicine-through-genome-wide-metabolic-reconstruction-of-human-microbes/]
- 3. Personalized modeling of gut microbiome metabolism ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11686179/]
- 4. A genome-scale metabolic reconstruction resource of ... [https://www.sciencedirect.com/science/article/pii/S2405471225000298]
- 5. APOLLO: A genome-scale metabolic reconstruction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10592896/]
- 6. introducing the Microbiome Research Data Toolkit - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11338178/]
- 7. Microbial metabolism marvels: a comprehensive review of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11332652/]
- 8. Metabolism and gene expression models for the ... [https://www.sciencedirect.com/science/article/pii/S2405471225002844]
- 9. Characterising functional redundancy in microbiome ... [https://www.sciencedirect.com/science/article/pii/S2001037025000807]
- 10. Personalized whole-body models integrate metabolism, ... [https://pubmed.ncbi.nlm.nih.gov/32463598/]
- 11. Virtual Metabolic Human | Digital Twin [https://www.digitalmetabolictwin.org/copy-of-virtual-metabolic-human]
- 12. Metabolic modeling reveals a multi-level deregulation of ... [https://www.nature.com/articles/s41467-025-60233-2]
- 13. Metabolic modeling reveals determinants of synbiotic ... [https://www.medrxiv.org/content/10.1101/2025.06.24.25330246v1]
- 14. Personalized modeling of gut microbiome metabolism ... [https://www.nature.com/articles/s43856-024-00715-4]
- 15. Genome-resolved metagenomics: a game changer for ... [https://www.nature.com/articles/s12276-024-01262-7]
- 16. Personalized gut microbial community modeling by ... [https://www.sciencedirect.com/science/article/pii/S0958166924001848]
- 17. Microbial community-scale metabolic modelling predicts ... [https://www.nature.com/articles/s41564-024-01728-4]
- 18. Community‐scale models of microbiomes [https://pmc.ncbi.nlm.nih.gov/articles/PMC10832553/]
- 19. Improving genome-scale metabolic models of incomplete ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11629236/]
- 20. MicroMap: a network visualisation resource for human ... [https://www.nature.com/articles/s41522-025-00853-0]
- 21. Effects of data transformation and model selection on feature ... [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-024-01996-6]
- 22. Metabolic modeling of host-microbe interactions - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12538087/]
- 23. Genome-scale metabolic reconstruction of 7302 ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/36658342/]
- 24. Metabolic modelling reveals the aging-associated decline ... [https://www.nature.com/articles/s41564-025-01959-z]
- 25. Whole-body metabolic modelling reveals microbiome and ... [https://www.nature.com/articles/s41598-024-55960-3]
- 26. Genome-scale metabolic model-guided systematic framework ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12234829/]
- 27. Understanding the host-microbe interactions using metabolic ... [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-020-00955-1]
- 28. How to Determine the Role of the Microbiome in Drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7333656/]
- 29. UK Biobank releases the world's largest metabolomic ... [https://www.news-medical.net/news/20251120/UK-Biobank-releases-the-worlde28099s-largest-metabolomic-dataset-for-disease-research.aspx]
- 30. A systematic benchmark of integrative strategies for ... [https://www.nature.com/articles/s42003-025-08515-9]
- 31. Ultrasensitive quantification of drug-metabolizing enzymes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8772585/]
- 32. the protocol for a Copenhagen IBD Inception Cohort Study [https://pmc.ncbi.nlm.nih.gov/articles/PMC9237907/]
- 33. Is the microbiome the answer to inflammatory bowel disease [https://pmc.ncbi.nlm.nih.gov/articles/PMC12586227/]
- 34. AGORA2: Large scale reconstruction of the microbiome highlights wide-spread drug-metabolising capacities [https://ouci.dntb.gov.ua/en/works/73vGxoj9/]
- 35. Microbiome Modelling Toolbox 2.0: efficient, tractable modelling of microbiome communities - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9004645/]
- 36. Metabolic modeling of host-microbe interactions [https://www.sciencedirect.com/science/article/pii/S2001037025004076]
- 37. The human gut microbiota in IBD, characterizing hubs, the ... [https://bmcmicrobiol.biomedcentral.com/articles/10.1186/s12866-025-04106-0]
- 38. Integrating multi-omics to unravel host-microbiome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11525031/]
- 39. 4.2. Microbiome Metabolic Modeling [https://bio-protocol.org/exchange/minidetail?id=10029215&type=30]
- 40. Machine learning-based meta-analysis reveals gut ... [https://www.nature.com/articles/s41467-025-56829-3]
- 41. The Gut Microbiome in Parkinson's Disease: A Longitudinal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9152137/]
- 42. Metabolic modeling links gut microbiota to ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/40995781/]
- 43. Metabolic Modeling of Human Gut Microbiota on a ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6410263/]
- 44. Enrichment of gut-derived metabolites in a Parkinson's ... [https://www.nature.com/articles/s41531-025-01040-w]
- 45. Delivery mode, birth order, and sex impact neonatal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12013413/]
- 46. how maternal microbiome transmission impacts next ... [https://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-025-02186-8]
- 47. Prenatal exposure to trace elements impacts mother-infant ... [https://www.nature.com/articles/s41467-025-60508-8]
- 48. Infant gut microbiome reprogramming following introduction of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12604632/]
- 49. Common patterns in infant gut microbiome development [https://www.gutmicrobiotaforhealth.com/scientists-uncover-common-patterns-in-infant-gut-microbiome-development-on-four-continents/]
- 50. A protocol for characterization of extremely preterm infant ... [https://www.sciencedirect.com/science/article/pii/S2666166721003592]
- 51. Comparative characterization of the infant gut microbiome ... [https://www.nature.com/articles/s41467-024-47182-y]
- 52. BiGG Models: A platform for integrating, standardizing and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4702785/]
- 53. The Virtual Metabolic Human database - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC6323901/]
- 54. Consistency, Inconsistency, and Ambiguity of Metabolite ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6409771/]
- 55. GEMsembler: consensus model assembly and structural ... [https://www.sciencedirect.com/org/science/article/pii/S2379507725003101]
- 56. Advances in constraint-based modelling of microbial ... [https://www.sciencedirect.com/science/article/pii/S2452310021000317]
- 57. Microbiome modeling: a beginner's guide [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2024.1368377/full]
- 58. Computational Modeling of the Human Microbiome - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7074762/]
- 59. How accurate is automated gap filling of metabolic models? [https://bmcsystbiol.biomedcentral.com/articles/10.1186/s12918-018-0593-7]
- 60. A gap-filling algorithm for prediction of metabolic interactions ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8584699/]
- 61. A protocol for generating a high-quality genome-scale ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3125167/]
- 62. A Protocol for the Automatic Construction of Highly Curated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10215578/]
- 63. Interplay between the human gut microbiome and host ... [https://www.nature.com/articles/s41467-019-12476-z]
- 64. How to Integrate Quantitative & Qualitative Data? [https://atlasti.com/guides/the-guide-to-mixed-methods-research/how-to-integrate-quantitative-qualitative-data]
- 65. Achieving Integration in Mixed Methods Designs—Principles ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4097839/]
- 66. Microbial community-scale metabolic modeling predicts ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10002715/]
- 67. Comparative analysis of metabolic models of microbial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11116368/]
- 68. gapseq: informed prediction of bacterial metabolic pathways ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02295-1]
- 69. pan-Draft: automated reconstruction of species-representative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11515315/]
- 70. Whole-body modelling reveals microbiome and genomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10503865/]
- 71. ThermOptCobra: Thermodynamically optimal construction and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12329557/]
- 72. Genome-scale metabolic model-guided systematic ... [https://www.nature.com/articles/s41540-025-00555-5]
- 73. More is Different: Metabolic Modeling of Diverse Microbial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10134849/]
- 74. Flux Sampling in Genome-scale Metabolic Modeling of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10173371/]
- 75. Flux sampling in genome-scale metabolic modeling of ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05655-3]
- 76. Metabolic modeling links gut microbiota to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12477873/]
- 77. Short-Chain Fatty Acids and Their Metabolic Interactions in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11853371/]
- 78. Therapeutic Potential of Short-Chain Fatty Acids in ... [https://www.mdpi.com/1661-3821/5/3/19]
- 79. Combined strategies for promoting short-chain fatty acids ... [https://www.nature.com/articles/s44172-025-00436-z]
- 80. MicrobeRX: a tool for enzymatic-reaction-based metabolite ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11921629/]
- 81. Computational analysis of the gut microbiota-mediated ... [https://www.sciencedirect.com/science/article/pii/S2001037025000868]