ATP/ADP and NADPH/NADP+ Ratios: A Comparative Guide for Biomedical Research and Drug Development
This article provides a comprehensive analysis of ATP/ADP and NADPH/NADP+ ratios as critical indicators of cellular bioenergetics and redox state.
ATP/ADP and NADPH/NADP+ Ratios: A Comparative Guide for Biomedical Research and Drug Development
Abstract
This article provides a comprehensive analysis of ATP/ADP and NADPH/NADP+ ratios as critical indicators of cellular bioenergetics and redox state. We explore foundational concepts of these nucleotide ratios across diverse biological systems, from human clinical samples to plant and bacterial models. Methodological approaches for accurate measurement are reviewed, highlighting common pitfalls in interpretation. The content details how these ratios serve as sensitive biomarkers for pre-analytical sample quality, drug metabolism phenotypes, and pathological conditions including mitochondrial diseases and cancer. For researchers and drug development professionals, this synthesis offers a framework for utilizing these metabolic parameters in troubleshooting experimental systems, validating disease models, and informing therapeutic strategies that target metabolic pathways.
Fundamental Principles of Cellular Energy and Redox Ratios
Defining the Core Adenylate and Nicotinamide Cofactor Systems
Cellular metabolism relies on the precise interplay of two core nucleotide cofactor systems: the adenylate system and the nicotinamide system. The adenylate system, centered on ATP, ADP, and AMP, serves as the primary currency for cellular energy transfer [1]. The enzyme adenylate kinase (ADK) plays a crucial role in maintaining the balance between these nucleotides through the reversible reaction 2ADP ATP + AMP [1]. Meanwhile, the nicotinamide system, comprising NAD(P)+/NAD(P)H, functions as the principal carrier of reducing equivalents for redox metabolism and biosynthetic reactions [2] [3]. These systems are not isolated; their ratios interlink to form a sophisticated network for monitoring cellular energy status and responding to metabolic demands.
The Adenylate Cofactor System
Core Components and Homeostatic Role
Adenylate kinase (ADK) is a phosphotransferase enzyme essential for cellular energy homeostasis. It catalyzes the reversible transfer of a phosphoryl group between adenosine phosphates, constantly monitoring and adjusting intracellular levels of ATP, ADP, and AMP [1]. This reaction has an equilibrium constant close to 1, meaning the Gibbs free energy change (ΔG°) is approximately zero [1]. In resting muscle tissue from various vertebrates and invertebrates, the concentration of ATP is typically 7-10 times that of ADP and usually more than 100 times that of AMP [1]. This steep concentration gradient reflects the high-energy status of cells under normal conditions.
The metabolic state of the cell is encoded in the adenylate energy charge, which is calculated from the concentrations of ATP, ADP, and AMP. Adenylate kinase plays a decoding function in this system by generating AMP as a key signaling molecule when energy levels decline [1]. This generated AMP can then stimulate various AMP-dependent receptors and pathways, including glycolytic enzymes, K-ATP channels, and 5' AMP-activated protein kinase (AMPK) [1].
Structural Dynamics and Catalytic Mechanism
Adenylate kinase undergoes significant conformational changes during its catalytic cycle, transitioning between open, closed, and locally unfolded states [4] [5]. The enzyme features two mobile domains known as the LID (binding ATP) and NMP (binding AMP), which undergo open-close motions relative to a static CORE domain [4]. These structural changes are essential for catalysis, as phosphoryl transfer occurs only after closure of these domains, which excludes water molecules and brings substrates into proximity for reaction [1].
Magnesium cofactor plays a critical role in the catalytic mechanism, activating two distinct molecular events: phosphoryl transfer (>10⁵-fold rate enhancement) and lid-opening (10³-fold enhancement) [5]. The catalytic mechanism involves a highly conserved arginine residue (Arg88 in E. coli ADK) that binds to the phosphate group of AMP [1]. Mutation of this residue (R88G) results in a 99% loss of catalytic activity, highlighting its essential role in phosphoryl transfer [1]. A network of positive, conserved residues stabilizes the buildup of negative charge on the phosphoryl group during transfer [1].
Table 1: Key Features of Adenylate Kinase Isozymes
| Isoform | Localization | Tissue Specificity | Km for AMP | NTP Preference |
|---|---|---|---|---|
| ADK1 | Cytosol | Ubiquitous | High (weak binding) | ATP |
| ADK7 | Cytosol | Skeletal muscle | Very low (tight binding) | ATP |
| ADK8 | Cytosol | Not in skeletal muscle | Very low (tight binding) | ATP |
| Mitochondrial GTP:AMP phosphotransferase | Mitochondria | Multiple tissues | N/A | GTP, ITP |
Metabolic Monitoring and Shuttling
Adenylate kinase functions as a sensitive monitor of cellular energy status by continually monitoring and altering the levels of ATP, ADP, and AMP under different metabolic stresses [1]. Through the adenylate kinase shuttle, the enzyme channels high-energy phosphoryls between mitochondrial and myofibrillar compartments, making ATP available to sites of high energy consumption and removing generated AMP [1]. This process involves sequential phosphotransfer relays along collections of ADK molecules, resulting in propagation of phosphoryl groups without apparent global changes in metabolite concentrations [1].
The Nicotinamide Cofactor System
Core Components and Redox Functions
The nicotinamide cofactor system consists of two primary redox pairs: NAD+/NADH and NADP+/NADPH. These cofactors differ primarily by a single phosphate group on the adenosine ribose moiety of NADP+, yet serve distinct metabolic roles [3]. NAD+/NADH primarily participates in catabolic reactions, accepting electrons during substrate oxidation to generate ATP, while NADP+/NADPH mainly serves as an electron donor for reductive biosynthesis and antioxidant defence [2].
The redox function of these cofactors resides in the nicotinamide ring, which can accept or donate a hydride ion (a proton with two electrons) [6]. In astrocytes, basal specific contents of NADPx (NADP+ + NADPH) and NADx (NAD+ + NADH) are approximately 0.64 ± 0.09 nmol/mg protein and 2.91 ± 0.40 nmol/mg protein, respectively, with reduced co-substrates accounting for 37 ± 14% and 28 ± 10% of the total pools [2].
Interconversion and Regulation
NAD kinase (NADK) catalyzes the phosphorylation of NAD+ to NADP+, representing the sole enzymatic pathway for NADP+ synthesis [2]. In cultured astrocytes, NADK has a specific vmax activity of approximately 1 nmol/(min·mg protein) with KM values of 1.30 ± 0.19 mM for NAD+ and 2.71 ± 0.18 mM for ATP [2]. During oxidative stress, astrocytes demonstrate a rapid doubling of their NADPx pool at the expense of the NADx pool through NADK-mediated phosphorylation of NAD+ [2]. This mechanism provides a crucial regulatory link that enhances antioxidative capacity during metabolic challenge.
Table 2: Comparison of Natural and Biomimetic Nicotinamide Cofactors
| Cofactor | Redox Potential (E°) | Primary Metabolic Role | Stability | Cost |
|---|---|---|---|---|
| NAD+/NADH | -320 mV [7] | Catabolic reactions, energy production | Moderate [8] | High [8] |
| NADP+/NADPH | -320 mV [7] | Anabolic reactions, antioxidant defence | Moderate [8] | High [8] |
| NMN+/NMNH | Similar to NADP+ [7] | Engineered orthogonal metabolism | Varies | Lower [7] |
| BNA+/BNAH | Varies with substituents [6] | Biocatalysis with specific enzymes | Moderate to low [8] | Low [8] |
Biomimetic Cofactors and Engineering Applications
Synthetic nicotinamide cofactor biomimetics (NCBs) have emerged as cost-effective alternatives to natural cofactors for biocatalytic applications [8]. These include both fully synthetic molecules (retaining only the nicotinamide-related group) and semi-synthetic analogs (structurally closer to natural cofactors) [8]. A significant engineering achievement involves the computational redesign of glucose dehydrogenase from Bacillus subtilis (Bs GDH) to utilize NMN+ instead of NADP+ [7]. The engineered triple mutant (I195R-A93K-Y39Q) exhibits a 1,000-fold increase in catalytic efficiency toward NMN+ and a 10⁷-fold specificity switch from NADP+ to NMN+ [7].
These engineered cofactor systems enable the creation of orthogonal metabolic pathways that operate in parallel to native metabolism without cross-talk [7]. Such systems have demonstrated the ability to support diverse redox chemistries in vitro with high total turnover numbers (~39,000) and to channel reducing power in E. coli whole cells specifically from glucose to target compounds like the pharmaceutical intermediate levodione [7].
Experimental Analysis of Cofactor Systems
Quantitative Assessment of Cofactor Pools
Enzymatic cycling assays provide a sensitive and specific method for quantifying the six redox co-substrates (GSH, GSSG, NAD+, NADH, NADP+, NADPH) in biological samples [2]. These assays exploit the substrate specificity of enzymes like glutathione reductase, lactate dehydrogenase, and glucose-6-phosphate dehydrogenase to measure oxidized and reduced forms of each redox pair.
For accurate determination of NADPH and NADP+ levels in astrocyte cultures, the following protocol can be employed [2]:
- Cell Extraction: Rapidly extract metabolites from cultured astrocytes using acidic conditions (sulfosalicylic acid) to preserve redox states.
- Neutralization: Adjust pH to 7.0-7.5 for enzymatic assays.
- Specific Detection:
- For NADPH: Use glutathione reductase and DTNB in the presence of GSSG.
- For NADP+: Convert to NADPH using glucose-6-phosphate dehydrogenase, then measure as above.
- Quantification: Compare to standard curves generated with known concentrations of authentic standards.
This approach revealed that in untreated astrocyte cultures, the basal specific content of NADPx is 0.64 ± 0.09 nmol/mg protein, with the reduced form (NADPH) accounting for 37 ± 14% of the total pool [2].
Crystallographic Analysis of Conformational Dynamics
X-ray crystallography has provided detailed insights into the conformational changes adenylate kinase undergoes during catalysis [4] [5]. The following methodology outlines the approach for determining structures of differently liganded states:
- Protein Purification: Express recombinant adenylate kinase in E. coli and purify using affinity and size-exclusion chromatography [4].
- Crystallization: Employ sitting-drop vapor diffusion with 20 mg/ml protein concentration. For ligand-bound structures, include substrates (AMP) or inhibitors (Ap5A) in the reservoir solution [4].
- Cryoprotection: Flash-cool crystals in liquid nitrogen with appropriate cryoprotectants.
- Data Collection: Collect diffraction data at synchrotron sources (e.g., Pohang Accelerator Laboratory beamline 7A at 100K) [4].
- Structure Determination: Process data with XDS, perform molecular replacement with PHENIX, and refine structures with Coot and PHENIX [4].
This approach has revealed that adenylate kinase samples multiple conformational states even in crystalline environments and that the presence of substrates shifts the equilibrium toward closed conformations [4].
Monitoring Cofactor Dynamics During Oxidative Stress
Experimental analysis of cofactor responses to oxidative stress involves:
- Induction of Oxidative Stress: Expose cultured astrocytes to 100 µM H₂O₂ in the presence of a pentose-phosphate pathway inhibitor (G6PDi-1) [2].
- Time-Course Sampling: Collect samples at multiple time points (e.g., 0, 5, 15, 30, 60 minutes) to track transient changes.
- Redox Pair Analysis: Simultaneously quantify all three redox pairs (GSx, NADx, NADPx) using enzymatic cycling assays.
- Inhibition Studies: Use specific inhibitors like thionicotinamide (precursor of the NADK inhibitor thio-NADP) to confirm mechanistic pathways [2].
This experimental paradigm demonstrated that oxidative stress triggers a rapid but transient oxidation of GSH to GSSG, accompanied by a doubling of the NADPx pool at the expense of NADx through NADK-mediated phosphorylation [2].
Comparative Visualization of Cofactor Systems
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for Cofactor Research
| Reagent/Chemical | Research Function | Example Application | Key Characteristics |
|---|---|---|---|
| Ap5A (P1,P5-di(adenosine-5')-pentaphosphate) | Transition-state analog inhibitor of adenylate kinase | Trapping enzyme in closed conformation for crystallography [4] | Mimics substrate structure but resistant to hydrolysis |
| Thionicotinamide | Precursor for NADK inhibitor thio-NADP | Investigating NADK role in oxidative stress response [2] | Competes with nicotinamide in NADP+ synthesis |
| G6PDi-1 | Glucose-6-phosphate dehydrogenase inhibitor | Blocking pentose phosphate pathway to study NADPH dynamics [2] | Specific inhibitor of primary NADPH regeneration pathway |
| BNAH (1-benzyl-1,4-dihydronicotinamide) | Synthetic nicotinamide cofactor biomimetic | Studying flavin-dependent oxidoreductases without NAD(P)H cross-talk [6] | Lower cost, modified redox properties compared to NADH |
| DTNB (5,5'-dithiobis-(2-nitrobenzoic acid)) | Thiol quantification reagent | Measuring glutathione levels in redox pair analysis [2] | Forms yellow-colored complex with thiol groups |
| Phenazine ethosulfate (PES) | Electron coupler in enzymatic assays | Enabling enzymatic cycling for sensitive cofactor detection [2] | Mediates electron transfer in detection systems |
NADH Reductive Stress as a Driver of Metabolic Reprogramming in Disease
For decades, the paradigm of oxidative stress has dominated the study of redox biology in disease pathogenesis. However, emerging evidence reveals that reductive stress—a shift in cellular redox balance toward an excessively reduced state—represents an equally critical driver of pathology. Central to this process is the NADH/NAD+ redox couple, which serves as a fundamental regulator of cellular energy metabolism [9]. NADH reductive stress, characterized by the pathological accumulation of NADH, is now recognized not merely as a passive consequence of metabolic dysfunction but as an active regulatory signal that drives profound metabolic reprogramming across diverse diseases [10] [11].
This review examines NADH reductive stress as a comparative pathological mechanism, focusing on quantitative alterations in ATP/ADP, NADPH/NADP+, and NADH/NAD+ ratios across experimental and disease conditions. We synthesize current understanding of how NADH accumulation disrupts mitochondrial function, impairs ATP production, and triggers compensatory metabolic shifts that fuel disease progression in mitochondrial disorders, cancer, and metabolic diseases [12] [11].
Mechanisms of NADH Reductive Stress and Metabolic Dysregulation
Fundamental Redox Couples in Cellular Homeostasis
Cellular metabolism is governed by three central redox pairs that maintain biochemical equilibrium: the NADH/NAD+ couple primarily regulates energy metabolism; the NADPH/NADP+ pair supports reductive biosynthesis and antioxidant defense; and the GSH/GSSG couple maintains thiol homeostasis and oxidative stress response [9] [2]. Under physiological conditions, these systems maintain a dynamic balance that supports efficient energy production while minimizing reactive oxygen species generation.
The NADH/NAD+ ratio serves as a particularly sensitive indicator of cellular redox status. NAD+ functions as a crucial electron acceptor during catabolic processes, while NADH serves as the primary electron donor for oxidative phosphorylation [9]. When this ratio becomes excessively elevated, NADH reductive stress occurs, creating a metabolic bottleneck that impairs mitochondrial function and triggers compensatory metabolic reprogramming [10].
Consequences of NADH/NAD+ Ratio Disruption
Excessive NADH accumulation disrupts mitochondrial efficiency through multiple mechanisms. NADH reductive stress impairs the electron transport chain (ETC), leading to decreased ATP production and increased electron leakage that generates reactive oxygen species (ROS) [11]. This creates a paradoxical situation where both reductive and oxidative stress coexist, further damaging cellular components.
In the endoplasmic reticulum (ER), NADH excess disrupts protein folding by altering disulfide bond formation, triggering ER stress and activating the unfolded protein response (UPR) [11]. This ER stress contributes to insulin resistance and compromised cellular homeostasis, particularly in metabolic diseases. The resulting metabolic inflexibility forces cells to implement alternative pathways for NAD+ regeneration, including increased lactate production and shifts in glutamine metabolism [10] [13].
Table 1: Primary Consequences of NADH Reductive Stress on Cellular Organelles
| Cellular Component | Impact of NADH Reductive Stress | Functional Consequences |
|---|---|---|
| Mitochondria | Impairs ETC function, reduces membrane potential | Decreased ATP production, increased ROS generation |
| Endoplasmic Reticulum | Disrupts disulfide bond formation, protein folding | ER stress, unfolded protein response activation |
| Cytosol | Alters metabolic flux, increases lactate production | Metabolic reprogramming, altered phosphorylation potential |
| Nucleus | Impacts sirtuin activity, gene expression | Altered epigenetic regulation, impaired DNA repair |
Quantitative Analysis of Redox Ratios Across Disease Models
ATP/ADP Ratios and Phosphorylation Potential
The ATP/ADP ratio represents a fundamental indicator of cellular energy status, with direct implications for energy-intensive processes including biosynthesis, ion transport, and signal transduction. NADH reductive stress directly compromises mitochondrial ATP production by creating an over-reduced environment that impedes electron flow through the ETC [11]. Experimental models demonstrate that the P/O ratio (ATP produced per oxygen atom reduced) declines under reductive stress conditions, with theoretical values falling from approximately 2.5 with NADH-linked substrates under normal conditions to significantly lower values during impaired ETC function [14].
In cancer cells, metabolic reprogramming toward aerobic glycolysis (the Warburg effect) may represent an adaptive response to reductive stress, allowing maintenance of ATP production despite mitochondrial impairment [15]. This metabolic shift enables continued proliferation but results in inefficient ATP generation per glucose molecule metabolized, necessitating increased glucose uptake—a hallmark observed across multiple cancer types including ovarian cancer and lung adenocarcinoma [13] [16].
NADH/NAD+ Ratios in Pathological Conditions
Direct measurements of NADH/NAD+ ratios provide compelling evidence for the role of reductive stress in disease pathogenesis. In a pilot study of Leigh syndrome, patient-derived fibroblasts displayed significantly elevated NADH levels compared to healthy controls (p = 0.04), despite comparable total NAD(H) pools [12]. This NADH elevation was replicated in a Ndufs4 knockout mouse model of Leigh syndrome (p = 0.002), confirming that complex I deficiency drives NADH accumulation and reductive stress [12].
Similar patterns emerge in cancer models, where oncogenic drivers and hypoxia-inducible factors promote metabolic shifts that increase NADH production while limiting its oxidation [13] [15]. The resulting reductive stress creates a permissive environment for tumor progression by supporting biosynthetic processes and enhancing resistance to oxidative damage.
Table 2: Comparative NADH/NAD+ Ratios and ATP Parameters Across Experimental Models
| Experimental System | NADH/NAD+ Ratio Alteration | ATP-Related Changes | Experimental Evidence |
|---|---|---|---|
| Leigh Syndrome Patient Fibroblasts | Significant NADH elevation (p = 0.04) | Not directly measured | LC-MS/MS quantification [12] |
| Ndufs4 KO Mouse (Leigh Model) | Significant NADH increase (p = 0.002) | Impaired mitochondrial ATP production | Genetically engineered model [12] |
| Ovarian Cancer Models | Increased NADH/NAD+ inferred from metabolic shifts | Hybrid glycolysis/OXPHOS phenotype | Metabolic flux studies [13] |
| Astrocyte Oxidative Stress | NAD+ phosphorylation to NADP+ | Compromised energy metabolism | Enzymatic cycling assays [2] |
| LUAD Metabolic Reprogramming | NADH accumulation driving reductive stress | Increased glycolytic flux | Single-cell RNA sequencing [16] |
NADPH/NADP+ Dynamics in Reductive Stress
The NADPH/NADP+ couple functions primarily in reductive biosynthesis and antioxidant defense, maintaining glutathione in its reduced form (GSH) to support cellular detoxification. Under conditions of NADH reductive stress, interconnected redox systems lead to concomitant changes in NADPH pools [2]. Research in astrocyte models demonstrates that oxidative stress triggers NAD kinase (NADK)-mediated phosphorylation of NAD+ to NADP+, effectively expanding the NADP(H) pool at the expense of NAD(H) to support antioxidant defense mechanisms [2].
This adaptive response highlights the interconnected nature of cellular redox systems, where perturbations in one compartment inevitably influence others. In metabolic disorders induced by overnutrition, simultaneous elevations in both NADH and NADPH have been documented, creating a doubly reductive environment that disrupts both energy metabolism and signaling pathways [11].
Experimental Models and Methodologies for Redox Assessment
LC-MS/MS-Based NAD(H) Quantification
The precise measurement of NADH and NAD+ pools presents significant technical challenges due to their rapid interconversion and compartmentalization within cells. A recently developed streamlined LC-MS/MS method enables precise quantification of these analytes, providing superior accuracy compared to conventional enzymatic assays [12]. This approach was applied to fibroblasts from mitochondrial disease patients and mouse models, revealing significant NADH elevations despite normal total NAD(H) pools [12].
Protocol Summary: Cells or tissues are rapidly extracted using acid-based methods (for NAD+ preservation) or alkaline conditions (for NADH stabilization). Following neutralization, analytes are separated via reverse-phase chromatography and detected using multiple reaction monitoring (MRM). Internal standards (e.g., stable isotope-labeled NAD+ and NADH) enable precise quantification. This method successfully differentiates NADH/NAD+ ratios in patient-derived fibroblasts, demonstrating its utility for detecting reductive stress in human samples [12].
Enzymatic Cycling Assays for Compartmentalized Redox Pairs
While less specific than LC-MS/MS, enzymatic cycling assays provide sensitive detection of NAD(H) and NADP(H) pools in cellular compartments. In astrocyte studies, these assays quantified basal levels of redox co-substrates, revealing specific contents of 2.91 ± 0.40 nmol/mg protein for NADx (NADH + NAD+) with the reduced form accounting for 28 ± 10% of the total pool [2].
Protocol Summary: For NAD+ quantification, samples are treated with alcohol dehydrogenase and ethanol, converting NAD+ to NADH which is detected via fluorescent reporters. For NADP+ measurement, glucose-6-phosphate dehydrogenase converts NADP+ to NADPH with similar detection. These assays can be adapted to subcellular fractions through differential centrifugation, though cross-contamination remains a concern [2].
Genetic and Pharmacological Manipulation of Redox States
Experimental models employing genetic manipulation provide causal evidence for the role of specific genes in reductive stress. The Ndufs4 knockout mouse, a model of Leigh syndrome, demonstrates that complex I deficiency directly elevates NADH levels and produces severe neurological pathology [12]. Similarly, cancer models with oncogenic activation (e.g., KRAS, MYC) demonstrate how driver mutations promote reductive stress through metabolic reprogramming [13] [15].
Pharmacological approaches to manipulate redox states include NADK inhibition using thionicotinamide, which prevents the oxidative stress-induced phosphorylation of NAD+ to NADP+ in astrocyte models [2]. Conversely, NAD+ precursors (e.g., nicotinamide riboside) attempt to restore NAD+ pools and mitigate reductive stress, though their efficacy varies across disease models [9] [11].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Investigating NADH Reductive Stress
| Reagent/Category | Specific Examples | Research Application | Experimental Function |
|---|---|---|---|
| NAD+ Precursors | Nicotinamide riboside (NR), Nicotinic acid (NA) | Restoring NAD+ pools | Substrates for NAD+ biosynthesis via salvage and Preiss-Handler pathways [9] |
| NADK Inhibitors | Thionicotinamide | Preventing NADP+ synthesis | Inhibits NAD kinase-mediated phosphorylation of NAD+ to NADP+ [2] |
| Complex I Inhibitors | Rotenone, IACS-010759 | Modeling reductive stress | Induces NADH accumulation by impairing mitochondrial oxidation [12] |
| Metabolic Inhibitors | G6PDi-1, 2-deoxy-D-glucose (2-DG) | Pathway manipulation | Modulates substrate flux through glycolytic and pentose phosphate pathways [13] [2] |
| Genetically Encoded Biosensors | SoNar, Peredox | Live-cell imaging | Real-time monitoring of NADH/NAD+ ratios in living cells [9] |
| LC-MS/MS Standards | Stable isotope-labeled NAD+, NADH | Quantitative metabolomics | Internal standards for precise quantification of redox metabolites [12] |
Metabolic Reprogramming Across Disease Contexts
Mitochondrial Diseases
In Leigh syndrome and other mitochondrial disorders, complex I deficiency creates a fundamental defect in NADH oxidation, leading to severe reductive stress [12]. The elevated NADH/NAD+ ratio inhibits multiple dehydrogenase enzymes, including pyruvate dehydrogenase and α-ketoglutarate dehydrogenase, creating metabolic bottlenecks that impair both glycolytic and oxidative metabolism. Compensatory metabolic reprogramming in these conditions includes increased lactate production and activation of alternative NAD+ regenerating pathways, though these adaptations often prove insufficient to restore homeostasis [12].
Cancer
Cancer cells exhibit remarkable metabolic plasticity, dynamically shifting between glycolysis and oxidative phosphorylation depending on environmental conditions and therapeutic pressures [13]. In ovarian cancer, metabolic reprogramming encompasses enhanced aerobic glycolysis, glutamine addiction, and dysregulated lipid metabolism to support rapid proliferation, redox homeostasis, and metastatic potential [13]. NADH reductive stress contributes to the stabilization of hypoxia-inducible factors and oncogenic signaling pathways that further drive tumor progression and therapy resistance.
Metabolic and Cardiovascular Diseases
In conditions of overnutrition and obesity, excessive nutrient flux drives NADH accumulation through increased glycolytic and TCA cycle activity [11]. The resulting reductive stress impairs both mitochondrial function and ER protein folding, contributing to insulin resistance and cardiovascular dysfunction. Paradoxically, antioxidant supplementation can sometimes exacerbate reductive stress by further shifting the redox balance toward reduction, highlighting the delicate balance required for redox homeostasis [11].
Visualization of NADH Reductive Stress Pathways and Methodologies
NADH Reductive Stress Signaling Pathway
NADH Reductive Stress Pathway: This diagram illustrates the mechanistic progression from initial mitochondrial dysfunction through NADH accumulation to compensatory metabolic reprogramming and eventual therapeutic resistance.
Experimental Workflow for Redox Assessment
Redox Assessment Workflow: This experimental workflow outlines the key steps in quantifying redox ratios, from sample collection through analytical separation and data interpretation for pathological assessment.
The emerging recognition of NADH reductive stress as a fundamental driver of metabolic reprogramming represents a paradigm shift in our understanding of disease pathogenesis. Quantitative assessment of ATP/ADP, NADPH/NADP+, and NADH/NAD+ ratios across experimental models reveals consistent patterns of redox disruption that transcend individual disease boundaries. These alterations create a permissive environment for pathological processes by compromising cellular energy production, disrupting biosynthetic pathways, and activating stress response programs.
Future therapeutic strategies targeting NADH reductive stress may include NAD+ restoration therapies, modulation of NAD kinase activity, and metabolic interventions that rebalance redox couples without exacerbating oxidative damage [9] [2] [11]. The development of compartment-specific redox assessments and genetically encoded biosensors will further enhance our understanding of spatial regulation in reductive stress pathologies. As our methodological sophistication grows, so too will our ability to precisely target NADH reductive stress across the spectrum of human disease.
Comparative Basal Ratios Across Species and Cell Types
The quantification of basal ratios of key metabolic cofactors—particularly ATP/ADP, NADPH/NADP+, and NADH/NAD+—is fundamental to understanding cellular energy management, reductive biosynthesis, and antioxidant defense across different biological systems. These ratios represent the thermodynamic status of the cell, influencing metabolic flux, gene expression, and stress adaptation [17] [18]. For researchers in drug development and metabolic engineering, accurate determination of these parameters provides critical insights into disease mechanisms and cellular responses to therapeutic interventions. This guide objectively compares established methodologies for measuring these ratios, evaluates their performance across different species and cell types, and provides the experimental protocols necessary for implementation.
Core Concepts and Biological Significance
The ratios of ATP/ADP, NADPH/NADP+, and NADH/NAD+ are not uniform across cellular compartments, species, or cell types. Their values reflect the specialized metabolic functions of different biological systems.
- ATP/ADP Ratio: This ratio is the primary indicator of cellular energy charge. ATP serves as the universal energy currency, and its hydrolysis to ADP releases energy that drives countless cellular processes. Substantial spatial gradients of ATP concentration can exist within eukaryotic cells, particularly when ATP sources (like mitochondria) or sinks (like the nucleus) are spatially clustered [19].
- NADPH/NADP+ Ratio: This ratio reflects the cell's reductive capacity and is crucial for anabolic biosynthesis and antioxidative defense. NADPH serves as the principal electron donor for reductive biosynthesis and for enzymes like glutathione reductase that combat oxidative stress [2] [18]. In mammalian cells, free NADPH/NADP+ ratios have been reported to be between 10 and 100, with higher ratios found in mitochondria compared to the nucleus and cytosol [20].
- NADH/NAD+ Ratio: This ratio is a central indicator of the cell's catabolic and redox state. NADH is a key product of catabolic pathways such as glycolysis and the TCA cycle, and it donates electrons to the mitochondrial electron transport chain for ATP production [17] [18]. The free NAD+/NADH ratio has been reported to be 100–600 in the cytosol and 4–10 in the mitochondria of mammalian cells [20].
Table 1: Functional Specialization of Key Metabolic Cofactors
| Cofactor Pair | Primary Cellular Role | Key Functions | Major Generating Pathways |
|---|---|---|---|
| ATP/ADP | Energy Currency | Provides energy for cellular work (biosynthesis, transport, motility) | Glycolysis, Oxidative Phosphorylation |
| NADPH/NADP+ | Reductive Biosynthesis & Defense | Electron donor for anabolic reactions and antioxidant systems (e.g., glutathione) | Pentose Phosphate Pathway (PPP), Malic Enzyme |
| NADH/NAD+ | Catabolic Redox Couple | Electron carrier in catabolic processes; delivers electrons to ETC for ATP production | Glycolysis, Ticarboxylic Acid (TCA) Cycle |
The diagram below illustrates the core metabolic pathways and cellular compartments involved in maintaining the balance of these key cofactor ratios.
Figure 1. Metabolic Pathways Regulating Cofactor Ratios. This diagram illustrates the core metabolic pathways in different cellular compartments that generate and consume ATP, NADPH, and NADH, highlighting their interconnected roles in energy production, biosynthesis, and redox homeostasis. Abbreviations: LDH, Lactate Dehydrogenase; ROS, Reactive Oxygen Species.
Comparative Data of Basal Ratios
Reported basal ratios for these cofactors vary significantly depending on the cell type, species, and methodological approach used for measurement.
Table 2: Comparative Basal Ratios and Concentrations Across Cell Types and Species
| Cell Type / Species | Parameter | Measured Value | Notes / Method | Reference |
|---|---|---|---|---|
| Cultured Rat Astrocytes | NADPH/NADP+ (Total Pool) | ~0.59 (37% reduced) | Specific content: 0.64 ± 0.09 nmol/mg protein. Measured by enzymatic cycling assays. | [2] |
| Cultured Rat Astrocytes | NADH/NAD+ (Total Pool) | ~0.39 (28% reduced) | Specific content: 2.91 ± 0.40 nmol/mg protein. Measured by enzymatic cycling assays. | [2] |
| Cultured Rat Astrocytes | GSH/GSSG (Total Pool) | ~32.3 (97% reduced) | Specific content: 44.7 ± 8.2 nmol/mg protein. | [2] |
| Mammalian Cells (General) | Free NADPH/NADP+ | 10 - 100 | Reported range for free (unbound) ratios. | [20] |
| Mammalian Cells (General) | Free Cytosolic NAD+/NADH | 100 - 600 | Reported range for free ratios in the cytosol. | [20] |
| Mammalian Cells (General) | Free Mitochondrial NAD+/NADH | 4 - 10 | Reported range for free ratios in mitochondria. | [20] |
| Mammalian Cells (General) | Free Cytosolic NAD+ | ~100 µM | Reported concentration of free NAD+. | [20] |
Methodologies for Ratio Quantification
Accurately determining these ratios is methodologically challenging. The choice of technique significantly impacts the results, as it determines whether total cellular pools, free (unbound) fractions, or compartment-specific ratios are measured.
Semisynthetic Fluorescent Biosensors
Principle: These are genetically encoded tools for mapping spatiotemporal dynamics of free (unbound) NAD+ and NADPH/NADP+ ratios in live cells. Sensing is based on controlling the spatial proximity of two synthetic fluorophores by analyte binding to the protein component of the sensor [20].
Key Advantages:
- Ratiometric & pH-insensitive: Allows for quantitative measurements independent of sensor concentration or pH fluctuations.
- High Dynamic Range: NADP-Snifit shows an 8.9-fold FRET ratio change.
- Subcellular Targeting: Can be localized to specific organelles (e.g., cytosol, nucleus, mitochondria) to measure compartment-specific ratios.
- Tunable Response Range: The sensor design allows for rational adaptation of the response range (c50).
Detailed Protocol (NADP-Snifit for NADPH/NADP+):
- Sensor Design: The sensor is a fusion protein containing human sepiapterin reductase (SPR), SNAP-tag, and Halo-tag.
- Labeling:
- The SNAP-tag is labeled with CP-TMR-SMX, a cell-permeable molecule containing sulfamethoxazole (a ligand for SPR) and a TMR fluorophore.
- The Halo-tag is labeled with SiR-Halo, a siliconrhodamine derivative that acts as a FRET acceptor for TMR.
- Mechanism: In the presence of NADP+, the tethered sulfamethoxazole binds to SPR, increasing FRET efficiency between TMR and SiR. NADPH does not support this interaction. Therefore, the equilibrium between the sensor's open and closed state is controlled by the NADPH/NADP+ ratio.
- Calibration: In vitro titration determines the half-maximal sensor response (r50), which for NADP-Snifit corresponds to an NADPH/NADP+ ratio of 30 ± 3.
- Live-Cell Imaging: Transfert cells with the NADP-Snifit construct, label with the synthetic fluorophores, and perform ratiometric fluorescence imaging (excitation at 560 nm) to determine free NADPH/NADP+ ratios dynamically [20].
Enzymatic Cycling Assays
Principle: This is a sensitive and specific biochemical method to quantify the total cellular levels (both free and protein-bound) of oxidized and reduced NAD(P) and glutathione after extraction from cells or tissues.
Key Advantages:
- High Sensitivity: Can detect low abundance cofactors (e.g., NADPx in astrocytes at 0.64 nmol/mg protein).
- Specificity: Relies on enzyme specificity (e.g., glucose-6-phosphate dehydrogenase for NADP+) for accurate quantification.
- Wide Applicability: Can be used on cell lysates, tissue homogenates, and biological fluids.
Detailed Protocol (for Astrocyte Redox Cofactors):
- Cell Culture and Treatment: Use confluent primary astrocyte-rich cultures. Wash cells twice with an incubation buffer (e.g., 20 mM HEPES, 145 mM NaCl, 5.4 mM KCl, pH 7.4).
- Rapid Extraction:
- For NADx and NADPx: Extract with perchloric acid to stabilize the oxidized forms. Neutralize the extract before assay.
- For GSx: Extract with sulfosalicylic acid.
- Specific Quantification:
- Total NAD+: Measure in an alkaline extract that destroys reduced forms.
- Total NADH: Measure in a separate, neutralized acidic extract.
- Total NADP+ and NADPH: Use similar principles with specific enzymatic systems.
- Enzymatic Cycling Reaction:
- For NADP+: Use a system containing Glucose-6-Phosphate (G6P) and yeast Glucose-6-Phosphate Dehydrogenase (G6PDH). NADP+ is reduced to NADPH, which then reduces a tetrazolium dye (e.g., MTT) via an intermediate electron acceptor (e.g., phenazine ethosulfate), generating a colored formazan product.
- The rate of formazan production, measured spectrophotometrically, is proportional to the concentration of NADP+ in the sample.
- Similar cycling reactions are set up for NAD+, NADH, and NADPH using specific enzymes and substrates.
- Data Calculation: Calculate concentrations by comparing the reaction rates of samples to standard curves generated with known concentrations of NADP+, NAD+, etc. [2].
The experimental workflow for these two primary methods is summarized below.
Figure 2. Workflow for Quantifying Cofactor Ratios. This diagram outlines the two primary methodological approaches for measuring metabolic cofactor ratios, guiding the choice between live-cell imaging of free pools and biochemical analysis of total cellular pools.
The Scientist's Toolkit: Essential Research Reagents
Successful experimentation requires a carefully selected set of reagents and tools, which vary depending on the chosen methodology.
Table 3: Essential Reagents for Cofactor Ratio Analysis
| Reagent / Tool | Function / Application | Key Considerations |
|---|---|---|
| NADP-Snifit / NAD-Snifit | Semisynthetic biosensors for measuring free NADPH/NADP+ and NAD+ in live cells. | Ratiometric, pH-insensitive, tunable dynamic range. Requires transfection and labeling. |
| SoNar / iNAP | Genetically encoded sensors for free NAD+/NADH and NADPH, respectively. | Based on cpYFP; large dynamic range but require short-wavelength excitation. |
| Enzymatic Cycling Assay Kits | Commercial kits for quantifying total NAD(P)(H) and GSH/GSSG from cell lysates. | Ensure specificity and sensitivity for the target analyte (e.g., NAD+ vs. NADP+). |
| G6PDi-1 | Inhibitor of glucose-6-phosphate dehydrogenase (G6PD). | Used to experimentally block the pentose phosphate pathway, a major source of NADPH. |
| Thionicotinamide | Precursor for synthesis of thio-NADP, an inhibitor of NAD kinase (NADK). | Used to probe the role of NADK-mediated phosphorylation of NAD+ to NADP+. |
| Dithiothreitol (DTT) | Reducing agent used in some biochemical assays. | Can disrupt protein disulfide bonds; use must be adapted to the specific assay context. |
| H₂O₂ | Inducer of oxidative stress. | Used to perturb cellular redox state and test the response of redox cofactor systems. |
Critical Considerations in Experimental Design
- Total vs. Free Pools: A critical distinction must be made between the total cellular pool of a cofactor and the free, unbound fraction. The majority of pyridine nucleotides are protein-bound, and only the free fraction is metabolically active and participates in signaling [20]. Biosensors typically report free ratios, while enzymatic assays on lysates measure total pools.
- Subcellular Compartmentalization: Ratios are not uniform throughout the cell. For example, the free NADPH/NADP+ ratio is higher in mitochondria than in the cytosol or nucleus [20]. Method selection should account for this compartmentalization.
- Compositional Nature of Data: Glycomics and other -omics data that report relative abundances are compositional. This means an increase in the relative abundance of one component (e.g., a specific glycan) mathematically necessitates a decrease in others. Applying traditional statistical tests to such data without proper transformation (e.g., Center Log-Ratio transformation) can generate a high rate of false-positive findings [21]. This principle is a critical consideration for any quantitative analysis of interrelated cellular components.
- Physicochemical Conditions: Biochemical assay buffers (e.g., PBS) often do not mimic the intracellular environment, which has high macromolecular crowding, different ionic concentrations (high K+, low Na+), and distinct viscosity. These differences can alter measured Kd values and enzyme kinetics, potentially explaining discrepancies between biochemical and cell-based assays [22]. Using cytoplasm-mimicking buffers for in vitro assays is recommended for greater physiological relevance.
Interplay Between ATP/ADP and NADPH/NADP+ in Redox Homeostasis
Redox homeostasis, the delicate balance between oxidative and reductive processes within cells, is fundamental to cellular health and function. This equilibrium is orchestrated by several key redox couples, most notably the interlocking systems of ATP/ADP and NADPH/NADP+ [9] [23]. The ATP/ADP ratio represents the primary energy currency of the cell, governing energy-transfer reactions, while the NADPH/NADP+ couple serves as the central redox buffer, providing reducing power for biosynthetic processes and antioxidant defense [24] [25]. These systems do not operate in isolation; their interplay is critical for maintaining metabolic stability, especially under conditions of stress. In biological systems, from algae to mammalian cells, the phosphorylation of ADP to ATP is often coupled to the consumption of reducing equivalents, and the generation of NADPH from NADP+ requires energy input [26]. Disruption of this intricate balance is a hallmark of numerous pathological states, including cancer, neurodegenerative diseases, and metabolic disorders [27] [23] [25]. This guide objectively compares the dynamics of ATP/ADP and NADPH/NADP+ ratios across varied experimental and physiological conditions, providing researchers with a consolidated resource of quantitative data and methodologies to advance therapeutic targeting of redox pathways.
Quantitative Comparison of Ratios Across Biological Contexts
The ATP/ADP and NADPH/NADP+ ratios are highly dynamic and vary significantly depending on cell type, energy demand, and oxidative stress conditions. The table below synthesizes quantitative data from multiple experimental models to facilitate direct comparison.
Table 1: Comparative Analysis of ATP/ADP and NADPH/NADP+ Ratios Across Experimental Conditions
| Cell Type / Condition | ATP/ADP Ratio | NADPH/NADP+ Ratio | Key Experimental Observations | Citation |
|---|---|---|---|---|
| Cultured Rat Astrocytes (Basal) | Not Specified | ~0.59 (37% NADPH of total NADPx) | Total NADPx pool: 0.64 ± 0.09 nmol/mg protein. | [2] |
| C. reinhardtii (Dark, Aerobic) | ~15.0 | ~1.5 | High energy charge and reducing power in the dark under aerobic conditions. | [26] |
| C. reinhardtii (Dark, Anaerobic) | ~5.0 | ~3.5 | Transition to anaerobiosis decreases ATP/ADP but increases NADPH/NADP+. | [26] |
| C. reinhardtii (Light, Aerobic - Low Light) | Increased from dark state | Decreased from dark state (~1.0) | Illumination increases ATP, but NADPH drops under light limitation. | [26] |
| C. reinhardtii (Light, Aerobic - Saturating Light) | Increased from dark state | Regained ~1.5 | Both energy and reducing power are high under optimal light. | [26] |
| Astrocytes under H₂O₂ stress | Not Specified | Transiently Decreased | Oxidative stress rapidly oxidizes NADPH to NADP+; total NADPx pool doubles at expense of NADx pool. | [2] |
The data reveals that the NADPH/NADP+ ratio is generally maintained well below the ATP/ADP ratio across systems, reflecting its role as a sensitive redox indicator rather than a high-capacity energy store [26] [2]. During energy stress, such as the shift to anaerobiosis in C. reinhardtii, the ATP/ADP ratio is highly vulnerable, dropping precipitously, while the NADPH/NADP+ ratio can actually increase, suggesting complex, compartment-specific regulatory mechanisms [26]. Furthermore, the response to oxidative stress is characterized by a rapid but often transient oxidation of the NADPH pool, accompanied by a metabolic rewiring where the total NADP(H) pool can be expanded through the phosphorylation of NAD+ to meet antioxidant demands [2].
Experimental Protocols for Ratio Quantification
Accurate measurement of these metabolic ratios is technically challenging due to the rapid turnover of metabolites and the compartmentalization of pools within the cell. The following protocols, derived from cited studies, provide reliable methodologies.
Enzymatic Cycling Assays for NADPH/NADP+ in Astrocytes
This sensitive and specific protocol is used to determine the absolute levels and redox state of the NADP(H) pool in cultured primary cells [2].
Key Research Reagents:
- Cell Lysis: Acid extraction (e.g., with sulfosalicylic acid) to rapidly quench metabolism and preserve redox states.
- Enzyme Cocktail: Glucose-6-phosphate dehydrogenase (G6PDH) is critical for the cycling reaction.
- Substrates: Glucose-6-phosphate (G6P), Phenazine Ethosulfate (PES) or Phenazine Methosulfate (PMS) as redox cyclers, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) as a terminal electron acceptor.
- Buffer: Tris-based buffer at optimal pH for G6PDH activity.
Step-by-Step Workflow:
- Rapid Quenching & Metabolite Extraction: Cultured astrocytes are swiftly washed with a cold buffer and immediately extracted with ice-cold acid (e.g., sulfosalicylic acid). This step denatures enzymes and locks metabolites in their current state.
- Neutralization: The acid extract is neutralized to a pH suitable for enzymatic activity.
- Specific Measurement of NADP+: An aliquot of the neutralized extract is incubated with G6PDH and its substrate, G6P. Any NADP+ present is reduced to NADPH during this reaction.
- Enzymatic Cycling & Signal Amplification: The total NADPH generated (from the initial extract plus that formed from NADP+) is then quantified in a cycling assay. The NADPH reduces the redox cycler (PES/PMS), which in turn reduces MTT to a purple formazan product.
- Quantification: The rate of formazan formation, measured by its absorbance at 570 nm, is directly proportional to the NADPH concentration. Initial NADPH and NADP+ levels are calculated from measurements with and without the G6PDH step.
Table 2: Key Research Reagent Solutions for NADP(H) Quantification
| Reagent | Function | Specific Example |
|---|---|---|
| Sulfosalicylic Acid | Rapid metabolic quenching and protein precipitation. | 5-10% solution in water. |
| Glucose-6-Phosphate Dehydrogenase (G6PDH) | Enzyme that specifically reduces NADP+ to NADPH for quantification. | From yeast, e.g., from Roche Diagnostics. |
| Phenazine Ethosulfate (PES) | Redox cycler that shuttles electrons from NADPH to MTT. | Sigma-Aldrich. |
| MTT | Terminal electron acceptor; forms measurable formazan dye upon reduction. | Sigma-Aldrich. |
In Vivo Analysis of Ratios in Algal Cells During Dark-Light Transitions
This protocol investigates the dynamic interplay of energy and redox states in response to light, the primary energy source for photosynthetic organisms [26].
Key Research Reagents:
- Biological Model: Intact cells of Chlamydomonas reinhardtii.
- Culture Conditions: Controlled aerobiosis/anaerobiosis, defined light intensities (saturating vs. low).
- Inhibitors/Uncouplers: Compounds like G6PDi-1 (PPP inhibitor) or carbonyl cyanide m-chlorophenyl hydrazone (CCCP, uncoupler) to dissect metabolic contributions.
Step-by-Step Workflow:
- Cell Preparation and Conditioning: Algal cells are cultured and dark-adapted to establish a baseline metabolic state. They are then subjected to controlled environments (aerobic vs. anaerobic).
- Controlled Illumination: Cells are exposed to precise light intensities (e.g., low vs. saturating photon flux), and the transition is monitored over time.
- Rapid Sampling and Metabolite Extraction: Samples are taken at critical time points (e.g., during the lag phase before O₂ evolution, at steady state) and rapidly extracted with acid.
- Enzymatic Quantification of ATP, ADP, and NADP: Metabolites in the extract are quantified using specific enzymatic assays linked to a change in absorbance of a cofactor, similar to the method described for astrocytes. The NADPH level is often calculated as the difference between total NADP(H) and measured NADP+.
- Correlation with Physiological Readouts: Metabolite data are directly correlated with real-time measurements of photosynthetic activity, such as O₂ evolution and chlorophyll fluorescence (Fm), to link ratios to functional outputs.
The following diagram illustrates the core logic and workflow for investigating these metabolic ratios.
Figure 1: Experimental workflow for ratio analysis, depicting the sequence from cell preparation to data correlation.
Regulatory Mechanisms and Signaling Pathways
The interplay between ATP/ADP and NADPH/NADP+ is governed by a network of metabolic pathways and enzymes that sense and respond to the cellular energy and redox status.
Key Metabolic Nodes Interconnecting the Ratios
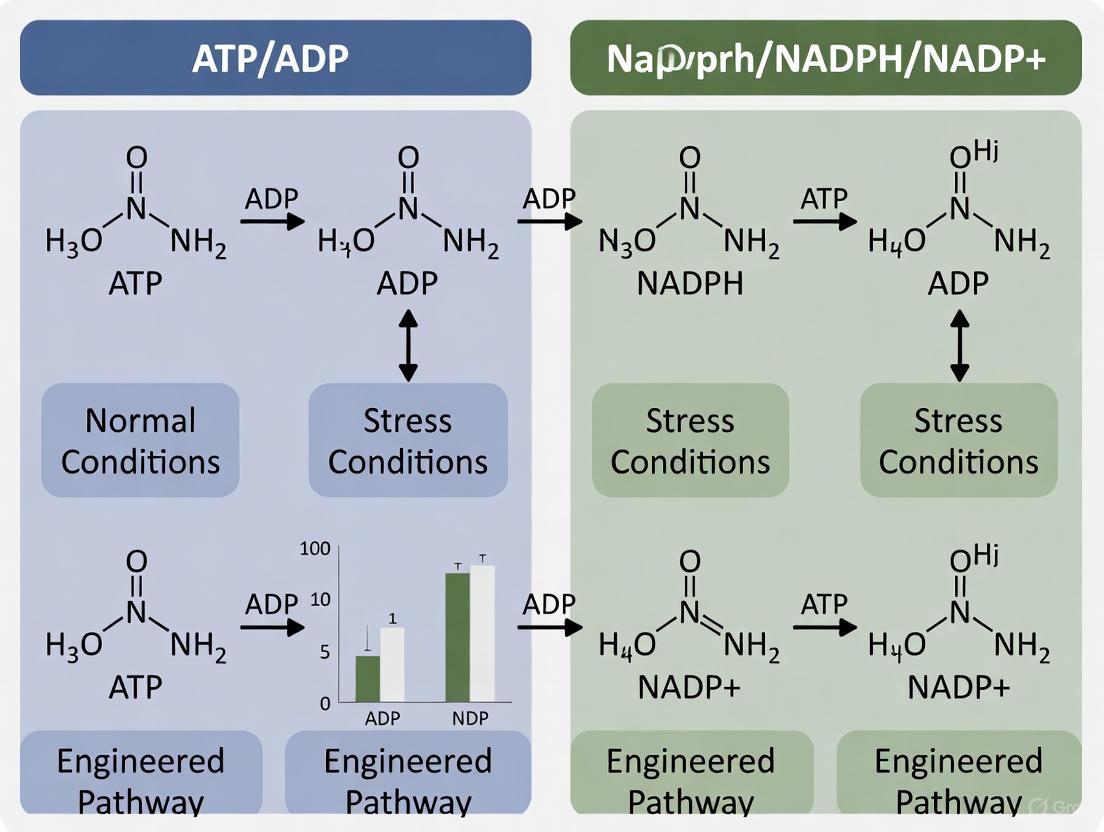
- NAD Kinase (NADK): This enzyme is a critical link, catalyzing the phosphorylation of NAD+ to NADP+ using ATP as the phosphate donor [24] [25] [2]. This reaction directly consumes ATP to create the precursor for NADPH. Under oxidative stress, observed in astrocytes, NADK activity is stimulated, doubling the cellular NADP(H) pool at the expense of NAD(H) to fuel antioxidant defense [2].
- Pentose Phosphate Pathway (PPP): As the major source of cytosolic NADPH, the oxidative branch of the PPP is regulated by the energy and redox state of the cell. Glucose-6-phosphate dehydrogenase (G6PD), the rate-limiting enzyme, is sensitive to the NADPH/NADP+ ratio, providing feedback inhibition [25]. The pathway also produces ribose-5-phosphate for nucleotide synthesis, linking NADPH production to biosynthetic demands.
- Nicotinamide Nucleotide Transhydrogenase (NNT): Located in the mitochondrial inner membrane, NNT couples the proton gradient generated by ATP hydrolysis to drive the reduction of NADP+ by NADH, effectively converting the energy from the proton motive force into reducing power in the form of NADPH [9] [25]. This creates a direct thermodynamic link between the ATP/ADP ratio (via the proton gradient) and the mitochondrial NADPH/NADP+ ratio.
- Malic Enzyme (ME1) and IDH1: These enzymes generate NADPH in the cytosol and mitochondria, respectively, and their activity is integrated into central carbon metabolism. They provide NADPH for lipid biosynthesis and antioxidant defense, with their flux influenced by the availability of mitochondrial substrates and cellular energy status [25].
The diagram below summarizes these key regulatory interactions.
Figure 2: Key enzymes and pathways connecting ATP/ADP and NADPH/NADP+ homeostasis.
The Circuitry of Redox and Energy Sensing
The regulatory network functions as an integrated circuit where energy status dictates redox management and vice-versa. A high ATP/ADP ratio reflects energy surplus, which can be directed by NNT to enhance the NADPH/NADP+ ratio, preparing the cell for reductive biosynthesis or pre-empting oxidative stress [9] [25]. Conversely, energy depletion (low ATP/ADP) compromises the cell's ability to maintain a reduced NADPH pool, increasing susceptibility to oxidative damage. During active oxidative stress, the rapid consumption of NADPH by glutathione and thioredoxin systems creates a demand signal, which is met by increasing NADPH production through PPP flux and, critically, by expanding the total NADP(H) pool via NADK activation, a process that itself consumes ATP [25] [2]. This creates a feed-forward loop where defending the redox state is prioritized, even at a cost to the energy charge of the cell.
The efficient allocation of cellular energy is a fundamental constraint governing plant metabolism, growth, and productivity. Metabolic pathways consume adenosine triphosphate (ATP) and nicotinamide adenine dinucleotide phosphate (NADPH) in distinct stoichiometries, creating a complex energy budget that must be balanced against the fixed output of the light reactions [28]. Understanding the specific energy demands of major metabolic processes—including the C3 cycle, photorespiration, the tricarboxylic acid (TCA) cycle, and biosynthesis of starch and sucrose—is therefore critical for both fundamental physiology and bioengineering efforts aimed at improving crop yields [29] [28].
This guide provides a systematic comparison of pathway-specific energy demands, synthesizing quantitative data from recent isotopically non-stationary metabolic flux analysis (INST-MFA) studies and physiological investigations. It is structured within the broader thesis of comparing ATP/ADP and NADPH/NADP+ ratios across experimental conditions, providing researchers with consolidated data, validated methodologies, and essential tools for advancing research in plant bioenergetics.
Comparative Energetics of Major Metabolic Pathways
The ATP:NADPH demand ratio is a crucial parameter for understanding metabolic balancing. The light reactions of photosynthesis produce ATP and NADPH in a constrained ratio of approximately 1.28 ATP per 2 NADPH via linear electron flow, which can create a deficit relative to the demands of downstream metabolism [28]. The following analysis details the consumption patterns of these energy currencies by the central metabolic pathways.
Table 1: Energy Demands of Central Carbon Metabolic Pathways
| Metabolic Pathway | ATP Consumed (per C consumed) | NADPH Consumed (per C consumed) | ATP:NADPH Demand Ratio | Primary Cellular Compartment(s) |
|---|---|---|---|---|
| C3 Cycle (Calvin-Benson) | 3 [29] | 2 [29] | 1.50 [28] | Chloroplast |
| Photorespiration | 3.5 [29] | 2 [29] | 1.75 [28] | Chloroplast, Peroxisome, Mitochondria |
| Starch & Sucrose Synthesis | Variable; requires ATP/UTP [29] | Not Major | N/A | Chloroplast, Cytosol |
| TCA Cycle | Net producer (GTP/ATP) [30] | Net producer (NADH/FADH2) [30] | N/A (Catabolic) | Mitochondria |
A meta-analysis of INST-MFA data reveals that while the C3 cycle and photorespiration account for the bulk of energy flux in illuminated leaves, other processes significantly influence the overall cellular energy balance [29]. Specifically, starch and sucrose synthesis imposes a notable additional ATP demand that is not coupled to NADPH consumption. This demand may help counterbalance the high ATP:NADPH requirement of photorespiration, potentially reducing the need for rapid activation of alternative ATP-generating processes like cyclic electron flow [29].
The TCA cycle operates primarily as a catabolic pathway in mitochondria, generating energy carriers (ATP/GTP, NADH, FADH2) rather than consuming them, and providing essential precursors for biosynthesis [30] [31].
Quantitative Flux Analysis and Energy Balancing
The relative flux through energy-demanding pathways is highly responsive to environmental conditions. The ratio of carboxylation to oxygenation by Rubisco (r), a key determinant of this flux, changes dramatically with CO₂ concentration and between photosynthetic types [32].
Table 2: Ratio of Rubisco Carboxylation to Oxygenation (r) Under Different CO₂ Conditions
| CO₂ Concentration (μbar) | Ratio (r) in C3 Plants | Ratio (r) in C4 Plants |
|---|---|---|
| 100 | 1.14 | 22.23 |
| 380 (Near-Ambient) | 4.33 | 70.73 |
| 550 | 6.26 | 85.97 |
| 800 | 9.11 | 87.11 |
This variation directly impacts the ATP:NADPH demand of the leaf. At low CO₂ levels, photorespiratory flux is higher in C3 plants, increasing the overall ATP:NADPH demand and exacerbating the ATP deficit. C4 plants, with their CO₂-concentrating mechanism, maintain a vastly superior carboxylation ratio, effectively suppressing photorespiration and its associated high energy demand [32]. Systematic modeling demonstrates that C4 metabolic networks exhibit higher robustness, better modularity, and higher CO₂ use efficiency compared to C3 networks [32].
To manage the inherent ATP deficit, plants deploy several energy-balancing mechanisms [28]:
- Cyclic Electron Flow (CEF) around Photosystem I: Produces ATP without net NADPH production.
- The Malate Valve: Exports reducing equivalents from the chloroplast as malate, consuming NADPH and regenerating NADP⁺.
- The Water-Water Cycle: Maintains electron flow when NADP⁺ is limited, consuming excess reductant.
Experimental Protocols for In Planta Energy Analysis
Monitoring NADPH and NADH/NAD+ Redox Dynamics with Fluorescent Sensors
Objective: To monitor dynamic changes in NADPH levels and the NADH/NAD+ ratio in specific subcellular compartments of Arabidopsis thaliana during photosynthesis and photorespiration [33].
Key Reagents:
- Sensors: Transgenic Arabidopsis lines expressing the high-affinity NADPH sensor iNAP1, the low-affinity NADPH sensor iNAP4, or the NADH/NAD+ sensor SoNar.
- Targeting: Sensors are directed to the cytosol, plastid stroma, or peroxisomes using organelle-specific targeting peptides (e.g., chloroplast transketolase transit peptide, TKTP, for plastids; peroxisomal targeting signal type 1, SRL, for peroxisomes) [33].
- Controls: Lines expressing a ligand-binding-abolished sensor (iNAPc) to account for pH changes.
Methodology:
- Plant Material: Use seedlings or leaf tissues from stable transgenic lines.
- Imaging: Utilize a confocal microscope configured for ratiometric imaging.
- Treatment: Apply light/dark transitions or metabolic inhibitors (e.g., a glycine decarboxylase inhibitor to block photorespiration).
- Data Acquisition: Measure sensor fluorescence intensities at appropriate excitation/emission wavelengths. The ratio of fluorescence emissions is used to determine NADPH concentration or the NADH/NAD+ ratio, minimizing artifacts from variable sensor expression [33].
Application: This protocol revealed that the photosynthetic increase in the stromal NADH/NAD+ ratio disappears when glycine decarboxylation is inhibited, highlighting that photorespiration is a major supplier of NADH to mitochondria during photosynthesis [33].
Determining Metabolic Flux Using INST-MFA
Objective: To quantitatively map carbon and energy flux through central metabolic pathways under different physiological conditions [29].
Key Reagents:
- Isotopic Tracer: ¹³CO₂ or ¹³C-labeled organic substrates (e.g., ¹³C-glucose).
- Plant Material: Leaves from species such as Arabidopsis thaliana, Nicotiana tabacum, or Camelina sativa.
- Analytical Platform: Gas Chromatography-Mass Spectrometry (GC-MS) or Liquid Chromatography-Mass Spectrometry (LC-MS).
Methodology:
- Labeling Pulse: Expose photosynthesizing leaves to a short pulse of ¹³CO₂.
- Rapid Sampling: Quench metabolism and collect tissue samples at precise time points (seconds to minutes) to capture non-steady-state label incorporation.
- Metabolite Extraction: Rapidly extract and process polar metabolites from the tissue.
- Mass Spectrometry: Analyze the mass isotopomer distribution of key metabolic intermediates.
- Computational Modeling: Use computational models to estimate metabolic flux networks that best fit the experimental isotopomer data [29].
Application: INST-MFA has been used to evaluate the flux of energy across different pathways and compartments, revealing the significant contribution of starch and sucrose synthesis to the overall cellular ATP demand [29].
Pathway Diagrams and Energy Flux Visualization
Photorespiration and Its Energetic Interface with Primary Metabolism
Diagram 1: Photorespiration and the Malate Shuttle. This diagram illustrates the multi-compartmental photorespiratory pathway (C2 cycle) and its integration with the malate-oxaloacetate (OAA) shuttle, a key system for managing reducing equivalents. The pathway consumes ATP and NAD(P)H across chloroplasts, peroxisomes, and mitochondria [33] [28]. Key energy transactions include NADH production by mitochondrial Glycine Decarboxylase (GDC) and NADH consumption by peroxisomal Hydroxypyruvate Reductase (HPR). The malate valve exports excess chloroplast NADPH as malate, which can be oxidized in mitochondria to generate more NADH, helping to balance the ATP:NADPH ratio [33] [28].
The TCA Cycle in Anabolism, Catabolism, and Signaling
Diagram 2: The Multifunctional Role of the TCA Cycle. The TCA cycle is a metabolic hub with three primary functions: it provides carbon skeletons for biosynthesis (anabolism), generates ATP and reducing equivalents for energy (catabolism), and supplies metabolites for signaling [30] [31]. Anaplerotic reactions (dashed lines) replenish cycle intermediates drawn off for biosynthesis. Signaling metabolites like acetyl-CoA (for histone acetylation) and succinate (hypoxia response) allow mitochondria to directly regulate nuclear gene expression and cell fate [31].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Metabolic Flux and Energy Balance Research
| Reagent / Tool | Function / Assay | Key Utility |
|---|---|---|
| iNAP & SoNar Sensors | Genetically encoded sensors for NADPH (iNAP) and NADH/NAD+ ratio (SoNar) [33]. | Enable real-time, subcellular monitoring of pyridine nucleotide redox states in living plant tissues. |
| ¹³C-Labeled Substrates (e.g., ¹³CO₂) | Tracers for Isotopically Non-Stationary Metabolic Flux Analysis (INST-MFA) [29]. | Allow quantitative mapping of absolute flux rates through metabolic networks. |
| Glycine Decarboxylase (GDC) Inhibitors | Chemical inhibitors of the mitochondrial photorespiratory enzyme GDC. | Used to dissect the specific contribution of photorespiration to overall metabolism and energy dynamics [33]. |
| Flux Balance Analysis (FBA) Models | Constraint-based computational models of genome-scale metabolic networks [32]. | Enable in silico prediction of metabolic fluxes, robustness, and optimal yields under different genetic or environmental conditions. |
| Chloroplast & Mitochondrial Isolation Kits | Standardized protocols and reagents for organelle purification. | Provide material for ex vivo studies of organelle-specific metabolic functions and transport. |
Analytical Approaches and Clinical Applications in Metabolic Phenotyping
In the study of cellular metabolism, accurately quantifying key metabolites and their ratios—such as ATP/ADP and NADPH/NADP+—is fundamental to understanding energy production, redox homeostasis, and signaling pathways. These ratios serve as critical indicators of cellular status, reflecting the balance between anabolic and catabolic processes, and responding to stressors, nutrient availability, and disease states. The selection of an appropriate analytical technique is paramount, as it directly influences the reliability, specificity, and biological relevance of the data obtained. Two predominant methodologies employed in this domain are Enzyme-Linked Immunosorbent Assays (ELISA) and Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS). This guide provides an objective comparison of these platforms, supported by experimental data, to inform researchers and scientists in their methodological choices.
Technology Comparison: Core Principles and Performance
The following table summarizes the fundamental characteristics and performance metrics of ELISA and LC-MS/MS platforms.
Table 1: Core Characteristics and Performance of ELISA and LC-MS/MS
| Feature | ELISA | LC-MS/MS |
|---|---|---|
| Basic Principle | Antibody-antigen interaction for detection [34]. | Physical separation followed by mass-based detection [34] [35]. |
| Technique Complexity | Simple, often a single-step assay [34]. | Multistep and complex technique [34]. |
| Cost-Effectiveness | Relatively inexpensive [34]. | More expensive due to instrumentation and expertise [34]. |
| Sensitivity | Good for moderate concentrations [34]. | Excellent for trace-level detection [34]. |
| Specificity | Can be affected by cross-reactivity or antibody interference [34] [35]. | Highly specific; can distinguish between molecular isoforms [34] [35]. |
| Multiplexing Capability | Limited; typically measures one analyte per assay. | High; enables simultaneous quantification of multiple targets [36]. |
| Dynamic Range | Declared ranges are suitable for standard concentrations (e.g., 7-2000 mg/L for lactose) [37]. | Wide dynamic range, ensuring accuracy across diverse sample matrices [34]. |
| Applicability to NAD(P)(H) | Enzyme cycling assays are common but show significant inter-method variability [38]. | Considered a gold standard, providing high sensitivity and resolution for the NAD+ metabolome [38]. |
Experimental Data and Methodological Protocols
Case Study: Quantifying Residual Lactose in Milk
A direct comparative study analyzing residual lactose in lactose-free milk demonstrated the practical performance differences between these methods [37] [39].
- Enzymatic Assay Performance: The study found that enzymatic kits failed to reliably quantitate residual lactose in lactose-free milk. A primary reason is their inability to differentiate between glucose/galactose originating from lactose hydrolysis and those from other sources, such as free monosaccharides or lactulose. This leads to significant errors, especially when lactose concentrations are very low [37].
- LC-MS/MS Performance: In contrast, an developed LC-MS/MS method using a formate adduct allowed for the precise quantitation of both lactose and lactulose at high levels of precision and repeatability. It effectively distinguished between these similar disaccharides, a task where the enzymatic and HPLC-RI methods proved inadequate [37] [39].
Table 2: Experimental Results from Lactose-Free Milk Analysis
| Method | Performance in Low-Lactose Analysis | Key Limitation | Reported Strengths |
|---|---|---|---|
| Enzymatic Assay | Inadequate for reliable quantification [37]. | Cannot differentiate lactose-derived sugars from other sources; error increases at low concentrations [37]. | Simplicity and cost-effectiveness for standard concentrations [37] [34]. |
| HPLC with RI Detection | Inadequate; lacks sufficient sensitivity [37]. | Limit of Detection (LOD) reported at ~250 mg/L [37]. | Straightforward analysis for high-concentration sugars [37]. |
| LC-MS/MS | Excellent precision and repeatability [37] [39]. | Requires specialized expertise and instrumentation [34]. | High specificity and sensitivity; can quantify and characterize multiple species simultaneously [37] [34]. |
Detailed Experimental Protocols
Protocol for NAD+ Metabolome Analysis via LC-MS/MS
The following workflow is adapted from methods used for comprehensive NAD+ metabolome characterization [40] [38].
- Sample Collection and Quenching: Rapidly quench metabolism immediately upon collection using flash-freezing in liquid nitrogen or chilled organic solvents (e.g., methanol at -80°C) to preserve the in vivo metabolite levels [41] [38].
- Metabolite Extraction:
- Use a biphasic liquid-liquid extraction with a methanol/chloroform/water system.
- Polar metabolites (like NAD+) are extracted into the methanol/water phase, while lipids partition into the chloroform phase [41].
- Critical Step: Add isotopically labeled internal standards (e.g., ( ^2H4 )-NAM, ( ^{13}C5 )-adenosine) to the extraction solvent before sample processing to correct for variability and enable accurate quantification [40] [41] [38].
- LC-MS/MS Analysis:
- Chromatography: Employ two separation methods for comprehensive coverage.
- Alkaline Separation: For metabolites with a ribose sugar (e.g., NAD+, NADP+). Use a Hypercarb column with solvents like 7.5 mM ammonium acetate with 0.05% ammonium hydroxide (Solvent A1) and 0.05% ammonium hydroxide in acetonitrile (Solvent B1) [40].
- Acid Separation: For metabolites without a sugar moiety [40].
- Mass Spectrometry: Operate the mass spectrometer in multiple reaction monitoring (MRM) mode for high sensitivity and specificity. Quantify metabolites by comparing their peak areas to those of the internal standards using a calibration curve [40] [38].
- Chromatography: Employ two separation methods for comprehensive coverage.
Protocol for NAD(P)H Analysis via Enzyme Cycling Assay
Enzyme cycling assays are a common, albeit less specific, method for quantifying NAD(P)H levels [38].
- Sample Preparation:
- Enzymatic Reaction:
- The assay relies on a proprietary enzyme that catalyzes the reduction of a probe by NAD(P)H.
- The reaction is coupled to a reporter system. For example, in some kits, the reduced probe is detected by its fluorescence (e.g., Ex/Em = 535/587 nm) [37].
- Detection and Quantification:
Diagram 1: NADPH in Metabolic Pathways (Max 760px)
Essential Research Reagent Solutions
The following table outlines key reagents and their functions in these analytical workflows.
Table 3: Key Research Reagents for Metabolic Quantification
| Reagent / Kit | Function / Application | Considerations |
|---|---|---|
| Isotopically Labeled Internal Standards (e.g., ( ^2H4 )-NAM, ( ^{13}C5 )-NAD+) [40] [38] | Essential for accurate LC-MS/MS quantification; corrects for matrix effects and extraction losses. | Crucial for normalizing pre-analytical and analytical variability. |
| Commercial Enzyme Cycling Kits (e.g., Amplite Fluorometric NADP+/NADPH Assay) [37] | Provide optimized reagents for spectrophotometric or fluorometric detection of NAD(P)(H). | Batch-to-batch variability can affect results [34]. |
| Hypercarb LC-MS/MS Column [40] | A porous graphitic carbon stationary phase for separating polar metabolites like NAD+ and related compounds. | Specialized for metabolomics; requires specific solvent conditions (e.g., alkaline buffers). |
| Specific Inhibitors (e.g., Apigenin for CD38, 3-Aminobenzamide for PARPs) [40] | Used in etheno-NAD+ assays to inhibit specific NAD+-consuming enzymes and study their individual activities. | Allows for functional dissection of the NAD+ metabolome. |
Diagram 2: LC-MS/MS Workflow (Max 760px)
The choice between HPLC-MS/MS and enzymatic assays is not merely a technical preference but a strategic decision that shapes research outcomes. LC-MS/MS stands out for applications demanding high specificity, sensitivity, and the ability to multiplex, making it the gold standard for precise quantification of metabolic ratios like NADPH/NADP+ and for characterizing complex metabolomes [34] [38]. Its capability to directly measure and distinguish between molecular isoforms and modifications provides a level of biochemical insight that is often unattainable with antibody-based methods [34] [35].
Conversely, enzymatic assays, including ELISA, offer a cost-effective and simpler alternative that is sufficient for detecting moderate analyte concentrations in high-throughput settings where extreme specificity is not the primary concern [34]. However, researchers must be cognizant of their limitations, including potential cross-reactivity and significant inter-method variability, as highlighted by the meta-analysis of NAD(P)(H) quantification [38]. For advanced research into cellular energy and redox states—a core aspect of the broader thesis on ATP/ADP/NADPH/NADP+ ratios—the precision and comprehensive data provided by LC-MS/MS make it the unequivocally superior technology.
Metabolite Ratios as Quality Indicators in Pre-Analytical Processing
In clinical diagnostics and biomedical research, blood samples represent one of the most frequently used biospecimens for metabolomic analysis. However, the chemical composition of human plasma and serum is highly dynamic and profoundly influenced by pre-analytical variation, introducing non-biological fluctuations that can compromise diagnostic accuracy and research validity [42] [43]. Among the most critical pre-analytical variables is the time-to-centrifugation (TTC)—the duration between blood collection and centrifugation for plasma or serum separation. During this period, ongoing cellular metabolism in erythrocytes, leukocytes, and platelets continuously alters the metabolite profile, making the resulting samples non-representative of in vivo conditions if not properly controlled [43].
The implementation of Standard Operating Procedures (SOPs) for the pre-analytical phase is imperative for advancing reliable metabolomics research. However, in large-scale epidemiological or clinical studies, strict adherence to optimal handling conditions can be challenging, particularly when transport to central laboratories is required [44]. Consequently, researchers require intrinsic quality indicators (QIs)—biomarkers capable of retrospectively revealing a sample's handling history and qualifying its suitability for downstream analysis. While individual metabolites have been proposed as QIs, their substantial interindividual variability often limits diagnostic performance. This limitation has prompted investigation into metabolite ratios, which demonstrate enhanced robustness by reflecting enzymatic conversion rates and metabolic pathway activities that are directly influenced by pre-analytical conditions [42] [44].
This guide objectively compares the performance of established and emerging metabolite ratios as intrinsic quality markers for pre-analytical processing, with particular emphasis on their utility within broader research contexts involving ATP/ADP and NADPH/NADP+ ratio investigations.
Key Metabolite Ratios as Pre-Analytical Quality Indicators
Nucleoside Ratios for Time-to-Centrifugation Assessment
Targeted metabolite profiling studies have identified specific nucleoside ratios that exhibit significant diagnostic performance for detecting prolonged time-to-centrifugation.
Hypoxanthine/Inosine (HI-Ratio) and Hypoxanthine/Guanosine (HG-Ratio): These ratios demonstrate remarkable sensitivity to pre-centrifugation delays at room temperature. In serum samples, both the HI-ratio and HG-ratio show high diagnostic performance (Sensitivity/Specificity > 80%) for discriminating samples with a TTC > 1 hour [42]. The underlying biochemical mechanism involves the continuous enzymatic degradation of adenosine nucleotides: ATP → ADP → AMP → Adenosine → Inosine → Hypoxanthine. As hypoxanthine accumulates and guanosine/inosine decrease, the ratios increase, providing a sensitive measure of cellular energy metabolism disruption ex vivo.
Validation Performance: These nucleoside ratios have been successfully validated in independent sample sets from patients with rheumatic and cardiovascular diseases (n=70 for serum, n=49 for EDTA plasma), confirming their reliability across diverse patient populations [42].
Lysophospholipid/Phospholipid Ratios for Room Temperature Exposure
The conversion of phosphatidylcholines (PCs) to lysophosphatidylcholines (lysoPCs) represents another well-characterized signature of sample degradation.
LysoPC/PC Ratio: Studies subjecting serum samples to pre-storage handling delays found especially pronounced increases in lysoPCs with corresponding decreases in PCs. The ratio between these molecular classes serves as a robust measure to distinguish between 'good' and 'bad' quality samples, with significant changes observable after only 12 hours of storage delay at room temperature [44]. This conversion is primarily mediated by phospholipase enzymes released from blood cells, activity that continues ex vivo.
Temperature Dependence: The degradation signature is markedly less pronounced when samples are maintained on wet ice compared to room temperature, highlighting the critical importance of temperature control during pre-analytical handling [44].
Eicosanoid Metabolites as Sensitive Quality Indicators
Recent discoveries have identified specific eicosanoids as sensitive markers for extended pre-centrifugation delays.
Hydroxyeicosatetraenoic Acids (HETEs): Metabolites including 12-HETE, 15-(S)-HETE, and 8-(S)-HETE have been identified as quality indicators for pre-centrifugation delays exceeding 2 hours [42]. These eicosanoids, derived from arachidonic acid metabolism, demonstrate significant concentration changes during prolonged sample handling.
Validation in Patient Cohorts: Several eicosanoids, including 12-HETE, 12-oxo-HETE, 8-(S)-HETE, and 12-(S)-HEPE, along with the HI- and HG-ratios, have been validated in independent patient sample sets, confirming their utility as QIs in real-world clinical contexts [42].
Amino Acid Ratios in Pre-Analytical Variation
Specific amino acid metabolism pathways also respond characteristically to pre-analytical stresses.
Ornithine/Arginine (OA-Ratio): This ratio has been proposed as a quality indicator in both serum and EDTA plasma [42]. The conversion of arginine to ornithine is catalyzed by arginase enzymes released from blood cells during prolonged handling, causing a measurable shift in this ratio that reflects sample history.
Pathway Enrichment: Over-representation analysis of metabolites significantly affected by TTC has identified enrichment in the "arginine and ornithine metabolism" pathway, supporting the biological plausibility of this ratio as a quality indicator [42].
Comparative Performance Analysis of Quality Indicator Ratios
Table 1: Comparative Performance of Key Metabolite Ratios as Pre-Analytical Quality Indicators
| Metabolite Ratio | Affected Pre-Analytical Variable | Threshold for Detection | Diagnostic Performance | Matrix | Key Metabolic Pathway |
|---|---|---|---|---|---|
| Hypoxanthine/Inosine (HI-Ratio) | Time-to-Centrifugation (TTC) | >1 hour | Sensitivity/Specificity >80% [42] | Serum | Purine Catabolism |
| Hypoxanthine/Guanosine (HG-Ratio) | Time-to-Centrifugation (TTC) | >1 hour | Sensitivity/Specificity >80% [42] | Serum | Purine Catabolism |
| LysoPC/PC Ratio | Room Temperature Exposure | >12 hours | Significant concentration changes [44] | Serum | Phospholipid Metabolism |
| Ornithine/Arginine (OA-Ratio) | Time-to-Centrifugation (TTC) | 0.5h vs 2h [42] | Validated in patient samples [42] | Serum, EDTA Plasma | Urea Cycle/Arginine Metabolism |
| 12-HETE | Time-to-Centrifugation (TTC) | >2 hours | Validated in patient samples [42] | Serum | Arachidonic Acid Metabolism |
Table 2: Experimental Protocols for Assessing Metabolite Ratio Quality Indicators
| Experimental Aspect | HI- and HG-Ratios [42] | LysoPC/PC Ratio [44] |
|---|---|---|
| Sample Collection | Blood collected from healthy volunteers (n=10) and patient cohorts (n=40, n=70) | Fasting serum samples from 19 healthy male volunteers |
| Pre-Analytical Variation | Variable TTC (0.5h, 2h) at room temperature | Pre-storage handling delays (12, 24, 36h) at RT, wet ice, dry ice |
| Processing Protocol | Centrifugation for plasma/serum preparation; UHPLC-MS/MS analysis | Clotting for 30min at RT; centrifugation at 2000×g for 15min at 15°C |
| Analytical Platform | Targeted metabolomics via UHPLC-MS/MS; validation by targeted MS/MS | Targeted metabolomics quantifying 127 metabolites |
| Data Analysis | Metabolite ratio calculation; pathway enrichment analysis; diagnostic performance evaluation | Statistical analysis of concentration changes; ratio calculation between metabolite classes |
Integration with ATP/ADP and NADPH/NADP+ Ratio Research
The use of metabolite ratios as quality indicators aligns methodologically with the investigation of ATP/ADP and NADPH/NADP+ ratios in bioenergetic research. Both applications leverage ratio-based approaches to discern functional metabolic states, albeit with different objectives: quality indicators aim to detect pre-analytical artifacts, while ATP/ADP and NADPH/NADP+ ratios typically reflect in vivo bioenergetic status.
Methodological Parallels in Ratio-Based Assessment
Research into NADPH and NADH/NAD+ redox states utilizes genetically encoded fluorescent sensors (e.g., iNAP, SoNar) to monitor dynamic changes in these ratios across subcellular compartments in response to environmental perturbations such as illumination [45]. Similarly, the evaluation of pre-analytical quality indicators employs mass spectrometry-based platforms to quantify ratio changes resulting from ex vivo handling conditions. Both fields face the challenge of distinguishing biologically meaningful ratio changes from technical artifacts, though the direction of inference is reversed: in bioenergetics research, careful pre-analytical control enables valid biological discovery, whereas in quality indicator application, the ratio changes themselves become the signal that reveals pre-analytical variance.
Pre-Analytical Considerations for NADPH/NADP+ and ATP/ADP Measurements
The lability of energy metabolites necessitates stringent pre-analytical control for accurate ATP/ADP and NADPH/NADP+ ratio determination. The same pre-centrifugation delays that affect hypoxanthine ratios and lysophospholipid/phospholipid ratios would be expected to profoundly alter cellular energy states and redox balances ex vivo. For instance, the ATP/ADP ratio in algal cells dropped from approximately 15 to 5 when anaerobiosis was established [26], highlighting the sensitivity of energy charge to environmental conditions. Similarly, the NADPH/NADP ratio is highly dynamic, changing in response to light exposure and metabolic perturbations [45]. Consequently, establishing quality control measures using the metabolite ratios described herein is particularly crucial for research focusing on cellular bioenergetics.
Signaling Pathways in Pre-Analytical Metabolite Degradation
The metabolite ratio changes observed during pre-analytical handling reflect specific, ongoing biochemical pathways activated during ex vivo sample processing. The diagram below illustrates the key metabolic pathways involved.
Diagram 1: Metabolic pathways activated during pre-analytical processing, leading to characteristic ratio changes used as quality indicators. Abbreviations: HI-Ratio (Hypoxanthine/Inosine), HG-Ratio (Hypoxanthine/Guanosine), LysoPC (Lysophosphatidylcholine), PC (Phosphatidylcholine), OA-Ratio (Ornithine/Arginine), HETEs (Hydroxyeicosatetraenoic Acids), TTC (Time-to-Centrifugation).
Experimental Workflow for Quality Indicator Validation
The development and validation of metabolite ratios as reliable quality indicators follows a structured experimental workflow, from discovery to application.
Diagram 2: Experimental workflow for the discovery and validation of metabolite ratios as quality indicators (QIs) for pre-analytical processing. TTC: Time-to-Centrifugation.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 3: Essential Research Reagents and Platforms for Metabolite Ratio Analysis
| Reagent/Platform | Function in Analysis | Specific Examples/Properties |
|---|---|---|
| UHPLC-MS/MS Systems | High-resolution separation and quantification of metabolites in complex biological samples | Enables targeted metabolomics; detection of 600+ metabolites in serum/plasma [42] |
| Stable Isotope Standards | Internal standards for quantification accuracy; correct for technical variation | Isotope-labeled metabolites for absolute quantification [42] |
| Quality Control Pools | Monitor analytical performance across batches; assess technical variance | Pooled serum samples from all study participants [44] |
| Cryoprotective Tubes | Maintain sample integrity during storage; prevent freeze-thaw artifacts | Pre-cooled cryotubes for aliquot storage [44] |
| Targeted Metabolomics Kits | Standardized panels for specific metabolite classes; improve reproducibility | Commercially available kits for nucleosides, eicosanoids, etc. |
| Pathway Analysis Software | Biological interpretation of metabolite changes; identify enriched pathways | MetaboAnalyst, KEGG, ChemRICH for pathway enrichment [42] |
Metabolite ratios represent robust intrinsic markers for assessing pre-analytical sample quality, with specific ratios offering sensitive detection of time-to-centrifugation delays and temperature excursions. The hypoxanthine/inosine and hypoxanthine/guanosine ratios demonstrate particularly strong diagnostic performance for TTC >1 hour, while lysophospholipid/phospholipid ratios effectively identify prolonged room temperature exposure. These ratio-based approaches offer enhanced robustness compared to individual metabolites by reflecting enzymatic activities and pathway fluxes that continue ex vivo.
For researchers investigating ATP/ADP and NADPH/NADP+ ratios, implementing these metabolite ratio quality indicators provides a critical quality assurance layer, ensuring that measured bioenergetic states reflect in vivo biology rather than pre-analytical artifacts. As metabolomics continues to advance as a tool for biomarker discovery and metabolic phenotyping, the integration of such intrinsic quality markers into standard operating procedures will be essential for generating reliable, reproducible data, particularly in multi-center studies where pre-analytical conditions may vary.
CYP450 Metabolic Phenotyping for Personalized Drug Dosing
Cytochrome P450 (CYP450) enzymes constitute a superfamily of heme-containing enzymes that play a pivotal role in the metabolism of approximately 80% of all clinically used drugs [46]. These enzymes facilitate the oxidation of lipophilic substrates, converting them into more hydrophilic metabolites for elimination from the body [47] [48]. The activity of these enzymes exhibits substantial interindividual variability, which significantly contributes to heterogeneous therapy outcomes, including lack of efficacy or adverse drug reactions [49] [46]. CYP450 metabolic phenotyping refers to the process of determining an individual's actual metabolic capacity for specific CYP enzymes in vivo. This approach provides a functional assessment that integrates the net effect of genetic polymorphisms, environmental exposures, and physiological factors on enzyme activity, offering valuable insights for personalized drug dosing that genotyping alone cannot reliably predict [49].
The clinical relevance of CYP450 phenotyping is underscored by the fact that variants in genes encoding drug-metabolizing enzymes influence drug pharmacokinetics and pharmacodynamics, leading to altered drug response [50]. Traditionally, pharmacogenomic effects are categorized using a star allele system that assigns individuals to different metabolizer phenotypes: poor, intermediate, normal, rapid, and ultrarapid metabolizers [50]. However, a significant challenge in clinical practice is phenoconversion, where the measured phenotype differs from the genotype predicted based on genetic testing alone [50] [49]. This discrepancy highlights the limitations of relying solely on genetic information and emphasizes the value of phenotypic assessments that capture the integrated effects of both genetic and non-genetic factors on drug metabolism.
Comparative Analysis of Phenotyping Methodologies
Probe Drug-Based Phenotyping Approaches
Probe drug-based phenotyping involves administering selective substrates for specific CYP enzymes and quantifying pharmacokinetic parameters that reflect metabolic activity. The "Geneva Cocktail" represents a comprehensive phenotyping approach that simultaneously assesses multiple CYP enzymes and P-glycoprotein activity using low doses of specific probes [49]. This methodology employs caffeine (50 mg) for CYP1A2, bupropion (150 mg) for CYP2B6, omeprazole (10 mg) for CYP2C19, flurbiprofen (50 mg) for CYP2C9, dextromethorphan (10 mg) for CYP2D6, midazolam (1 mg) for CYP3A4, and fexofenadine (120 mg) for P-gp [49]. Metabolic activities are determined using metabolite/probe concentration ratios measured 2 hours after cocktail administration, while P-gp activity is assessed through the area under the curve (AUC) of fexofenadine concentration at 3 and 6 hours post-administration [49].
Table 1: Standard Probe Drugs for CYP Phenotyping
| CYP Enzyme | Probe Drug | Dose Range | Primary Metric | Clinical Applications |
|---|---|---|---|---|
| CYP1A2 | Caffeine | 50-100 mg | Metabolic ratio (MR) | Psychotropic drug optimization |
| CYP2B6 | Bupropion | 150 mg | MR (hydroxybupropion/bupropion) | Antidepressant dosing |
| CYP2C9 | Flurbiprofen | 50 mg | MR (4'-hydroxyflurbiprofen/flurbiprofen) | Warfarin, NSAID therapy |
| CYP2C19 | Omeprazole | 10-100 mg | AUC0-24 or MR | Clopidogrel, PPI therapy |
| CYP2D6 | Dextromethorphan | 10 mg | MR (dextrorphan/dextromethorphan) | Codeine, tramadol, antidepressants |
| CYP3A4 | Midazolam | 1-75 mg | AUC∞ or clearance | Benzodiazepines, immunosuppressants |
Microdosing approaches represent an innovative development in phenotyping methodologies, administering doses at 1/100th of the anticipated therapeutic dose with a maximum of 100 μg [46]. This approach minimizes potential side effects and pharmacological activity while maintaining the ability to assess metabolic capacity. Current evidence supports the use of microdosing for phenotyping CYP2C19 (using 100 μg omeprazole with AUC0-24 as the metric) and CYP3A (using 0.1-75 μg midazolam with AUC∞ or clearance as metrics) [46]. Limited sampling strategies further enhance the clinical utility of microdosing, with AUC0-10 and AUC2-4 identified as viable metrics for midazolam phenotyping [46].
Genotyping Versus Phenotyping: Comparative Data
Genetic testing for CYP polymorphisms provides valuable information about an individual's inherited metabolic potential, with population-scale sequencing data revealing substantial ethnogeographic differences in CYP gene variability [51]. Analysis of 141,614 individuals across 12 populations demonstrated that rare alleles contribute between 1.5% and 17.5% to the overall genetically encoded functional variability in CYP enzymes [51]. However, the correlation between genotype and observed metabolic phenotype is imperfect, with phenoconversions occurring in 30-60% of cases [49]. This discrepancy arises because phenotypic assessments capture the integrated effects of genetic polymorphisms, drug-drug interactions, environmental exposures, and physiological factors on enzyme activity.
Table 2: Comparison of CYP Phenotyping Methodologies
| Methodology | Key Features | Advantages | Limitations | Clinical Validation |
|---|---|---|---|---|
| Genotyping | Identifies star alleles and copy number variations | Simple sample collection; no drug administration | Does not capture environmental influences or phenoconversion | CPIC guidelines for multiple drug-gene pairs |
| Therapeutic/Subtherapeutic Phenotyping | Uses milligram-range probe doses | Established metrics and interpretation guidelines | Risk of side effects; requires medical supervision | Validated procedures for major CYP enzymes |
| Microdosing Phenotyping | ≤100 μg probe doses | Minimal side effects; suitable for vulnerable populations | Limited validation for some CYP enzymes | Established for CYP2C19 and CYP3A |
Real-world evidence from the All of Us Research Program demonstrates the clinical utility of CYP phenotyping, validating known pharmacogenomic interactions for drugs including propranolol, metoprolol, citalopram, tramadol, and sertraline with CYP2D6; paroxetine, esomeprazole, venlafaxine, and omeprazole with CYP2C19; warfarin with CYP2C9; and tacrolimus with CYP3A5 [50]. However, not all expected associations were confirmed, suggesting that pharmacogenomic analyses may be affected by factors such as phenoconversion, limited dose adjustments by clinicians, or inherent dataset noise [50].
Experimental Protocols for CYP Phenotyping
Geneva Cocktail Phenotyping Protocol
The Geneva Cocktail approach provides a standardized methodology for comprehensive CYP phenotyping [49]. The experimental workflow begins with an overnight fast before cocktail administration, and participants must delay their morning drug intake until after the first blood sample collection. When assessing CYP1A2 activity with caffeine, patients are asked to avoid caffeine-containing products (coffee, cola beverages, tea) for 24 hours before phenotyping to prevent interference with plasma measurements [49]. The cocktail administration is supervised by medical personnel, and blood samples are collected into lithium heparin tubes at specified time points. Samples are stored at room temperature (<25°C) before shipment to the laboratory within 24 hours, or frozen between -15°C and -25°C if processing is delayed beyond three weeks [49].
Quantification of probe drugs and their metabolites is performed using HPLC-MS/MS methods with established analytical parameters [49]. CYP activities are classified as decreased, normal, or increased based on metabolic ratios (MRs) determined 2 hours after cocktail administration, using ranges previously established in healthy populations [49]. For CYP2D6, activities are categorized as ultrarapid metabolizer (UM), extensive metabolizer (EM), intermediate metabolizer (IM), or poor metabolizer (PM) [49]. The phenotypic results are integrated with the patient's clinical presentation and medication history to guide therapeutic decisions, including drug switching, dosage adjustments, or alternative treatment selection.
Microdose Phenotyping Protocol
Microdose phenotyping follows a similar workflow but utilizes substantially lower probe doses to minimize pharmacological effects [46]. For CYP2C19 phenotyping, a single 100 μg dose of omeprazole is administered, with blood sampling over 24 hours to determine the AUC0-24 [46]. For CYP3A phenotyping, 0.1-75 μg of midazolam is administered, with metrics including AUC extrapolated to infinity (AUC∞) or clearance [46]. Limited sampling strategies have been validated for midazolam microdosing, with AUC0-10 and AUC2-4 identified as practical alternatives to comprehensive sampling [46].
The sensitivity of microdose phenotyping methods is validated through drug-drug interaction studies that quantify the phenotyping metric under three conditions: baseline (absence of influencing factors), during inhibition (co-administration with a CYP inhibitor), and during induction (co-administration with a CYP inducer) [46]. A phenotyping method is considered sensitive when the metric significantly changes during both inhibition and induction compared to baseline [46]. Pharmacokinetic scalability from microdose to therapeutic levels is assessed using the twofold criterion, where microdose pharmacokinetics are considered predictive if mean values fall within twofold of therapeutic dose parameters [46].
The Impact of Exposome on CYP450 Activity
The exposome, defined as the sum of all internal and external exposures throughout a person's lifetime, significantly influences CYP450 function and contributes to phenoconversion [47] [48]. External exposome components include dietary substances, lifestyle choices, environmental pollutants, and medications, while internal factors encompass gut microbiota, hormone fluctuations, oxidative stress, inflammation, and disease states [47] [48]. Understanding these influences is essential for interpreting phenotyping results and optimizing personalized dosing strategies.
Dietary components significantly modulate CYP activity, with grapefruit juice serving as a potent inhibitor of CYP3A4, affecting the bioavailability of numerous cardiovascular drugs [47] [48]. Conversely, cruciferous vegetables containing indole-3-carbinol can induce CYP1A enzymes, potentially altering the metabolism of substrates including antidepressants and antipsychotics [47]. Lifestyle factors such as tobacco smoking induce CYP1A1 and CYP1A2 activity through exposure to polycyclic aromatic hydrocarbons, while chronic alcohol consumption can either suppress or stimulate different CYP enzymes depending on the pattern and duration of consumption [47] [48].
The internal exposome components further complicate the prediction of CYP activity based solely on genetic information. Gut microbiota-derived metabolites can either inhibit or induce specific CYP enzymes, while inflammatory states and infectious diseases can downregulate CYP expression through cytokine-mediated pathways [47] [48]. Disease-related physiological changes, particularly hepatic and renal impairments, directly affect drug metabolism and clearance, necessitating phenotypic assessment rather than reliance on genotypic predictions alone [48]. This complex interplay between environmental exposures and biological systems underscores why phenoconversion occurs so frequently and highlights the clinical value of phenotyping approaches that capture the net effect of these influences on drug metabolism.
Research Reagent Solutions for CYP Phenotyping
Table 3: Essential Research Reagents for CYP Phenotyping Studies
| Reagent Category | Specific Examples | Research Application | Technical Considerations |
|---|---|---|---|
| Selective CYP Probes | Caffeine (CYP1A2), Bupropion (CYP2B6), Omeprazole (CYP2C19), Flurbiprofen (CYP2C9), Dextromethorphan (CYP2D6), Midazolam (CYP3A4) | Phenotyping specific CYP enzyme activities | Cross-reactivity with other enzymes should be minimal; dose selection critical for safety and sensitivity |
| Analytical Standards | Parent compounds and major metabolites for each probe | HPLC-MS/MS quantification | Stable isotope-labeled internal standards recommended for optimal precision |
| CYP Inhibitors | Furafylline (CYP1A2), Ticlopidine (CYP2B6), Fluconazole (CYP2C9), Fluoxetine (CYP2C19), Quinidine (CYP2D6), Ketoconazole (CYP3A) | Method validation through inhibition studies | Selectivity and potency vary; consider clinical relevance when choosing inhibitors |
| CYP Inducers | Omeprazole (CYP1A2), Rifampicin (broad inducer), Phenobarbital (CYP2B, CYP3A) | Method validation through induction studies | Induction requires multiple doses over days rather than single administration |
| Biological Matrices | Human plasma, liver microsomes, recombinant CYP enzymes | In vitro studies and analytical validation | Inter-individual variability in human tissue samples; storage conditions critical |
The implementation of CYP phenotyping in research settings requires careful selection of probe drugs based on their specificity, sensitivity, and safety profiles [49] [46]. The "Geneva Cocktail" offers a balanced approach with probes specifically selected for minimal interaction and side effects at the administered doses [49]. For analytical quantification, HPLC-MS/MS represents the gold standard methodology, providing the necessary sensitivity and specificity for simultaneous quantification of multiple probes and metabolites, particularly important for cocktail approaches [49]. Method validation must establish precision, accuracy, linearity, and stability under appropriate storage conditions, with quality control samples ensuring consistent performance across analytical runs.
CYP450 metabolic phenotyping represents a powerful tool for advancing personalized medicine by providing functional assessment of drug metabolism capacity that integrates genetic, environmental, and physiological influences. While genotyping identifies inherited metabolic potential, phenotyping captures the net effect of all factors affecting enzyme activity, including the frequently overlooked exposome components [47] [48]. Clinical implementation of phenotyping has demonstrated value in optimizing pharmacotherapy, particularly in complex cases involving therapeutic failure or adverse drug reactions [49].
Future developments in CYP phenotyping will likely focus on standardizing methodologies across institutions, validating limited sampling strategies to enhance clinical feasibility, and expanding the application of microdosing approaches to additional CYP enzymes [46]. Integration of phenotyping data with electronic health records, as demonstrated by the All of Us Research Program, offers exciting opportunities for large-scale correlation of metabolic phenotypes with clinical outcomes across diverse populations [50]. Furthermore, advancing our understanding of how specific exposome components influence CYP activity will refine the interpretation of phenotyping results and enhance the precision of personalized dosing recommendations.
As precision medicine evolves, CYP phenotyping stands as a crucial methodology for addressing the significant interindividual variability in drug response, potentially reducing the incidence of adverse drug reactions and therapeutic failures while optimizing treatment efficacy across diverse patient populations.
Assessing Mitochondrial Bioenergetics Beyond Static ATP Measurements
Mitochondrial bioenergetics transcends the simplistic measurement of cellular ATP levels. A comprehensive understanding requires the real-time assessment of energy flux, oxidative phosphorylation (OXPHOS) efficiency, and the redox state of critical cofactors across diverse physiological conditions. Modern approaches now simultaneously monitor multiple parameters—including ATP synthesis rates, oxygen consumption, and NAD(P)H dynamics—to capture the metabolic flexibility and functional capacity of mitochondrial networks within their native cellular context [52] [53]. This guide compares cutting-edge methodologies that enable researchers to move beyond static snapshots and capture the dynamic nature of bioenergetic processes, which is particularly crucial for understanding complex disease pathologies and drug responses.
Comparative Analysis of Methodologies for Dynamic Bioenergetic Assessment
The following table summarizes the capabilities of key advanced techniques for evaluating mitochondrial function.
Table 1: Comparison of Methodologies for Dynamic Bioenergetic Assessment
| Methodology | Key Measured Parameters | Temporal Resolution | Throughput Potential | Primary Applications |
|---|---|---|---|---|
| Enzyme-Linked Respiratory Clamp [52] | Simultaneous JATP (ATP synthesis rate) and JO2 (O2 consumption rate) | Real-time, steady-state | Medium (Purified systems) | Direct calculation of OXPHOS efficiency (ATP/O ratio); Assessment under clamped ADP conditions. |
| NAD(P)H Autofluorescence & FLIM [53] | Mitochondrial redox state (combined NADH & NADPH); FLIM separates NADH from NADPH signals. | Real-time, high resolution | Low to Medium | Investigating metabolic shifts (e.g., glycolysis vs. OXPHOS); Linking redox state to cellular function. |
| Seahorse XF Technology (Inferred from context) | Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) | Near real-time | High (Multi-well plates) | Profiling cellular metabolic phenotypes; Drug toxicity screening. |
Detailed Experimental Protocols for Key Assays
Protocol 1: Simultaneous Real-Time Measurement of ATP Synthesis and Oxygen Consumption
This enzyme-coupled "respiratory clamp" technique allows the direct, real-time quantification of OXPHOS efficiency (ATP/O ratio) in permeabilized cell systems or isolated mitochondria [52].
Principle: The system couples mitochondrial ATP production to the stoichiometric reduction of NADP+ to NADPH, whose fluorescence is measured simultaneously with polarigraphic O2 consumption.
Workflow:
- System Calibration: Generate an ATP standard curve in the presence of the enzyme-coupled system and respiratory substrates to quantify JATP from NADPH fluorescence.
- Sample Preparation: Use permeabilized muscle fiber bundles (PmFBs) or isolated mitochondria. PmFBs are preferred as they retain the native mitochondrial reticulum and energy transfer systems [52].
- Assay Setup: Load samples into a chamber integrating fluorometry and respirometry. The assay buffer must contain:
- Pyruvate (5 mM), Malate (0.5 mM), Succinate (5 mM) as respiratory substrates.
- The enzyme-coupled system: Hexokinase II (1 U/mL), Glucose-6-phosphate dehydrogenase (2.5 U/mL), D-glucose (2.5 mM), and NADP+ (2.5 mM).
- Data Acquisition: Initiate reactions with successive ADP additions (e.g., 15, 200, 2000 μM). Simultaneously record NADPH fluorescence (JATP) and O2 concentration (JO2).
- Data Analysis: Calculate the ATP/O ratio at each steady-state flux from the ratio of JATP to JO2. The study demonstrated that ATP/O ratios increase with energy demand, from <1 at low ADP to >2 at maximal ADP-stimulated respiration [52].
Diagram 1: Enzyme-coupled respiratory clamp workflow.
Protocol 2: Investigating Mitochondrial Redox State via NAD(P)H Fluorescence Lifetime Imaging (FLIM)
This method leverages the innate fluorescence of reduced NADH and NADPH to assess the mitochondrial redox state, with FLIM providing superior molecular discrimination [53].
Principle: NADH and NADPH are fluorescent in their reduced states but not when oxidized. Their fluorescence lifetimes are sensitive to protein binding, allowing FLIM to distinguish free from enzyme-bound NADH and to separate the signals of NADH from those of NADPH.
Workflow:
- Sample Preparation: Live cells or fresh tissue slices are required to maintain metabolic activity. No exogenous labeling is needed.
- Image Acquisition: Use a multi-photon microscope equipped with FLIM capability. Excite samples at ~740 nm and collect emission at 450-470 nm.
- Lifetime Decay Analysis: Fit the fluorescence decay curve at each pixel to a multi-exponential model. The average fluorescence lifetime and the fraction of protein-bound NADH are calculated.
- Metabolic Perturbation: Treat cells with inhibitors (e.g., oligomycin, rotenone) or nutrients to induce metabolic shifts and monitor consequent changes in NAD(P)H lifetime and intensity.
- Data Interpretation: An increase in the protein-bound NADH fraction indicates a more reduced mitochondrial matrix and a shift toward oxidative metabolism. FLIM can dissect complex contributions to the total NAD(P)H signal, providing deeper insight into the underlying metabolic pathways [53].
Metabolic Pathways and NAD+ Metabolism in Bioenergetics
The cofactor NAD+ is a central node in mitochondrial metabolism, serving as a crucial redox carrier and a signaling molecule.
- NAD+ Biosynthesis and Function: NAD+ is synthesized via the de novo pathway from tryptophan or recycled via the salvage pathway from nicotinamide (NAM). It is critical for the TCA cycle and ETC, and also acts as a substrate for sirtuins and PARPs, linking energy status to cellular signaling [54] [55].
- Redox Couples and Bioenergetics: The NAD+/NADH ratio primarily reflects the catabolic and energy-producing capacity of the cell. In contrast, the NADP+/NADPH ratio is maintained in a more reduced state and is crucial for anabolic biosynthesis and antioxidant defense [53] [2]. During oxidative stress, cells can rapidly convert NAD+ to NADP+ via NAD kinase (NADK) to bolster antioxidant capacity [2].
Diagram 2: NAD+ metabolism and redox pairs.
The Scientist's Toolkit: Essential Research Reagents
Table 2: Key Reagents for Mitochondrial Bioenergetic Assays
| Reagent / Assay Component | Function in Experiment | Example Application |
|---|---|---|
| Permeabilizing Agents (Saponin) | Selectively perforates the plasma membrane, allowing external application of substrates and ADP to mitochondria within their native cytoskeletal network. | Preparation of permeabilized fiber bundles (PmFBs) for physiological assessment of OXPHOS [52]. |
| Enzyme Cocktail (HK/G-6-PDH) | Core of the "respiratory clamp"; HK consumes ATP, and G-6-PDH generates NADPH in a 1:1 stoichiometry with ATP, enabling its fluorescent quantification. | Real-time JATP measurement in the enzyme-linked respiratory clamp assay [52]. |
| NAD+ Precursors (NMN, NR) | Boost intracellular NAD+ levels by feeding into the salvage pathway, allowing researchers to test the metabolic impact of enhanced NAD+ bioavailability. | Investigating the role of NAD+ metabolism in aging, disease models, and mitochondrial function [55]. |
| Fluorescence Dyes (JC-1, MitoSox Red) | JC-1 assesses mitochondrial membrane potential (ΔΨm). MitoSox Red detects mitochondrial superoxide production. | Complementary measures of mitochondrial health and ROS production [56]. |
| NADK Inhibitor (Thionicotinamide) | Inhibits NAD kinase, the enzyme that phosphorylates NAD+ to form NADP+, allowing study of this specific metabolic conversion. | Probing the interplay between NAD+ and NADP+ pools during oxidative stress [2]. |
Isotopically Nonstationary Metabolic Flux Analysis (INST-MFA) is a powerful computational and experimental methodology for estimating in vivo reaction fluxes in metabolic networks by analyzing transient isotope labeling patterns. Unlike traditional steady-state 13C-MFA that requires isotopic equilibrium, INST-MFA leverages data from the dynamic period following the introduction of an isotope tracer, before the system reaches isotopic steady state [57]. This approach is particularly valuable for studying pathway energetics, as it provides enhanced sensitivity for quantifying reversible reactions and exchange fluxes that are central to energy metabolism [57] [58]. For researchers investigating ATP/ADP and NADPH/NADP+ ratios across different physiological conditions, INST-MFA offers a unique window into the dynamic regulation of energy charge and redox balance within living systems.
The fundamental principle underlying INST-MFA is that metabolic pathways rearrange substrate atoms in predictable ways. By administering a stable isotope tracer (such as 13C-labeled glucose or 13CO2) and monitoring the patterns of isotope incorporation into downstream metabolites over time, researchers can infer the operational fluxes through interconnected metabolic pathways [57]. This process has been likened to adding colored dye into a network of interconnected tanks to determine flow rates based on how the color spreads through the system [57]. The technique requires solving differential equations that describe the time-dependent labeling of network metabolites while iteratively adjusting flux and metabolite pool size parameters to achieve the best fit with experimental measurements [59] [57].
Fundamental Principles and Comparative Advantages
Theoretical Foundations of INST-MFA
INST-MFA relies on several core mathematical and computational components. The approach requires solving systems of ordinary differential equations (ODEs) that describe the temporal evolution of mass isotopomer distributions (MIDs) for metabolites within the network [59]. These ODEs are constructed based on the network stoichiometry, atom transitions in biochemical reactions, and the current estimates of metabolic fluxes and pool sizes. The inverse problem of estimating fluxes from labeling data is typically solved using iterative optimization algorithms that minimize the difference between simulated and measured labeling patterns [59] [60]. For large networks, the concept of Elementary Metabolite Units (EMUs) has been developed to reduce computational complexity by grouping isotopomers that exhibit equivalent labeling states [59].
INST-MFA imposes specific requirements on experimental design and data collection. Time-resolved measurements of isotope labeling are essential, typically obtained using mass spectrometry techniques such as GC-MS or LC-MS/MS [61]. The frequency and duration of sampling must be carefully designed to capture the labeling kinetics without missing important transient metabolic states. For studies focusing on energy metabolism, additional measurements of energy cofactors and their ratios may be incorporated to constrain flux solutions further [62]. The optimal experimental time frame depends on the turnover rates of metabolite pools, which can vary significantly between different cellular compartments and metabolic pathways.
Comparative Analysis of INST-MFA Versus Alternative Flux Analysis Methods
Table 1: Comparison of INST-MFA with Other Metabolic Flux Analysis Approaches
| Method | Principle | Data Requirements | Applicability to Energetics | Limitations |
|---|---|---|---|---|
| INST-MFA | Fitting transient isotope labeling data using ODEs | Time-course mass isotopomer distributions, metabolite pool sizes (optional) | Excellent for reversible reactions, exchange fluxes, and energy cofactor cycling [57] [58] | Computationally intensive, requires dense time-series data |
| Stationary 13C-MFA | Fitting isotopic steady-state labeling patterns | Mass isotopomer distributions at isotopic steady state | Good for net fluxes, limited for exchange fluxes [60] | Cannot be used for autotrophic systems or when isotopic steady state is not achievable |
| Flux Balance Analysis (FBA) | Optimization of objective function subject to stoichiometric constraints | Genome-scale model, uptake/secretion rates (optional) | Good for predicting optimal flux distributions under assumed cellular objectives [60] [63] | Requires assumption of cellular objective, does not use experimental labeling data |
| Metabolic Flux Ratio Analysis (METAFoR) | Analysis of 13C-1H correlation patterns from NMR | 2D NMR spectra of hydrolyzed cell protein | Provides relative flux ratios at metabolic branch points [64] | Limited to central carbon metabolism, requires specialized NMR equipment |
INST-MFA provides several distinct advantages for studying pathway energetics compared to alternative approaches. First, it offers enhanced sensitivity for quantifying exchange fluxes and reversible reactions, which are often fundamental to energy metabolism [57] [58]. Second, it can be applied to autotrophic systems where the sole carbon source is CO2, a scenario where steady-state MFA fails because all metabolites become fully labeled at isotopic steady state [59] [61]. Third, INST-MFA enables simultaneous estimation of both metabolic fluxes and metabolite pool sizes, providing a more comprehensive view of metabolic state [57]. This is particularly valuable for energy metabolism studies, as it allows researchers to relate flux changes to variations in energy charge and redox state.
Methodological Implementation and Experimental Design
Experimental Workflow for INST-MFA
The standard workflow for implementing INST-MFA involves several critical steps that must be carefully executed to ensure reliable flux estimates. First, the biological system is cultivated under metabolic steady-state conditions, ensuring that metabolic fluxes and pool sizes remain constant throughout the experiment [57]. Next, an isotope tracer is introduced at time zero, typically through a rapid medium switch or gas phase change for autotrophic systems [61]. Following tracer introduction, samples are collected at multiple time points to capture the kinetics of isotope incorporation into intracellular metabolites. These samples are immediately quenched to arrest metabolic activity, typically using cold methanol or other extraction solvents [61]. Metabolites are then extracted using appropriate methods, with nonaqueous fractionation techniques often employed to separate metabolites from different cellular compartments [61]. The extracted metabolites are analyzed using mass spectrometry to determine their mass isotopomer distributions (MIDs) over time [61].
For researchers specifically interested in energy metabolism, additional measurements of ATP/ADP and NADPH/NADP+ ratios can be incorporated into the experimental design. These measurements typically involve specialized assays such as the BacTiter-Glo assay for ATP quantification and NADP/NADPH-Glo assay for NADPH/NADP+ ratios [65]. In the ATP assay, cell culture is harvested during early exponential phase, mixed with an equal volume of detection reagent, incubated for 5 minutes, and measured using a luminescence plate reader [65]. The NADPH/NADP+ assay requires cell lysis under specific alkaline conditions, followed by neutralization and detection using specialized reagents [65]. These measurements provide additional constraints for flux estimation and enable direct correlation between flux changes and energy metabolism.
Figure 1: Experimental and Computational Workflow for INST-MFA Studies
Computational Analysis and Flux Estimation
The computational component of INST-MFA involves several interconnected steps. First, a metabolic network model must be defined, including stoichiometry, atom transitions, and compartmentation [59]. For studies focusing on energy metabolism, special attention should be paid to reactions involving ATP hydrolysis, oxidative phosphorylation, and redox cofactor regeneration. Next, the model is used to simulate the time-dependent labeling patterns expected for a given set of flux values and pool sizes [59]. Nonlinear optimization is then performed to identify the flux and pool size parameters that minimize the difference between simulated and experimental labeling data [60]. Statistical analysis follows to evaluate the goodness-of-fit and determine confidence intervals for the estimated parameters [60].
Several software tools are available to facilitate INST-MFA computations. The most widely used platforms include INCA and OpenMebius, both implemented in MATLAB [57]. These tools automate the generation of metabolite and isotopomer balances from user-defined reactions and atom transitions, significantly simplifying the model building process [57]. They also provide capabilities for importing experimental data, performing flux estimation, and conducting statistical analysis. For Bayesian approaches to flux inference, which are gaining popularity for handling model uncertainty, specialized implementations are available that enable Markov Chain Monte Carlo (MCMC) sampling of the flux solution space [66].
Research Reagent Solutions for INST-MFA
Table 2: Essential Research Reagents and Tools for INST-MFA Experiments
| Reagent/Tool | Function | Example Application | Considerations |
|---|---|---|---|
| 13C-labeled substrates | Isotope tracer for metabolic labeling | [U-13C6]glucose for heterotrophic systems, 13CO2 for autotrophic systems [64] [61] | Purity (>98% 13C), position-specific labeling for pathway elucidation |
| Quenching solutions | Rapid arrest of metabolic activity | Cold methanol for microbial systems, liquid nitrogen for tissues | Must effectively stop metabolism without causing metabolite leakage |
| Extraction solvents | Metabolite extraction from cells/tissues | Methanol/water/chloroform for comprehensive metabolite coverage | Selectivity for different metabolite classes, compatibility with MS analysis |
| Mass spectrometry systems | Measurement of mass isotopomer distributions | GC-MS, LC-MS/MS for time-course labeling analysis | Sensitivity, resolution, and fragmentation patterns for MID determination |
| INST-MFA software | Computational flux estimation | INCA, OpenMebius [57] | Support for network definition, ODE solving, parameter estimation |
| Cofactor assay kits | Quantification of energy charge and redox state | BacTiter-Glo for ATP, NADP/NADPH-Glo [65] | Sensitivity, specificity, compatibility with cell type |
Applications in Pathway Energetics and Case Studies
Investigation of Cancer Cell Energetics
INST-MFA has provided remarkable insights into the metabolic basis of aerobic glycolysis in cancer cells, a phenomenon known as the Warburg effect. A comprehensive INST-MFA study across 12 human cancer cell lines revealed that the total ATP regeneration flux did not correlate with cellular growth rates, challenging conventional assumptions about energy metabolism in proliferating cells [62]. When combined with flux balance analysis (FBA), these INST-MFA results indicated that cancer cells rewire their metabolic networks to balance ATP production with thermal constraints, providing a novel explanation for the prevalence of inefficient aerobic glycolysis in cancer metabolism [62]. The INST-MFA data showed that cancer cells maintain a specific balance between glycolytic and oxidative phosphorylation fluxes that minimizes heat generation while sustaining biosynthetic precursor production, demonstrating how INST-MFA can reveal fundamental principles of metabolic regulation that are not apparent from steady-state approaches.
Analysis of Photosynthetic and Autotrophic Metabolism
INST-MFA has proven particularly valuable for studying energy metabolism in photosynthetic organisms, where traditional steady-state MFA is not applicable due to complete labeling of all metabolites when 13CO2 is the sole carbon source [61]. In cyanobacteria and plants, INST-MFA has been used to quantify fluxes through the Calvin cycle, photorespiratory pathway, and associated energy-generating processes [57] [61]. These studies have revealed how photosynthetic organisms allocate reducing power (NADPH) and ATP between carbon fixation, photoprotection, and maintenance processes under different environmental conditions. For example, INST-MFA studies of Synechococcus sp. PCC 7002 provided detailed insights into how cyanobacteria balance light capture, carbon fixation, and energy dissipation under high-light conditions [57]. Similarly, INST-MFA applications in Arabidopsis thaliana have elucidated the metabolic adjustments that occur during cold acclimation, including changes in starch and organic acid accumulation that define photosynthetic acclimation to temperature stress [63].
Figure 2: Simplified Cancer Metabolism Showing ATP/NADPH Production Pathways
Local INST-MFA Approaches for Targeted Analysis
For researchers focusing on specific aspects of energy metabolism, local INST-MFA approaches offer simplified alternatives to comprehensive network analysis. Three main local approaches have been developed: kinetic flux profiling (KFP), non-stationary metabolic flux ratio analysis (NSMFRA), and ScalaFlux [59]. KFP relies on a system of ordinary differential equations for the unlabeled fraction of metabolites and can estimate the total flux through a specific metabolite [59]. NSMFRA estimates relative local fluxes at metabolic branch points where pathways converge, using simulated labeling data based on Hill kinetics for metabolites without measured isotopomers [59]. ScalaFlux is designed to estimate fluxes for any reaction or subnetwork with sufficient labeling data and automatically constructs the necessary ODEs from the network structure [59].
Table 3: Comparison of Local INST-MFA Approaches for Energetics Studies
| Method | Data Requirements | Flux Output | Applicability to Energy Metabolism |
|---|---|---|---|
| Kinetic Flux Profiling (KFP) | Unlabeled (M+0) isotopomer fractions only | Total flux through a metabolite | Limited to specific network motifs; extended KFP (eKFP) can be adapted to more motifs [59] |
| Non-stationary Metabolic Flux Ratio Analysis (NSMFRA) | All isotopomer fractions, atom transitions | Relative local fluxes at branch points | Excellent for determining flux partitioning at energy branch points (e.g., pyruvate node) [59] |
| ScalaFlux | All isotopomer fractions, atom transitions, network structure | Absolute fluxes for any subnetwork | Highly flexible; can be applied to energy-generating pathways like TCA cycle or pentose phosphate pathway [59] |
These local approaches are particularly useful for drug development professionals studying how therapeutic interventions affect specific energy-producing pathways in disease models. For example, local INST-MFA could be used to quantify how an investigational drug affects flux through glycolysis versus oxidative phosphorylation in cancer cells, providing mechanistic insights that complement traditional measurements of ATP levels and viability assays.
Future Directions and Concluding Perspectives
The field of INST-MFA continues to evolve with several emerging trends that will further enhance its utility for studying pathway energetics. Bayesian approaches to flux inference are gaining popularity as they enable more robust handling of measurement uncertainty and model selection [66]. Unlike conventional best-fit approaches, Bayesian methods can quantify the probability of different flux solutions and perform multi-model inference, which is particularly valuable when studying complex and poorly constrained energy metabolism networks [66]. Additionally, there is growing interest in integrating INST-MFA with other omics technologies and constraint-based modeling approaches such as flux balance analysis [60] [62]. Such integration enables researchers to place INST-MFA-derived fluxes into a broader systems biology context, potentially revealing how transcriptional and translational regulation impacts energy metabolism.
Another promising direction is the expansion of INST-MFA to study compartmented energy metabolism in eukaryotic systems. Many energy-related processes, such as oxidative phosphorylation in mitochondria and glycolysis in the cytosol, involve intricate cross-compartment metabolite exchange that has been challenging to quantify. Recent methodological advances in nonaqueous fractionation and subcellular metabolomics are beginning to enable INST-MFA applications that can resolve fluxes between cellular compartments [61]. These developments will be particularly important for understanding how diseases such as cancer and metabolic disorders alter the spatial organization of energy metabolism within cells.
In conclusion, INST-MFA represents a powerful methodology for investigating dynamic pathway energetics that offers significant advantages over stationary approaches for many applications. Its ability to quantify exchange fluxes, resolve compartmented metabolism, and estimate metabolite pool sizes makes it uniquely suited for studying the complex regulation of ATP production, NADPH regeneration, and cellular energy charge. As computational tools become more accessible and experimental protocols more standardized, INST-MFA is poised to become an increasingly central technique in both basic metabolic research and applied drug development.
Interpreting Ratio Dynamics and Overcoming Analytical Challenges
Why Cellular ATP Levels Alone Misrepresent Bioenergetic Health
In bioenergetic research, the quantification of cellular adenosine triphosphate (ATP) has long been a standard metric for assessing cellular energy status. However, a growing body of evidence underscores that ATP levels in isolation provide a static and often misleading snapshot, failing to capture the dynamic and compartmentalized nature of cellular energy production and flux. This guide synthesizes current research to demonstrate that a comprehensive understanding of bioenergetic health necessitates a multi-parameter approach, including the assessment of adenine nucleotide ratios (ATP/ADP), nicotinamide adenine dinucleotide phosphate ratios (NADPH/NADP+), and kinetic measurements of energy flux. We objectively compare these parameters across various physiological and disease contexts, providing structured experimental data and protocols to equip researchers and drug development professionals with a robust framework for accurate bioenergetic assessment.
Cellular bioenergetics is the cornerstone of physiological function, powering everything from fundamental housekeeping processes to specialized cellular tasks. The prevailing focus on ATP concentration as the primary indicator of energetic health is increasingly recognized as insufficient. ATP levels can remain stable due to robust homeostatic mechanisms, even when the underlying capacity for energy production is severely compromised. Static ATP measurements cannot distinguish between a high-turnover, energetically robust state and a low-turnover, compromised state [67]. This limitation is critical in disease research, where mitochondrial dysfunction often manifests as a reduction in energy flux and reserve capacity long before a decline in baseline ATP levels.
A more accurate assessment requires a systems-level view of the bioenergetic network. This includes:
- Energy Charge: The balance between ATP, ADP, and AMP (ATP/ADP ratio).
- Redox State: The ratios of NAD+/NADH and NADP+/NADPH, which reflect the cellular reducing capacity and are crucial for anabolic reactions and antioxidant defense.
- Metabolic Flux: The rates of ATP production and consumption, measured via oxygen consumption rate (OCR) and extracellular acidification rate (ECAR).
- Compartmentalization: The distinct bioenergetic pools within the cytosol, nucleus, and mitochondria.
This article will dissect these components, providing comparative data and methodologies to advance the field beyond the simplistic paradigm of ATP quantification.
Quantitative Comparisons of Bioenergetic Parameters
To illustrate the necessity of a multi-faceted approach, the following tables synthesize quantitative data on key bioenergetic parameters across different experimental and disease conditions.
Table 1: Bioenergetic Parameters in Human Brain Aging and Disease Models
| Condition / Model | ATP Level | ATP/ADP Ratio | NAD+ Level & NAD+/NADH | NADPH/NADP+ Status | Key Findings |
|---|---|---|---|---|---|
| Healthy Human Brain (Young) [68] | Baseline | Baseline | tNAD, NAD+, NAD+/NADH: High | Not Reported | Positive correlation between NAD+ and ATP levels; positive correlation between NAD+/NADH and ATP production rate. |
| Aged Human Brain [68] | Unchanged or mildly decreased | Not Reported | ↓ tNAD, ↓ NAD+, ↓ NAD+/NADH; ↑ NADH | Not Reported | Total NAD and redox ratio decrease with age, impacting energy production capacity without a proportional drop in ATP. |
| Bipolar Disorder (Patient Serum) [69] | Not Measured | Not Measured | Not Measured | Implied by altered TCA metabolites | Significantly elevated serum TCA cycle metabolites (succinate, α-KG, malate) are negatively correlated with cognitive performance. |
| Cancer Cell Model [70] | ↓ Cytosolic ATP/ADP | ↓ | ↑ Mitochondrial NADH/NAD+; ↑ NADPH/NADP+ | ↑ | Non-electrogenic ATP/ADP exchange lowers cytosolic ATP/ADP, stimulating glycolysis (Warburg Effect) and increasing NADPH for biosynthesis. |
| The arrows indicate a significant increase (↑) or decrease (↓) relative to a healthy or control baseline. |
Table 2: Energetic Costs and Output in Model Systems
| Organism / Process | ATP Production per Glucose | Reported ATP Cost per Amino Acid | Methodological Notes |
|---|---|---|---|
| Theoretical Maximum (OxPhos) | ~36 ATP | N/A | Standard biochemical textbook value. |
| Microbial Models (E. coli) [71] | ~100 ATP (if flawed model is used) | ~23.5 ATP (Flawed Model) [71] | A widely used modern model contains a severe error, overestimating real costs by up to 200-fold. |
| Established Microbial Costing [71] | Consistent with ~36 ATP | ~1.2 ATP (Robust Model) [71] | Robust, established microbiological methods provide this realistic estimate. |
| Mammalian Cells (Flawed Model) [71] | ~240 ATP (Untenable) | ~30 ATP (Flawed Model) [71] | The same severe error applied to mammals, leading to untenable conclusions about ATP yield and cost. |
Experimental Protocols for Comprehensive Assessment
Protocol 1: Assessing Brain Energetics and NAD Metabolism via 31P-MRS
This non-invasive method allows for the simultaneous quantification of phospho-metabolites and enzymatic flux rates in the living human brain [68].
Key Workflow Steps:
- Participant Preparation: Participants are fasted overnight and refrain from strenuous exercise prior to the scan to standardize metabolic state.
- Data Acquisition: Scans are performed on a high-field (e.g., 7 Tesla) MR scanner. A pulse-acquire sequence is used to measure 31P resonance signals from metabolites including PCr, γ-ATP, α-ATP, β-ATP, Pi, NAD+, and NADH.
- Saturation Transfer Experiments: To measure kinetic rates, saturation transfer is applied. This includes:
- Saturation of γ-ATP to measure the forward rate constant of creatine kinase (k~CK~).
- Saturation of Pi to measure the forward rate constant of ATP synthase (k~ATP~).
- Quantification: Acquired spectra are analyzed using fitting algorithms like LCModel. Metabolite levels are expressed as a percentage of the total 31P signal. Rate constants are calculated based on the magnetisation transfer effects.
Supporting Reagents:
- Equipment: 7 Tesla MR scanner, 1H quadrature surface coil, single-loop 31P coil.
- Software: Sim4Life for RF simulation, LCModel for spectral analysis, FAST(EST)MAP for B~0~ shimming.
Protocol 2: Quantifying Serum TCA Cycle Metabolites via LC-MS/MS
This protocol is ideal for identifying bioenergetic dysfunction associated with psychiatric and neurological disorders using patient serum [69].
Key Workflow Steps:
- Sample Collection: Blood is drawn from fasted subjects and centrifuged to isolate serum.
- Metabolite Extraction: Proteins are precipitated from the serum, and the supernatant containing metabolites is collected.
- Chromatographic Separation: The extract is injected into an UPLC system to physically separate TCA cycle intermediates (e.g., citrate, α-ketoglutarate, succinate, malate) based on their chemical properties.
- Mass Spectrometric Detection: Metabolites are ionized and detected based on their mass-to-charge ratio (m/z) using a tandem mass spectrometer (MS/MS). This provides high specificity and sensitivity.
- Data Analysis: Peak areas for each metabolite are quantified and compared to internal standards and calibration curves to determine absolute concentrations. Statistical analysis (e.g., Spearman correlation) links metabolite levels to clinical scores.
Supporting Reagents:
- Equipment: UPLC system coupled to a tandem mass spectrometer (e.g., UPLC-MS/MS).
- Chemicals: Authentic chemical standards for all TCA cycle metabolites, internal standards (isotope-labeled), mass spectrometry-grade solvents (acetonitrile, methanol).
- Software: Vendor-specific MS data acquisition software (e.g., MassLynx, Xcalibur), SPSS for statistical analysis.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Reagents and Tools for Bioenergetic Research
| Reagent / Tool | Primary Function | Application Example |
|---|---|---|
| Seahorse XF Analyzer | Simultaneously measures OCR and ECAR in live cells. | Profiling mitochondrial respiration and glycolytic rate in real-time [67]. |
| Oroboros O2k Respirometer | High-resolution respirometry to study mitochondrial function in isolated mitochondria or cells. | Detailed analysis of electron transport chain function and coupling efficiency [67]. |
| LC-MS/MS Systems | Quantifies specific metabolites (TCA intermediates, nucleotides) with high sensitivity. | Measuring serum levels of succinate, α-KG, and malate in bipolar disorder patients [69]. |
| 31P-MRS at High Magnetic Field | Non-invasive measurement of phosphorus metabolites (ATP, PCr, NAD+) and enzyme flux in vivo. | Correlating brain NAD+ levels with ATP production rates in healthy aging [68]. |
| Fluorescent Biosensors (e.g., genetically encoded) | Visualizing subcellular ATP/ADP, NAD+/NADH ratios in real-time. | Revealing compartment-specific NAD+ concentrations in cytosol, nucleus, and mitochondria [72]. |
| CD38 Inhibitor (e.g., 78c) | Potent and specific inhibition of the NAD+-consuming enzyme CD38. | Used to study age-related NAD+ decline and to boost cellular NAD+ levels [72]. |
Integrated Signaling Pathways in Health and Disease
Bioenergetic parameters do not exist in isolation; they form an integrated network that communicates with major signaling pathways. The following diagram illustrates how a primary insult, such as mitochondrial dysfunction, disrupts this network, leading to diverse pathological outcomes. This integrates mechanisms discussed across cancer [70], bipolar disorder [69], and neurodegeneration [68].
The evidence is clear: relying solely on cellular ATP levels to gauge bioenergetic health is a flawed practice that can obscure true mitochondrial pathophysiology. A static ATP concentration can mask significant underlying deficiencies in energy flux, redox balance, and metabolic resilience. As detailed in this guide, a rigorous assessment requires a multi-parametric strategy that includes dynamic flux measurements (OCR, k~ATP~), adenine nucleotide ratios (ATP/ADP), and redox cofactor ratios (NAD+/NADH, NADPH/NADP+). The experimental protocols and tools provided offer a roadmap for researchers to accurately dissect bioenergetic status across diverse conditions, from cancer and psychiatric disorders to aging. Adopting this comprehensive framework is essential for developing effective therapeutic strategies that genuinely target the core metabolic disruptions in human disease.
In research focusing on energy and redox metabolism, the accurate quantification of ATP, ADP, NADPH, and NADP+ is foundational. The ratios of these metabolites—ATP/ADP and NADPH/NADP+—serve as critical indicators of cellular energy status and reducing power across diverse biological fields, from photosynthesis to diabetes research [26] [73]. However, the integrity of this data is highly susceptible to pre-analytical variables, primarily the sample preparation steps of centrifugation and temperature control. This guide objectively compares the effects of these variables on analyte stability, drawing on experimental data to underscore their impact and provide robust methodological recommendations.
Quantitative Comparison of Pre-Analytical Variable Effects
The following tables synthesize experimental data on how centrifugation and temperature influence the measurement of adenosine and nicotinamide nucleotides.
Table 1: Impact of Centrifugation Parameters on ATP Measurement in Red Blood Cells [74]
| Centrifugation Force (x g) | Centrifugation Duration (min) | Number of Centrifugation Steps | Effect on ATP Measurement |
|---|---|---|---|
| 90 - 16,000 | 1 - 10 | 1 | Increased extracellular ATP concentration, correlating with increased force and duration. |
| 2,300 | 5 | Multiple (Cumulative) | Each subsequent centrifugation step released a comparable amount of ATP, indicating a cumulative effect. |
| 500 - 2,000 | 5 - 10 | 3 - 4 (Typical in literature) | Released ATP quantities are on the same order as the lower range of values measured in mechanotransduction experiments. |
Table 2: Effects of Temperature on NADPH/NADP+ Redox State and ATP Levels [75] [76] [77]
| System / Cell Type | Temperature Condition | Observed Effect on Metabolites |
|---|---|---|
| Stored Human Red Blood Cells | Transition from 4°C storage to 37°C | Dramatic alterations in NADP(H) intracellular content and redox state; increased susceptibility to oxidative stress [77]. |
| Bean Leaves (Phaseolus vulgarus L.) | Increase from 30°C to 35°C | Oxidation of the NADPH/NADP+ pool, limiting carbon assimilation; suggested limitation in reducing power supply [75]. |
| Plant Host Cells during Bacterial Infection (Ralstonia solanacearum) | Not Specified (Ambient) | Effector protein RipAF1 interaction with FNR reduced host NADPH and ATP levels, promoting infection [76]. |
Detailed Experimental Protocols for Investigating Pre-Analytical Variables
To ensure reproducibility and data integrity, below are detailed methodologies for key experiments cited in this guide.
This protocol systematically evaluates the impact of centrifugation force, duration, and repetition on ATP measurement.
- Sample Collection: Collect human whole blood via venipuncture into heparin tubes. Perform experiments within 6 hours of collection.
- Variable Centrifugation: Aliquot whole blood and subject it to different centrifugation conditions.
- Force Gradient: Centrifuge aliquots at room temperature across a range of forces (e.g., 90 g to 16,000 g) for a fixed duration.
- Duration Gradient: Centrifuge aliquots at a fixed force (e.g., 2,300 g) for varying durations (e.g., 1 to 10 minutes).
- Repetition Test: Subject a single sample to multiple consecutive centrifugation steps (e.g., 2,300 g for 5 minutes each), resuspending the pellet in buffer between steps.
- Post-Centrifugation Analysis: Following centrifugation, immediately measure ATP concentration in the supernatant and/or the packed RBC fraction using a validated assay (e.g., luciferase-based). In parallel, measure free hemoglobin in the supernatant as an indicator of hemolysis.
- Data Correlation: Correlate the measured ATP concentrations with centrifugation parameters (force, time, repetitions) and hemolysis levels.
This protocol assesses the metabolic consequences of rewarming stored RBCs to physiological temperature.
- Sample Preparation and Storage: Store therapeutic-grade human RBC units under standard blood bank conditions at 4°C for varying durations.
- Temperature Challenge: After storage, aliquot the RBCs and incubate them at different temperatures:
- 4°C Control: Maintain an aliquot at storage temperature.
- 37°C Test: Incubate an aliquot at body temperature (37°C) for a defined period.
- 40°C Test: Incubate an aliquot at a febrile temperature (40°C) for a defined period.
- Metabolite Extraction and Analysis: After incubation, rapidly lyse the cells. Use acid extraction to stabilize labile metabolites.
- Pathway Modulation: To probe mechanism, modulate the NADPH-producing pentose phosphate pathway (PPP) during the temperature challenge (e.g., with substrates or inhibitors) and compare the outcomes to controls.
Signaling Pathways and Experimental Workflows
Diagram: Pre-Analytical Stressors and Their Impact on Nucleotide Ratios
The diagram below illustrates the logical pathway through which pre-analytical variables like centrifugation and temperature disrupt cellular state and lead to inaccurate measurement of key nucleotide ratios.
The Scientist's Toolkit: Research Reagent Solutions
This table details key reagents and materials essential for conducting rigorous research into energy and redox metabolites, along with their primary functions in this context.
Table 3: Essential Reagents for ATP/ADP and NADPH/NADP+ Research
| Reagent / Material | Function in Research |
|---|---|
| Enzymatic Cycling Reagents (e.g., Glucose-6-Phosphate Dehydrogenase, Glutamate Dehydrogenase) | Amplify signals for sensitive spectrophotometric or fluorometric detection of NADPH and NADP+ in low-concentration samples [73]. |
| Genetically Encoded Biosensors (e.g., NAPstar family) | Enable real-time, subcellular resolution monitoring of NADPH/NADP+ ratios in live cells, overcoming limitations of destructive extraction methods [78]. |
| Chemical Inhibitors/Uncouplers (e.g., Carbonyl cyanide m-chlorophenyl hydrazone) | Probe the contribution of specific pathways (e.g., cyclic electron flow) to ATP synthesis and NADP+ reduction in studies of photosynthesis [26]. |
| Membrane-Permeable Metabolite Analogs (e.g., Dimethyl α-ketoglutarate) | Deliver specific metabolites into the cytosol to investigate their role in signaling pathways, such as fuel-induced insulin secretion [73]. |
| Luciferase-Based ATP Assay Kits | Provide a highly sensitive and specific method for quantifying ATP concentrations from complex biological samples like cell extracts [74]. |
Distinguishing Between Compartment-Specific Pools and Whole-Cell Measurements
In eukaryotic cells, metabolism is organized into specialized compartments such as the cytosol and mitochondria, each maintaining distinct biochemical environments. Understanding the differences between compartment-specific pools and whole-cell measurements is fundamental for accurate interpretation of energy and redox states, particularly when researching ATP/ADP and NADPH/NADP+ ratios across experimental conditions. While whole-cell measurements provide a population-average snapshot, they often mask critical subcellular heterogeneity that drives cellular decision-making and metabolic adaptations [79] [80].
This comparison guide examines the methodological approaches, analytical capabilities, and limitations of both measurement strategies within the context of ATP/ADP/NADPH/NADP+ ratio research. We present experimental data demonstrating how these approaches yield fundamentally different interpretations of metabolic function, with particular relevance for researchers and drug development professionals investigating metabolic diseases, cancer, and biotherapeutic production.
Methodological Comparison: Technical Approaches and Workflows
Compartment-Specific Measurement Techniques
Compartment-specific metabolomics requires sophisticated techniques to physically or analytically separate subcellular compartments before measurement. The cornerstone method is differential fast filtration followed by rapid quenching, which preserves compartment-specific metabolite levels [80]. For labeling experiments, compartment-specific 13C metabolic flux analysis (13C MFA) utilizes isotopic non-stationary modeling with subcellular resolution, requiring precise measurement of labeling dynamics in cytosolic and mitochondrial pools separately [80].
Additional advanced techniques include:
- Immunoisolation of organelles using antibody-based separation
- Fluorescent biosensors genetically targeted to specific compartments
- Spatially-resolved mass spectrometry imaging approaches
Whole-Cell Measurement Techniques
Whole-cell measurements employ global metabolomic profiling from lysed cells, providing a composite view of metabolite concentrations across all compartments [80]. Standard 13C MFA applied to whole-cell extracts assumes metabolic steady-state throughout the cell and uses averaged labeling patterns for flux calculation [80]. These methods are technically less demanding but inherently conflate distinct subcellular pools.
Key Comparative Data: Quantitative Experimental Findings
ATP/ADP Ratio Discrepancies Across Compartments
Table 1: Compartment-Specific vs. Whole-Cell ATP/ADP Ratio Measurements
| Cell Type | Measurement Type | Cytosolic ATP/ADP | Mitochondrial ATP/ADP | Whole-Cell ATP/ADP | Experimental Conditions |
|---|---|---|---|---|---|
| CHO DP-12 [80] | Compartment-specific | 8.2 ± 0.7 | 5.1 ± 0.4 | 6.3 ± 0.5 | Exponential growth phase, glucose-limited |
| CHO DP-12 [80] | Compartment-specific | 6.8 ± 0.6 | 7.3 ± 0.6 | 7.1 ± 0.5 | Stationary phase, glucose-depleted |
| S. cerevisiae [79] | Computational prediction | Compartment-specific constraints drive metabolic strategies | N/A | Traditional FBA predictions | Glucose limitation vs. excess |
NADPH/NADP+ Pool Measurements and Metabolic Engineering Implications
Table 2: NADPH/NADP+ Redox State Comparisons
| Measurement Approach | Cytosolic NADPH/NADP+ | Mitochondrial NADPH/NADP+ | Whole-Cell NADPH/NADP+ | Key Findings |
|---|---|---|---|---|
| Compartment-specific 13C MFA [80] | Preferentially maintained reducing environment | More variable reducing state | Intermediate values | Compartmentalized isozymes serve different catabolic/anabolic needs |
| Whole-cell 13C MFA [80] | N/A | N/A | Averaged value | Cannot resolve malic enzyme flux distribution |
| Computational modeling (pcYeast) [79] | Mitochondrial constraint limits growth at glucose limitation | Cytosolic volume constraint dictates overflow metabolism at sugar excess | N/A | Identifies condition-dependent compartment constraints |
Experimental data from CHO cells demonstrates that pool sizes differed significantly between cytosol and mitochondria across all cultivation conditions [80]. Particularly for NADPH-dependent processes, compartment-specific analysis revealed that cytosolic malic enzyme (vME) flux could not be accurately estimated without compartment-specific malate labeling information, establishing it as an important metabolic engineering target for improving cell-specific IgG1 productivity [80].
Experimental Protocols for Compartment-Specific Analysis
Differential Fast Filtration Protocol for Subcellular Metabolomics
This protocol enables physical separation of cytosolic and mitochondrial fractions from cultured cells [80]:
Cell Culture: Grow CHO DP-12 cells in TC-42 medium supplemented with 42 mM d-glucose and 6 mM l-glutamine in controlled bioreactors (37°C, 150 rpm, 5% CO2, 40% dissolved oxygen).
Sampling: Extract culture samples using differential fast filtration at multiple time points throughout growth phase.
Fractionation:
- Rapid filtration through appropriate membrane systems
- Immediate quenching of metabolic activity
- Separate extraction of cytosolic and mitochondrial fractions
- Preservation of compartment integrity
Metabolite Analysis:
- Process samples using HPLC system coupled to triple quadrupole mass spectrometer
- Employ electrospray ionization source
- Quantify metabolites using multiple reaction monitoring (MRM)
13C Labeling Experiments:
- Use [U-13C6]-d-glucose as carbon tracer (75% 13C, 25% 12C)
- Monitor isotopic labeling dynamics in separate compartments
- Correct measured 13C labeling distributions for natural isotope abundances
Computational Modeling Protocol for Compartment-Specific Constraints
For predictive modeling of compartment-specific metabolism [79]:
Model Construction:
- Extend genome-scale metabolic model with protein synthesis and degradation reactions
- Incorporate compartment-specific protein pools (cytosol, mitochondria, membranes)
- Include 1523 proteins described by 16,304 reactions including translation factors
Constraint Application:
- Apply enzyme capacity constraints linking metabolic fluxes to enzyme synthesis
- Implement ribosome capacity constraints for cytosolic and mitochondrial translation
- Set compartment-specific proteome constraints based on experimental data
Flux Prediction:
- Use linear programming with fluxes as optimization variables
- Employ binary search algorithm to find maximum feasible growth rate
- Predict metabolic fluxes and corresponding protein expression
Visualizing Compartment-Specific Metabolic Workflows
Diagram Title: Experimental Workflow for Compartmental Metabolite Analysis
Diagram Title: Compartmentalized Metabolic Pathways and Measurement Regions
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Compartment-Specific Metabolism Studies
| Reagent/Material | Function | Example Application | Considerations |
|---|---|---|---|
| [U-13C6]-d-glucose | Metabolic tracer for 13C flux analysis | Enables tracking of carbon fate through compartmentalized pathways | Use 25-75% 13C labeling ratio; correct for natural isotope abundance [80] |
| Differential filtration apparatus | Physical separation of subcellular compartments | Rapid sampling while preserving compartment integrity | Requires optimized quenching protocols to prevent metabolite interconversion [80] |
| Triple quadrupole LC-MS/MS | Quantitative metabolite analysis | Simultaneous measurement of multiple metabolites in small samples | Use multiple reaction monitoring (MRM) for sensitivity; requires proper ionization controls [80] |
| pcYeast computational model | Genome-scale modeling with proteome constraints | Predicts compartment-specific metabolic strategies | Incorporates 1523 proteins, 16,304 reactions; requires calibration to specific strains [79] |
| Compartment-specific biosensors | Real-time monitoring of metabolite ratios | Live-cell imaging of ATP/ADP or NADPH/NADP+ dynamics | Requires genetic engineering; potential for perturbation of native metabolism |
| Antibody-based isolation kits | Immunoisolation of organelles | Purification of mitochondrial or other organellar fractions | Potential for cross-contamination; validation of purity essential |
The choice between compartment-specific and whole-cell measurement approaches depends significantly on research goals and technical capabilities. Compartment-specific analysis is essential when investigating metabolic engineering targets, understanding regulatory mechanisms underlying metabolic adaptations, or studying diseases with suspected subcellular metabolic dysfunction. The demonstrated inability to accurately determine cytosolic malic enzyme flux without compartment-specific labeling information underscores its necessity for precise metabolic engineering [80].
Whole-cell measurements remain valuable for initial screening, studies requiring high-throughput analysis, or when investigating global metabolic changes where subcellular redistribution is not the primary focus. However, researchers must recognize that whole-cell measurements of ATP/ADP and NADPH/NADP+ ratios represent weighted averages that may mask critical compartment-specific changes driving cellular physiology [79] [80].
For drug development professionals, these distinctions are particularly crucial when targeting metabolic enzymes or pathways, as compartment-specific pool sizes and fluxes may determine both drug efficacy and toxicity profiles. Future methodological advances will likely focus on increasing the spatial and temporal resolution of compartment-specific measurements while reducing technical complexity, ultimately making these powerful approaches more accessible to the broader research community.
Identifying Technical Artifacts in Bioluminescence and Colorimetric Assays
The accurate quantification of key metabolites and enzymatic activities is fundamental to research exploring ATP/ADP and NADPH/NADP+ ratios across varying physiological conditions. Bioluminescence and colorimetric assays represent two cornerstone technologies for these measurements, each with distinct mechanisms, advantages, and inherent limitations. Colorimetric assays, such as the Bradford and BCA protein assays, rely on the absorption of light by a sample at specific wavelengths, with the measured absorbance being proportional to analyte concentration according to Beer's Law [81]. In contrast, bioluminescence assays generate light through enzymatic reactions—such as the ATP-dependent oxidation of luciferin by firefly luciferase—and detect the emitted light without requiring an external excitation source [81] [82]. This fundamental difference in detection principle underlies many of the performance variations and potential technical artifacts associated with each method. The choice between these platforms can significantly impact data reliability, especially in complex experimental systems like drug screening and metabolic profiling where accurate determination of nucleotide ratios is critical.
This guide provides an objective comparison of these technologies, focusing on their application in measuring ATPase and kinase activities—processes central to cellular energy homeostasis. We will summarize performance data, detail standardized experimental protocols, and identify common technical artifacts to support researchers in selecting and implementing the most appropriate assay for their specific research context.
Performance Comparison: Key Metrics and Experimental Data
Direct comparison of colorimetric and bioluminescence technologies reveals critical differences in sensitivity, dynamic range, and susceptibility to interference, all of which can introduce artifacts into metabolic data.
Quantitative Performance Metrics
Table 1: Overall Comparison of Absorbance vs. Luminescence Assays [81]
| Criterion | Absorbance Assays | Luminescence Assays |
|---|---|---|
| Sensitivity | Low to moderate | High |
| Detection Limit | Nanomolar to micromolar range | Can reach femtomolar range |
| Dynamic Range | 2-3 orders of magnitude | 6-8 orders of magnitude |
| Background Interference | High (susceptible to sample color/turbidity) | Low (no excitation light required) |
| Assay Workflow | Often more steps, longer incubation | Generally simpler, fewer steps |
| Equipment Cost | Lower (standard plate readers) | Higher (requires luminometers) |
Table 2: Specific Protein Quantitation Assay Performance [83]
| Assay Method | Linear Range (μg/mL) | Reported LoD (μg/mL) | Key Interfering Substances |
|---|---|---|---|
| Bradford (Colorimetric) | 0 to ~670 | 9.24 - 30.5 | Polyphenols, detergents [84] [85] |
| ProteOrange (Fluorometric) | Up to 350 | 24.3 - 34.6 | Varies with dye chemistry |
| QuDye (Fluorometric) | 40 to 670 | 0.81 - 182 | Highly dependent on protein structure |
| Direct Detect (IR) | From 0.25 | N/A | Buffers with C=O bonds (e.g., urea) [85] |
Data from Comparative Studies
Kinase Activity Assays: A quantitative high-throughput screening (qHTS) study compared two bioluminescent methods for the kinase Clk4: an ATP-depletion assay (Kinase-Glo) and a product formation assay (ADP-Glo). While both formats showed high performance (Z′-factors ~0.9) and similar potency distributions for hundreds of active compounds, the underlying principles differ substantially. The ATP-depletion format requires high (~50%) substrate conversion to achieve a sufficient signal-to-background ratio, whereas the ADP detection format measures product formation directly and can be effective at lower conversion levels [82].
Protein Quantitation in Complex Samples: A comparison of the Bradford and Lowry colorimetric assays for measuring protein in soil extracts—which contain high levels of interfering polyphenolic compounds—found the Bradford assay highly susceptible to two confounding artifacts: 1) substantial colour inhibition caused by polyphenols, and 2) significant colour development directly from the polyphenols themselves. In contrast, a modified Lowry microplate method allowed for better distinction between colour development from protein and non-protein sources, providing more accurate quantification [84].
General Performance Considerations: As summarized in Table 1, luminescence assays generally provide superior sensitivity and a wider dynamic range due to minimal background interference. Absorbance assays, while more cost-effective and suitable for many applications, are more vulnerable to artifacts from sample impurities, color, and turbidity [81].
Experimental Protocols for Key Assays
ADP-Glo Bioluminescent Kinase Assay
The ADP-Glo assay is a luminescent method that measures ADP formation from kinase reactions, providing a direct readout of enzyme activity.
Principle: In a kinase reaction, ATP is converted to ADP. The ADP-Glo reagent first terminates the kinase reaction and depletes any remaining ATP. A second addition then converts ADP to ATP, which is detected using a luciferase/luciferin reaction, generating a luminescent signal proportional to the ADP concentration [82].
Detailed Protocol (1,536-well plate format) [82]:
- Reaction Setup: Dispense 2 μL/well of substrate-buffer solution (containing 100 μM peptide substrate, 1 μM ATP, and appropriate buffer salts) into a white assay plate.
- Compound Addition: Transfer 23 nL of compound solution (e.g., from a library dilution series) using a pin tool.
- Kinase Reaction Initiation: Add 1 μL/well of kinase-buffer solution (containing 25 nM Clk4 kinase in this example) for a total reaction volume of 3 μL.
- Incubation: Incubate the reaction at room temperature for a predetermined time (e.g., 4.5 hours).
- ADP Detection: Add 3 μL of ADP-Glo reagent to terminate the kinase reaction and deplete residual ATP. Incubate before adding the second reagent to convert ADP to ATP, followed by the luciferase/detection reagent.
- Signal Measurement: Detect luminescence using a plate reader (e.g., ViewLux) after a 5-minute incubation.
Bradford Colorimetric Protein Assay
The Bradford assay is a rapid colorimetric method for estimating protein concentration based on the binding of Coomassie Brilliant Blue G-250 dye to basic amino acids.
Principle: Under acidic conditions, the dye shifts from a cationic (red) form to an anionic (blue) form upon binding to protein, primarily through interactions with arginine and lysine residues. This shift causes a change in the absorption maximum from 465 nm to 595 nm [83] [85].
Detailed Protocol (96-well microplate format) [83]:
- Standard and Sample Preparation: Prepare a dilution series of a standard protein (e.g., BSA) in the same buffer as the unknown samples. The typical measurable range is 1-20 μg protein.
- Reagent Addition: Pipette 200 μL of Bradford reagent into each well containing 10 μL of standard or unknown sample.
- Incubation and Color Development: Incubate the mixture at room temperature for at least 5 minutes. The color is stable for approximately one hour.
- Absorbance Measurement: Measure the absorbance at 595 nm using a microplate reader. Subtract the background absorbance at 800 nm if necessary.
- Data Analysis: Generate a standard curve of absorbance versus protein concentration and interpolate the unknown sample concentrations from this curve.
Technical Artifacts and Interference Factors
Understanding and controlling for technical artifacts is crucial for obtaining reliable data, particularly when interpreting subtle changes in metabolite ratios.
Table 3: Common Artifacts and Mitigation Strategies
| Assay Type | Common Interfering Substances | Potential Artifact | Mitigation Strategy |
|---|---|---|---|
| Bradford Assay | Polyphenols, detergents (SDS, Triton) [84] [85] | Polyphenols inhibit colour development and produce colour directly [84]. | Use a modified Lowry method [84] or perform polyphenol removal. |
| BCA/Lowry Assay | Reducing agents (DTT, β-mercaptoethanol), chelators (EDTA) [85] | Artificially inflated signal due to direct reduction of Cu²⁺. | Dialyze samples to remove interfering substances. |
| Bioluminescence (General) | Colored or quenching compounds from compound libraries [82] | Signal absorption or quenching, leading to false negatives/inhibition. | Use a control to check for compound interference. |
| ATP-Depletion Assay | High compound conversion (>50%) [82] | Altered apparent compound potency. | Use product-formation assays (e.g., ADP-Glo) at lower conversion. |
| UV A280 Protein Quantitation | Non-protein UV-absorbing substances (nucleic acids) [85] | Overestimation of protein concentration. | Use FTIR (Direct Detect) or perform sample purification [85]. |
Impact on Data Interpretation
Artifacts can lead to significant misinterpretation of experimental results. For example, in the context of ATP/ADP ratio measurements:
- Inhibitor Potency Shifts: ATP-depletion assays run at high conversion rates can lead to shifts in the apparent IC₅₀ values of kinase inhibitors. While these shifts may be within the noise of a typical HTS, they can still affect the accurate ranking of compound potency [82].
- False Positives/Negatives in Screening: Compound libraries often contain colored or fluorescent molecules that can quench the luminescence signal in bioluminescence assays or absorb light in colorimetric assays, leading to false hits or missed actives [82] [81].
- Protein Quantitation Errors: The amino acid composition of the protein standard versus the target protein can greatly affect results in both Bradford and BCA assays. A protein rich in arginine will produce a stronger signal in a Bradford assay than a protein with low arginine content, even at the same concentration [85]. This is a critical consideration when measuring enzyme concentrations for kinetic studies of ATPases or kinases.
Visualization of Assay Workflows and Signaling Pathways
To better understand the technical context and potential points of artifact introduction, the following diagrams outline core signaling pathways and general assay workflows.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Reagent Solutions for Metabolic Assays
| Reagent / Material | Function / Application | Key Considerations |
|---|---|---|
| ADP-Glo / Kinase-Glo Kits | Bioluminescent detection of ADP/ATP for kinase/ATPase profiling. | Enables direct product (ADP) measurement, often with better initial rate kinetics than substrate (ATP) depletion [82]. |
| Firefly Luciferase (Various) | Core enzyme for bioluminescent ATP detection. | Ultra-Glo luciferase variant is engineered for stability and used in commercial kits [82]. |
| Nanoluc (Nluc) Luciferase | Small, stable luciferase for reporter assays and fusion proteins. | 19 kDa size, bright glow-type luminescence with furimazine substrate, useful for fusion proteins and immunoassays [86]. |
| Coomassie Brilliant Blue G-250 | Active dye in Bradford protein assay. | Binds basic amino acids (Arg, Lys); signal is protein-sequence dependent, a key source of artifact [83] [85]. |
| Bicinchoninic Acid (BCA) | Colorimetric chelator of Cu⁺ ions for protein quantitation. | Sensitivity varies with protein composition (dependent on Cys, Tyr, Trp). Compatible with some detergents [85]. |
| Folin-Ciocalteu Reagent | Component of the Lowry assay, reduced by protein-Cu⁺ complexes. | Also reduced by polyphenols, leading to interference in complex samples like soil extracts [84]. |
| D-Luciferin | Substrate for firefly luciferase in bioluminescence assays. | Requires ATP and Mg²⁺ for the light-producing reaction. Potassium salt offers high solubility [87]. |
| Nanobodies (VHH) | Small recombinant antibodies for detection probes. | ~15 kDa size, high stability, easily expressed as fusion proteins with reporters like Nluc for sensitive immunoassays [86]. |
Strategies for Differentiating High and Low Metabolic Turnover States
Cellular metabolism operates through finely tuned energy currencies and redox pairs that exist in dynamic equilibrium. The differentiation between high and low metabolic turnover states is fundamental to understanding cellular energetics, physiological adaptations, and pathological conditions. High metabolic turnover states are characterized by rapid adenosine triphosphate (ATP) synthesis and consumption, elevated rates of substrate utilization, and increased electron flux through redox systems. In contrast, low metabolic turnover states feature reduced energy demand, slower substrate cycling, and distinct alterations in key metabolic ratios. The critical biomarkers for distinguishing these states include the ATP:ADP ratio, NADPH:NADP+ ratio, and their interrelated dynamics with other cellular redox systems.
The assessment of these metabolic parameters provides valuable insights into cellular energy status, oxidative stress responses, and metabolic adaptation mechanisms. Researchers and drug development professionals require precise experimental strategies to quantify these states across various biological contexts, from athletic performance to disease pathology. This guide presents a comprehensive comparison of methodological approaches, quantitative benchmarks, and technical considerations for accurately differentiating metabolic states through key biochemical parameters.
Quantitative Comparison of Metabolic States
Key Metabolite Ratios and Concentrations Across Conditions
Table 1: Metabolic Signatures of High and Low Turnover States
| Metabolic Parameter | High Turnover State | Low Turnover State | Measurement Techniques | Biological Context |
|---|---|---|---|---|
| ATP:ADP Ratio | Decreased [88] | Increased | HPLC, enzymatic assays | Cellular energy charge |
| NADPH:NADP+ Ratio | Variable (context-dependent) | Variable (context-dependent) | Enzymatic cycling assays [2] | Redox capacity, antioxidant defense |
| Total NADPx Pool | Increased during oxidative stress [2] | Decreased | Enzymatic cycling assays [2] | Oxidative stress response |
| Adenylate Energy Charge | Lower | Higher | Calculation from ATP, ADP, AMP | Cellular energy status |
| Glycogen Utilization Rate | Intensity-dependent increase [89] | Reduced | Muscle biopsy, biochemical assay | Exercise intensity |
| Mitochondrial Efficiency | Increased proton leak | Reduced proton leak [90] | Respiration assays | Metabolic adaptation |
Table 2: Absolute Metabolite Concentrations in Different Physiological States
| Metabolite | Sedentary Subjects | Endurance Athletes | Strength Athletes | Oxidative Stress Conditions |
|---|---|---|---|---|
| Muscle Glycogen | 50-150 mmol·kg⁻¹ d.w. [89] | 400-800 mmol·kg⁻¹ d.w. [89] | 400-800 mmol·kg⁻¹ d.w. [89] | Not reported |
| Total NADPx | ~0.64 nmol/mg protein [2] | Not reported | Not reported | Doubled (vs. basal) [2] |
| NADPH (% of NADPx) | 37% [2] | Not reported | Not reported | Decreased during oxidation [2] |
| NAD+ (% of NADx) | 72% [2] | Not reported | Not reported | Decreased during stress [2] |
Temporal Dynamics in Metabolic State Transitions
Metabolic transitions between high and low turnover states occur across different timescales, from rapid seconds-minute fluctuations to chronic adaptations. During high-intensity exercise, the ATP:ADP ratio can decrease within seconds as ATP hydrolysis exceeds regeneration capacity [88]. This rapid change triggers immediate compensatory mechanisms including increased glycolytic flux and phosphocreatine breakdown. The NADPH:NADP+ ratio responds within minutes to oxidative stress, as demonstrated in astrocyte cultures where hydrogen peroxide exposure triggered NAD+ phosphorylation within 5-15 minutes [2].
Chronic metabolic adaptations develop over weeks to months of sustained physiological challenges. Endurance training induces mitochondrial biogenesis that enhances the capacity for ATP regeneration while maintaining favorable ATP:ADP ratios during submaximal exercise [91]. Similarly, prolonged energy restriction leads to reduced mitochondrial uncoupling and decreased proton leak, representing a shift toward increased metabolic efficiency in what was historically a conservation mechanism during famine conditions [90]. These temporal considerations are crucial for designing appropriate sampling protocols in metabolic research.
Experimental Protocols for Metabolic State Differentiation
Protocol for Comprehensive Redox Pair Analysis
This protocol enables simultaneous quantification of major redox pairs (NADx, NADPx, GSx) from cell cultures or tissue samples, based on established methodologies with modifications for enhanced precision [2].
Sample Preparation:
- Rapidly harvest cells or tissue (50-100 mg) and immediately freeze in liquid nitrogen
- Homogenize in ice-cold 6% (w/v) sulfosalicylic acid (1:10 w/v ratio)
- Centrifuge at 12,000 × g for 10 minutes at 4°C
- Collect supernatant for immediate analysis or store at -80°C under argon atmosphere
NAD+ and NADH Quantification:
- Principle: Enzymatic cycling assay using lactate dehydrogenase
- Reagent composition: 100 mM Tris-HCl (pH 8.0), 0.5 mM MTT, 1.6 mM phenazine ethosulfate, 0.8 mM pyruvate, 2.5% ethanol, 4 U/mL alcohol dehydrogenase
- Procedure: For NAD+, heat extract at 60°C for 30 minutes to destroy NADH; for NADH, use untreated extract
- Incubate at 37°C for 10-30 minutes, measure absorbance at 570 nm
- Calculate concentrations using standard curves (0-10 μM)
NADP+ and NADPH Quantification:
- Principle: Enzymatic cycling assay using glucose-6-phosphate dehydrogenase
- Reagent composition: 100 mM Tris-HCl (pH 8.0), 0.5 mM MTT, 1.6 mM phenazine methosulfate, 5 mM glucose-6-phosphate, 2 U/mL G6PDH
- Procedure: For NADP+, heat extract at 60°C for 30 minutes; for NADPH, use untreated extract
- Incubate at 37°C for 10-30 minutes, measure absorbance at 570 nm
- Calculate concentrations using standard curves (0-5 μM)
Glutathione (GSH and GSSG) Quantification:
- Total glutathione: Use DTNB recycling assay with glutathione reductase
- GSSG-specific: Derivatize GSH with 2-vinylpyridine before assay
- Calculate GSH as difference between total glutathione and GSSG
Quality Control:
- Include internal standards in each assay batch
- Maintain sample processing at 4°C unless specified
- Perform recovery experiments with spiked standards
- Use argon atmosphere to prevent oxidation during processing
Protocol for ATP Turnover and IMP Formation Assessment
This method evaluates high-energy phosphate metabolism and purine nucleotide degradation, particularly relevant for high-intensity muscle metabolism [92].
Muscle Sample Processing:
- Obtain muscle biopsies using percutaneous needle technique under local anesthesia
- Immediately freeze samples in liquid nitrogen (within 1-2 seconds of collection)
- Freeze-dry samples and dissect away connective tissue and blood
- Extract metabolites in 0.5 M perchloric acid, neutralize with 2.2 M KHCO₃
Metabolite Analysis:
- ATP, ADP, AMP: Quantify using HPLC with UV detection at 254 nm
- Inosine monophosphate (IMP): Separate using ion-pair chromatography
- Phosphocreatine: Enzymatic assay coupled with creatine kinase
- Lactate: Enzymatic spectrophotometric assay
Experimental Conditions for Metabolic Stress:
- Isometric contraction protocol: Electrical stimulation at 50-100 Hz
- Ischemic conditions: Apply pressure cuff proximal to muscle group
- Recovery phases: Monitor metabolite replenishment post-contraction
Data Interpretation:
- Calculate ATP:ADP ratio from concentration data
- Determine rate of IMP accumulation as indicator of AMP deaminase activation
- Correlate metabolic changes with contractile force production
Figure 1: Metabolic Pathways in High Turnover States. This diagram illustrates key regulatory nodes and metabolic fluxes distinguishing high from low metabolic turnover conditions.
The Scientist's Toolkit: Essential Research Reagents
Table 3: Essential Reagents for Metabolic State Research
| Reagent/Category | Specific Examples | Research Application | Key Functions |
|---|---|---|---|
| Enzymatic Cycling Assay Components | Phenazine ethosulfate, MTT, glucose-6-phosphate dehydrogenase [2] | NAD(P)H quantification | Enable sensitive detection of pyridine nucleotides via signal amplification |
| Metabolite Stabilizers | Sulfosalicylic acid, perchloric acid, liquid nitrogen [2] | Sample preservation | Instantaneous metabolic arrest, protein precipitation |
| Chromatography Systems | HPLC with UV detection, ion-pair chromatography | Nucleotide separation and quantification | High-resolution separation of ATP, ADP, AMP, IMP |
| Oxidative Stress Inducers | Hydrogen peroxide, menadione, β-lapachone [2] | Experimental perturbation | Controlled induction of oxidative challenge |
| Metabolic Pathway Inhibitors | G6PDi-1 (PPP inhibitor), respiratory chain inhibitors [2] | Pathway manipulation | Selective blockade of specific metabolic routes |
| Biological Matrices | Primary astrocyte cultures, muscle biopsy samples [2] [91] | Experimental models | Physiologically relevant systems for metabolic studies |
Technical Considerations and Data Interpretation
Methodological Challenges in Metabolic State Assessment
Accurate differentiation of metabolic states requires careful attention to technical challenges that can compromise data quality. Sample collection speed is critical, as metabolic turnover rates can rapidly change following physiological perturbations. Studies investigating exercise metabolism typically employ muscle biopsy techniques with freezing within 1-2 seconds of collection to obtain accurate metabolite measurements [89]. For cell culture studies, rapid aspiration and freezing methods are essential, particularly when assessing labile metabolites like NADPH.
Spatial compartmentalization presents another significant challenge. Metabolite gradients exist within eukaryotic cells, with ATP concentration variations occurring when sources or sinks are spatially clustered [19]. Similarly, glycogen distribution within muscle fibers shows distinct subcellular localization patterns (subsarcolemmal, intermyofibrillar, intramyofibrillar) that display different depletion and repletion kinetics [89]. These spatial considerations necessitate careful sampling strategies and interpretation of homogenate-based measurements.
Analytical specificity is paramount when assessing metabolite ratios. The structural similarity between NADH and NADPH necessitates specific enzymatic cycling assays that selectively detect one cofactor without cross-reactivity [2]. Similarly, the assessment of ATP:ADP ratios requires separation techniques that adequately resolve these nucleotides from other interfering compounds. HPLC methodologies with appropriate internal standards provide the necessary specificity for accurate quantification.
Context-Dependent Interpretation of Metabolic Ratios
The interpretation of metabolite ratios must consider the specific physiological context. The ATP:ADP ratio typically decreases during high metabolic turnover states as energy demand exceeds regeneration capacity [88]. However, in well-trained endurance athletes, the enhanced mitochondrial capacity may maintain higher ATP:ADP ratios despite high absolute ATP turnover rates [91]. This adaptation represents a metabolic efficiency that distinguishes chronically trained from untrained states.
The NADPH:NADP+ ratio reflects the redox balance and antioxidant capacity but demonstrates complex behavior across different conditions. During acute oxidative stress, this ratio may initially decrease as NADPH is consumed for glutathione reduction, followed by a compensatory increase as NAD kinase activation expands the NADP+ pool [2]. The dynamic response of this ratio over time provides more valuable information than single timepoint measurements.
The integration of multiple metabolic parameters provides the most robust assessment of metabolic states. Combining ATP:ADP ratios with measures of glycogen utilization, mitochondrial efficiency, and redox status creates a comprehensive metabolic signature that accurately differentiates high and low turnover conditions across various physiological and pathological contexts.
Cross-System Validation and Disease-Specific Ratio Alterations
Validation of Metabolic QIs in Independent Patient Cohorts
The validation of metabolic quality indicators (QIs) in independent patient cohorts is a critical step in translating preclinical research into clinically applicable tools. This process ensures that biomarkers and predictive models are not only statistically significant in a development cohort but also generalizable, reliable, and robust across diverse populations and settings. In the context of research comparing ATP, ADP, NADPH, and NADP+ ratios across conditions, rigorous validation provides the necessary evidence for their utility in drug development and clinical decision-making. This guide objectively compares approaches, performance metrics, and methodological considerations for validating metabolic QIs, providing researchers and drug development professionals with a framework for evaluating validation evidence.
Comparative Analysis of Validation Approaches & Performance
The validation of metabolic QIs employs distinct methodological approaches, each with specific strengths and applications. The table below summarizes performance data from published studies to enable direct comparison of their outputs.
Table 1: Performance Metrics of Validated Metabolic Biomarkers and Prediction Models
| QI / Model Name | Validation Cohort(s) | Key Performance Metrics | Reference |
|---|---|---|---|
| Machine Learning MetS Prediction (MLP-based) | CHARLS (China), KNHANES (Korea), UK Biobank, NHANES (USA) | AUC: 0.8556 - 0.9108; PRAUC: 0.6246 - 0.8264 | [93] |
| m-Metabolic Signature for PDAC | Multi-center cohorts (VD1 & VD2) | Sensitivity: 77.3%; Specificity: 89.6%; Overall Accuracy: 82.4% | [94] |
| 4-Year MetS Risk Prediction Model | Retrospective Chinese Cohort (Temporal Validation) | C-statistic: 0.782 (95% CI: 0.771-0.793); Brier Score: 0.164 | [95] |
| DNA Methylation EpiScores (for BMI) | Generation Scotland, HELIOS (Singapore), LBC1936 | Variance Explained (R²): 20.8% in HELIOS, 14.4% in LBC1936 | [96] |
Table 2: Comparison of Validation Strategies and Their Characteristics
| Validation Strategy | Definition | Key Strengths | Common Applications |
|---|---|---|---|
| Temporal Validation | Validation using data from a later time period in the same setting as the development cohort. | Assesses model performance over time; simpler logistics. | Validating clinical prediction models for local use [95]. |
| External/Geographic Validation | Validation using data from a different location or population. | Tests generalizability across diverse genetic and environmental backgrounds. | Evaluating machine learning models [93] and biomarker panels [94] for broad applicability. |
| Cross-Study Validation | Application of a model or signature to entirely independent studies, often using different protocols. | Provides the highest level of evidence for robustness. | Metabolomic signatures [94] and epigenetic scores [96]. |
Experimental Protocols for Key Validation Studies
Protocol for Metabolic Syndrome Prediction Model Validation
The external validation of a 4-year metabolic syndrome (MetS) risk prediction model exemplifies a robust temporal validation approach [95].
- Cohort Design: A retrospective cohort design was used, applying a temporal validation strategy. The model was developed on data from 2011-2014 and validated on data from a later period (2015-2018) from the same tertiary hospital.
- Participants: The validation included 7,681 participants aged 18 or older without MetS at baseline, who underwent consecutive health examinations for 4 years.
- Outcome & Predictors: The outcome was incident MetS, defined by the 2009 Joint Scientific Statement harmonizing criteria. The model incorporated five predictors: age, total cholesterol, serum uric acid, alanine transaminase, and body mass index.
- Statistical Analysis: Missing data were handled using multiple imputation by chained equations. Model performance was evaluated via discrimination (C-statistic), calibration (calibration plot, slope, and intercept), overall performance (Brier score), and clinical utility (decision curve analysis). The C-statistic of 0.782 indicated acceptable discrimination, and the calibration plot showed good agreement between predicted and observed risk.
Protocol for Metabolic Biomarker Signature Validation
The development and validation of a metabolic signature for differentiating pancreatic ductal adenocarcinoma (PDAC) from chronic pancreatitis demonstrate a high-throughput, cross-platform approach [94].
- Study Cohorts: The study evaluated 941 patients across three independent multicenter cohorts: an identification cohort and two validation cohorts (PDAC, n=356; chronic pancreatitis, n=304; non-pancreatic disease, n=281).
- Biomarker Analysis: Targeted quantitative plasma metabolite analysis was performed using liquid chromatography–tandem mass spectrometry (LC-MS/MS), a single-platform method that simplifies clinical application.
- Signature Development & Validation: A machine learning-aided algorithm identified a minimalistic metabolic (m-Metabolic) signature comprising just 4 metabolites used in combination with serum CA19-9. The signature's performance was rigorously tested in the two independent validation cohorts, demonstrating consistent accuracy above 82% and maintaining performance in patients with resectable tumors.
Protocol for Data Normalization in Cross-Cohort Metabolomics
Technical validation requires careful data processing to minimize cohort discrepancies. A comparative analysis tested seven data-driven normalization methods on quantitative metabolome data from rat models of hypoxic–ischemic encephalopathy [97].
- Methods Compared: The study evaluated normalization by total concentration, autoscaling, quantile normalization, Probabilistic Quotient Normalization (PQN), Median Ratio Normalization (MRN), Trimmed Mean of M-values (TMM), and Variance-Stabilizing Normalization (VSN).
- Quality Assessment: The quality of each normalization method was assessed by the performance of Orthogonal Partial Least Squares (OPLS) models built on training datasets and then applied to validation datasets. Performance was measured by explained variance (R2Y), predicted variance (Q2Y), and the sensitivity and specificity of the validation models.
- Findings: PQN, MRN, and VSN demonstrated higher diagnostic quality for OPLS models. The model based on VSN showed superior performance (86% sensitivity, 77% specificity) and uniquely highlighted pathways related to brain fatty acid oxidation and purine metabolism, underscoring the impact of normalization choice on biological interpretation.
Signaling Pathways and Workflows
The following diagrams illustrate key experimental workflows and the logical relationship between redox pairs relevant to metabolic QI validation.
Diagram 1: Validation Workflow for Metabolic QIs. This flowchart outlines the standard process for developing and validating metabolic quality indicators, highlighting the critical stage of independent testing and the different types of validation employed.
Diagram 2: Redox Pair Interplay Under Oxidative Stress. This diagram shows the biochemical interplay between key redox pairs (NADx, NADPx, GSx) in astrocytes during oxidative stress, a core concept in metabolic research. Experimental evidence shows oxidative stress triggers NADK-mediated phosphorylation of NAD+ to NADP+, supporting the antioxidative defense system [2].
The Scientist's Toolkit: Research Reagent Solutions
Successful validation of metabolic QIs relies on specific reagents and platforms. The following table details essential materials and their functions as derived from the cited experimental protocols.
Table 3: Essential Research Reagents and Platforms for Metabolic QI Validation
| Reagent / Platform | Specific Function | Experimental Context |
|---|---|---|
| Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) | High-throughput, quantitative analysis of plasma metabolites on a single platform. | Used to identify and validate the m-Metabolic signature for pancreatic cancer [94]. |
| Enzymatic Cycling Assays | Sensitive and specific measurement of redox co-substrates (e.g., NAD+, NADH, NADP+, NADPH, GSH, GSSG). | Employed to quantify basal levels and stress-induced changes in astrocyte redox pairs [2]. |
| Data Normalization Algorithms (PQN, MRN, VSN) | Correction of technical variance and batch effects in quantitative molecular data across cohorts. | Compared for effectiveness in minimizing cohort discrepancies in metabolomic data [97]. |
| Preclinical Animal Models (Canine) | Modeling human metabolic syndrome and cardiorenal pathophysiology in a non-rodent, translatable system. | Used to establish a model of diet-induced MetS independent of obesity for therapeutic testing [98]. |
| 18F-FDG PET/CT Imaging | Quantification of metabolic tumor volume (MTV), a prognostic metabolic biomarker. | Incorporated into the CAR-IMPI index for risk stratification in lymphoma patients receiving CAR-T therapy [99]. |
In the field of pharmacogenomics, genetic variations in drug-metabolizing enzymes significantly influence individual responses to medication. The cytochrome P450 2D6 (CYP2D6) enzyme, responsible for metabolizing approximately 25% of all commonly prescribed drugs, exhibits substantial phenotypic diversity within populations [100]. Patients can be categorized into distinct metabolic phenotypes—including ultrarapid (UM), extensive (EM), intermediate (IM), and poor metabolizers (PM)—based on the functional status of their CYP2D6 alleles [101] [100]. These phenotypes are not merely biochemical curiosities; they have profound clinical implications, potentially determining whether a drug will be effective, ineffective, or even toxic for a particular patient.
This guide provides a comparative analysis of ultrarapid versus poor metabolizer phenotypes, framing the discussion within broader research on cellular energy and redox states, specifically the ATP/ADP and NADPH/NADP+ ratios that are fundamental to metabolic regulation. We present objective experimental data, detailed methodologies, and analytical frameworks to equip researchers and drug development professionals with the tools needed to understand and investigate these critical pharmacogenetic variations.
Phenotype Definitions and Clinical Consequences
Genetic polymorphisms for drug-metabolizing enzymes give rise to population phenotypes with metabolic capabilities ranging from extremely poor to extremely fast [100]. More than 75 allelic variants of CYP2D6 have been identified, with enzyme activities spanning this entire spectrum [100].
Ultrarapid metabolizers possess multiple copies of functional CYP2D6 genes, leading to accelerated metabolism of substrate drugs. Poor metabolizers, in contrast, carry two non-functional or severely impaired alleles, resulting in significantly reduced or absent enzymatic activity [100] [102]. The clinical consequences of these phenotypes depend critically on whether the drug involved is a prodrug requiring metabolic activation or an active drug that is metabolized to an inactive form.
Table 1: Clinical Consequences of Metabolizer Phenotypes
| Drug Type | Metabolizer Phenotype | Effect on Metabolism | Potential Clinical Consequence |
|---|---|---|---|
| Prodrug (e.g., Codeine → Morphine) | Poor to Intermediate | Slow | Poor drug efficacy; accumulation of prodrug; risk of side effects [100] |
| Ultrarapid | Fast | Good drug efficacy; rapid therapeutic effect [100] | |
| Active Drug (e.g., Omeprazole → Inactive metabolite) | Poor to Intermediate | Slow | Good drug efficacy; accumulation of active drug; risk of side effects; may require lower dose [100] |
| Ultrarapid | Fast | Poor drug efficacy; therapeutic failure; may require higher dose [100] |
The real-world impact of these phenotypes is significant. A 2017 study found that ultrarapid metabolizers had a 69% higher risk of hospitalization and a 50% higher risk of an emergency department visit compared to extensive metabolizers [101]. For poor metabolizers, while no difference in hospitalization risk was found, there was a trend toward increased ED visits [101].
Quantitative Data and Comparative Analysis
Clinical Outcome Data
The following table summarizes key findings from a cohort study of 929 patients who underwent CYP2D6 testing, highlighting the differential risks associated with metabolizer status.
Table 2: Clinical Outcomes by CYP2D6 Phenotype [101]
| Phenotype | Risk of Hospitalization(HR vs. EM, 95% CI) | Risk of ED Visit(HR vs. EM, 95% CI) | Incidence of Hospitalization | Incidence of ED Visit |
|---|---|---|---|---|
| Ultrarapid (UM) | 1.69 (1.11 - 2.57) | 1.50 (1.05 - 2.14) | 47% | 62% |
| Extensive (EM) | 1.00 (Reference) | 1.00 (Reference) | 30% | 49% |
| Poor (PM) | 0.95 (0.58 - 1.56) | 1.38 (0.96 - 1.98) | Not Specified | Not Specified |
Abbreviations: HR, Hazard Ratio; CI, Confidence Interval; EM, Extensive Metabolizer.
Linking Metabolism to Cellular Energetics: NADPH/NADP+ and ATP/ADP Ratios
The metabolic capacity of an individual is fundamentally linked to cellular energy and redox states. The ratios of NADPH/NADP+ and ATP/ADP are critical parameters that reflect the redox balance and energy charge of a cell, influencing biosynthetic capacities and stress responses [103] [104].
Table 3: NADP+/NADPH and ATP/ADP Ratios Across Biological Systems
| Organism/Condition | Redox or Energy Ratio | Measured Value | Context and Significance |
|---|---|---|---|
| E. coli (Bacteria) | NADP+/NADPH Ratio | 1.2 [105] | Anabolic reduction charge of ~0.45; indicates redox poise for biosynthesis. |
| C. reinhardtii (Algae), Dark-adapted, Aerobic | NADPH/NADP Ratio | ~1.5 [26] [106] | Baseline redox state in the dark. |
| C. reinhardtii (Algae), Anaerobic | NADPH/NADP Ratio | ~3.5 [26] [106] | Increased reduction under anaerobic conditions. |
| C. reinhardtii (Algae), Dark-adapted, Aerobic | ATP/ADP Ratio | ~15 [26] [106] | High energy charge under aerobic conditions. |
| C. reinhardtii (Algae), Anaerobic | ATP/ADP Ratio | ~5 [26] [106] | Lower energy charge due to lack of respiratory ATP synthesis. |
| A. vinelandii (Bacteria), High O₂ | NADPH/NADP+ Ratio | Lower (3-fold decrease vs. low O₂) [103] | Correlates with high respiration and polymer synthesis. |
| A. vinelandii (Bacteria), Low O₂ | NADPH/NADP+ Ratio | Higher [103] | Response to oxygen limitation, shifting metabolic fluxes. |
These ratios are not static and respond dynamically to environmental conditions, such as oxygen availability. For instance, in Azotobacter vinelandii, a low oxygen transfer rate (OTR) led to a higher NADPH/NADP+ ratio, which correlated with a 6.6-fold increase in the metabolic flux through poly-3-hydroxybutyrate (P3HB) biosynthesis [103]. Conversely, under high OTR conditions, the NADPH/NADP+ ratio decreased threefold, and the flux was redirected through the pentose phosphate pathway [103]. This illustrates how redox state governs carbon flux in biological systems—a principle directly relevant to understanding the metabolic handling of drugs in humans with different CYP450 phenotypes.
Experimental Protocols and Methodologies
Genotyping and Phenotype Assignment
Accurate determination of CYP2D6 metabolizer status requires a robust genotyping protocol. The following workflow, derived from clinical studies, outlines the key steps [101]:
Title: CYP2D6 Genotyping Workflow
Key Steps:
- DNA Extraction: Genomic DNA is isolated from a patient's whole blood sample [101].
- Primary Genotyping: A cascade of tests is employed. This often begins with a multiplex PCR and allele-specific primer extension assay, such as the xTAG CYP2D6 Kit on the Luminex platform, to identify common variants [101].
- Copy Number Analysis: If necessary, a real-time PCR copy number assay is performed. This step is crucial for identifying gene duplications that cause the ultrarapid metabolizer phenotype [101].
- Sequencing: In selected samples, full-gene sequencing of the promoter, exons, and intron-exon boundaries may be required to resolve ambiguous results or detect rare variants [101].
- Phenotype Assignment: Finally, the combined genotype data is interpreted using established guidelines (e.g., from the Clinical Pharmacogenetics Implementation Consortium) to assign one of seven clinical phenotypes: Ultrarapid, Extensive to Ultrarapid, Extensive, Intermediate to Extensive, Intermediate, Poor to Intermediate, or Poor [101].
Quantifying Cellular Redox and Energy States
Measuring metabolites like ATP, ADP, NADP, and NADPH requires precise techniques to capture the rapid changes in these pools.
Representative Protocol for Metabolite Extraction and Measurement [26] [106]:
- Rapid Sampling and Quenching: Cells or tissue samples are rapidly harvested and immediately quenched in cold acid (e.g., perchloric acid) or liquid nitrogen to instantly halt metabolic activity and preserve the in vivo ratios of labile metabolites.
- Extraction: The quenched material is subjected to extraction procedures to obtain a clear supernatant containing the metabolites of interest, while neutralizing the pH as needed.
- Enzymatic Assays: The concentrations of metabolites are determined using specific, coupled enzymatic reactions. For example:
- ATP/ADP: Measured using assays coupled to glucose-6-phosphate dehydrogenase, which consumes NADP+.
- NADP/NADPH: The total pool (NADP + NADPH) can be measured directly. To distinguish the oxidized and reduced forms, separate aliquots of the extract are treated to selectively destroy either NADP+ or NADPH before the measurement.
- Calculation of Ratios: The concentrations are used to calculate the ATP/ADP and NADPH/NADP+ ratios, which serve as indicators of cellular energy charge and redox balance, respectively.
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents and materials essential for conducting research in pharmacogenetics and cellular metabolism.
Table 4: Essential Reagents for Metabolic Phenotyping Research
| Research Reagent / Solution | Function and Application |
|---|---|
| CYP2D6 Genotyping Assay Kits (e.g., xTAG CYP2D6 Kit) | Multiplexed detection of major CYP2D6 alleles for initial phenotype screening [101]. |
| TaqMan Copy Number Assays | Quantitative real-time PCR to determine gene copy number variations, critical for identifying UM phenotypes [101]. |
| Sanger Sequencing Reagents | Full-gene sequencing to identify rare variants or resolve complex genotypes [101]. |
| Enzymatic Assay Kits for ATP, ADP, NADP, NADPH | Spectrophotometric or fluorometric quantification of metabolite concentrations in cell extracts [26] [105]. |
| Acid Quenching Solutions (e.g., Perchloric Acid) | Rapid inactivation of metabolism to preserve the in vivo state of labile metabolites during extraction [26]. |
| Specific CYP2D6 Substrate/Inhibitor Compounds | In vitro tools for functional validation of metabolic activity in enzyme assays or cell models. |
Integrated Signaling and Metabolic Pathways
The clinical outcomes of metabolizer phenotypes are the result of complex interactions between drug pharmacokinetics and the body's fundamental biochemical pathways. The following diagram integrates these concepts, showing how genetic variation influences drug fate and intersects with core energy and redox metabolism.
Title: Drug Metabolism in a Metabolic Context
This integrated view underscores that an individual's response to a drug is not solely determined by the genetics of a single enzyme but is also modulated by the broader metabolic state of the cell, which provides the energy and reducing equivalents necessary for biotransformation processes.
Nicotinamide adenine dinucleotide (NAD) and nicotinamide adenine dinucleotide phosphate (NADP), along with their reduced forms NAD(P)H, are essential metabolic coenzymes in both prokaryotic and eukaryotic cells [18]. These molecules serve as central electron carriers in myriad biochemical reactions, with ATP serving as the primary energy currency for most biological activities [107]. The fundamental functional division between these redox couples is well-established: NADH primarily provides reducing power for catabolic reactions such as glycolysis and mitochondrial oxidative phosphorylation, while NADPH mainly drives anabolic reactions including lipid and nucleic acid synthesis, along with antioxidative reactions [18] [9]. This functional specialization is conserved across diverse organisms, though the specific demand ratios and regulatory mechanisms differ significantly between photosynthetic plants and heterotrophic mammals.
The presence of specialized organelles derived from endosymbiotic events has considerably complicated the functional division of NADH and NADPH systems in eukaryotes [18]. Mammalian cells, possessing only mitochondria from a single endosymbiotic event, maintain relatively straightforward NAD(P)H routing compared to photoautotrophic plants, which contain both mitochondria and chloroplasts from two distinct endosymbiotic events [18]. Chloroplasts in plants provide additional sources of reductants and introduce unique compartmentalization challenges, making the maintenance of redox balance for NAD(P)+/NAD(P)H considerably more complex than in heterotrophic eukaryotes [18] [108]. Understanding how these systems operate in both plant and mammalian contexts provides crucial insights into fundamental bioenergetic principles with implications for fields ranging from crop engineering to therapeutic development.
Comparative Analysis of ATP:NADPH Demand and Supply
ATP:NADPH Dynamics in Plant Systems
In photosynthetic organisms, the coordination between energy production and consumption is particularly critical during illumination when chloroplasts actively produce ATP and NADPH [107]. Linear electron flow (LEF) in chloroplasts generates ATP and NADPH at a constrained stoichiometry of approximately 1.28 ATP per NADPH, creating an inherent supply challenge for metabolic processes with higher ATP demands [29] [33]. The Calvin-Benson-Bassham (CBB) cycle requires ATP and NADPH at a 1.5 ratio, while photorespiration demands an even higher ratio of approximately 1.75 ATP per NADPH equivalent [29]. This discrepancy between supply and demand creates a potential ATP deficit that plants must address through alternative ATP-generating pathways [29].
Table 1: ATP:NADPH Demand Ratios of Major Metabolic Pathways in Plants
| Metabolic Pathway | ATP:NADPH Demand Ratio | Cellular Compartment | Primary Function |
|---|---|---|---|
| Linear Electron Flow | 1.28 (Supply) | Chloroplast | Light-dependent ATP/NADPH production |
| Calvin-Benson-Bassham Cycle | 1.50 | Chloroplast | Carbon fixation |
| Photorespiration | 1.75 | Chloroplast, Peroxisome, Mitochondria | 2-phosphoglycolate recycling |
| Starch/Sucrose Synthesis | Variable | Chloroplast, Cytosol | Carbohydrate storage & transport |
Recent metabolic flux analyses have revealed that the bulk of energy flux in illuminated leaves occurs through the C3 cycle and photorespiration [29]. However, the energy demand from these pathways does not exclusively determine the overall cellular ATP:NADPH demand ratio. Starch and sucrose synthesis contribute significantly to the overall energy budget and may help counterbalance the high ATP demand from photorespiration, potentially reducing the need for rapid adjustments in alternative ATP-generating processes [29]. This distributed energy consumption highlights the complex integration of metabolic pathways across cellular compartments in plants.
To manage the inherent ATP deficit from linear electron flow, plants employ several energy balancing strategies including cyclic electron flow (CEF), the malate valve, and the water-water cycle [29]. The malate-oxaloacetate (malate-OAA) shuttle plays a particularly important role in transporting reducing equivalents between chloroplasts, mitochondria, and peroxisomes, effectively redistributing the redox burden across organelles [33]. During photosynthesis, excess NADPH generated in chloroplasts can be exported to the cytosol as malate via NADP-dependent malate dehydrogenase, which simultaneously regenerates NADP+ as an electron acceptor while moving reducing power to other cellular compartments [33].
ATP:NADPH Dynamics in Mammalian Systems
Mammalian systems maintain a fundamentally different bioenergetic paradigm centered on mitochondrial oxidative phosphorylation as the primary ATP source [9]. The NAD+/NADH redox couple primarily regulates cellular energy metabolism through glycolysis and mitochondrial oxidative phosphorylation, while the NADP+/NADPH couple focuses on maintaining redox balance and supporting biosynthetic processes [9]. Unlike plants, mammals do not face the challenge of balancing energy production from two different energy-generating organelles with incompatible stoichiometries.
The pentose phosphate pathway serves as the major source of NADPH in most mammalian cells, providing reducing power for biosynthetic reactions and maintaining glutathione in its reduced state to combat oxidative stress [109]. Additional NADPH generation occurs through NADP-linked isoforms of malic enzyme, isocitrate dehydrogenase, and glutamate dehydrogenase, with different pathways predominating in various tissue types [109]. For instance, the isocitrate dehydrogenase mechanism appears to be the major source of NADPH in fat and possibly liver cells [109].
Table 2: NADPH-Generating Systems in Mammalian Cells
| NADPH Source | Primary Location | Key Enzymes | Tissue Prevalence |
|---|---|---|---|
| Pentose Phosphate Pathway | Cytosol | Glucose-6-phosphate dehydrogenase | Ubiquitous |
| Malic Enzyme | Cytosol, Mitochondria | ME1, ME2 | Tissue-dependent |
| Isocitrate Dehydrogenase | Mitochondria, Cytosol | IDH1, IDH2 | Adipose tissue, Liver |
| Mitochondrial Folate Cycle | Mitochondria | MTHFD1L, MTHFD2 | Rapidly proliferating cells |
Research has revealed that the relative demand for NAD+ versus ATP can significantly influence metabolic programming in mammalian cells. A 2021 study demonstrated that aerobic glycolysis, often associated with proliferating cells including cancers, reflects situations where cellular demand for NAD+ exceeds the rate of ATP turnover [110]. When NAD+ regeneration by mitochondrial respiration becomes constrained under these conditions, cells increasingly engage in fermentation despite available oxygen, highlighting how the balance between NAD+ and ATP requirements can shape overall metabolic flux [110].
Experimental Approaches and Methodologies
Monitoring NAD(P)H and ATP Dynamics In Planta
The challenge of monitoring in planta dynamic changes of NADP(H) and NAD(H) redox states at the subcellular level has historically been a major obstacle in plant bioenergetics studies [33]. Recent advances in genetically encoded fluorescent sensors have revolutionized this field by enabling real-time monitoring of these metabolites in living cells. The iNAP and SoNar sensors, engineered from bacterial Rex repressor proteins capable of sensing pyridine dinucleotide redox states, provide intense fluorescence, rapid response, high specificity, and large dynamic range [33].
Experimental Protocol for In Planta NAD(P)H Monitoring:
- Generate stable transgenic Arabidopsis lines expressing iNAP (NADPH sensor) or SoNar (NADH/NAD+ ratio sensor) fused with organelle-specific targeting peptides
- Target sensors to specific subcellular compartments (cytosol, plastid stroma, peroxisome) using appropriate signal sequences
- Image seedlings using confocal microscopy with appropriate excitation/emission settings (e.g., excitation at 488 nm)
- Conduct ratiometric measurements to compensate for varied sensor expression levels across tissues and compartments
- Apply light-dark transitions or metabolic inhibitors to perturb photosynthetic and photorespiratory pathways
- Quantify dynamic changes in NADPH levels or NADH/NAD+ ratios in response to experimental manipulations
This approach has revealed that NADPH levels are lower in the cytosol than in plastid stroma and peroxisomes, and that the photosynthetic increases in stromal NADPH and NADH/NAD+ ratio disappear when glycine decarboxylation is inhibited, highlighting the complex interplay between chloroplasts and mitochondria during photosynthesis [33].
Isotopically Non-Stationary Metabolic Flux Analysis (INST-MFA)
Isotopically non-stationary metabolic flux analysis (INST-MFA) has emerged as a powerful technique for quantifying metabolic flux through central metabolism in plants [29]. This method enables researchers to resolve cellular energy flux by coupling metabolic reactions to their associated ATP and NADPH demands, providing unprecedented insights into how individual pathways contribute to overall energy budgets.
Experimental Protocol for INST-MFA:
- Expose plant tissues to 13CO2 under controlled environmental conditions
- Collect time-series samples following isotopic labeling
- Extract metabolites and analyze mass isotopomer distributions using mass spectrometry
- Construct comprehensive metabolic network models incorporating stoichiometric constraints
- Compute flux distributions that best fit experimental labeling data
- Calculate ATP and NADPH demands for individual pathways based on flux values
- Integrate demands across pathways and compartments to determine overall cellular energy budgets
This methodology has been applied across various species including Arabidopsis thaliana, Nicotiana tabacum, and Camelina sativa under different environmental conditions (high/low light, varying day length, different photorespiratory levels), enabling characterization of broad trends in energy demand across pathways and compartments [29].
Research Reagent Solutions Toolkit
Table 3: Essential Research Reagents for NAD(P)H and ATP Studies
| Reagent/Category | Specific Examples | Function/Application | System |
|---|---|---|---|
| Genetically Encoded Fluorescent Sensors | iNAP, SoNar, cpYFP | Real-time monitoring of NADPH, NADH/NAD+ ratios, and ATP in living cells | Plant, Mammalian |
| Metabolic Inhibitors | Glydecarboxylase inhibitors, Rotenone, Oligomycin | Perturb specific metabolic pathways to study their contributions to energy budgets | Plant, Mammalian |
| Isotopic Tracers | 13CO2, U-13C-glucose | Track metabolic flux through central carbon metabolism using INST-MFA or similar approaches | Plant, Mammalian |
| Organelle-Specific Targeting Sequences | Chloroplast transit peptides, Peroxisomal targeting signals | Direct sensors to specific subcellular compartments for compartmentalized analysis | Plant |
| Enzyme Activity Assays | NADK, NADP+ phosphatase, NAMPT | Quantify activity of enzymes involved in NAD(P) biosynthesis and interconversion | Plant, Mammalian |
Pathway Diagrams and System Relationships
The following diagrams illustrate key signaling pathways and metabolic relationships governing ATP:NADPH balance in plant and mammalian systems.
Plant Energy Balancing Pathways
Mammalian NADPH and ATP Generation
Redox Poise Variations in Microbial vs. Mammalian Systems
The redox poise of a cell defines its internal redox state, reflecting the balance between reducing and oxidizing equivalents. This balance is predominantly governed by the ratios of key metabolite pairs, most notably NADPH/NADP+ and ATP/ADP. The NADPH/NADP+ couple is maintained at a relatively reducing steady-state to fuel biosynthetic reactions and antioxidant defenses, while the ATP/ADP ratio represents the energy currency that drives cellular work [111]. In living systems, the redox poise is not merely a static indicator but a dynamic regulator that influences metabolic pathway activity, transcriptional responses, and overall cellular function. Understanding the variations in this poise between microbial and mammalian systems is fundamental for research in bioenergetics, metabolic engineering, and drug development.
The principles of redox organization reveal an underlying simplicity across life forms. Critically, redox reactions enable the essential characteristics of life: energy extraction from the environment, support of metabolic and structural organization, dynamic responses to environmental threats, and mechanisms directing cellular differentiation [111]. This review provides a comparative analysis of redox poise by presenting quantitative data, detailing experimental methodologies for its determination, and visualizing the core regulatory networks in microbial and mammalian cells.
Comparative Quantitative Analysis of Redox Metabolites
The redox and energy states of cells can be quantitatively compared by measuring the concentrations of key metabolites and calculating their ratios. The tables below summarize experimental data from distinct systems.
Table 1: NAD(P)H/NAD(P)+ Ratios in Microbial and Mammalian Systems
| System / Organism | Growth Condition / Cell Type | CNADH/CNAD+ Ratio | CNADPH/CNADP+ Ratio | Citation |
|---|---|---|---|---|
| Geobacter sulfurreducens (Bacterium) | Anode or Fe(III)-citrate (extracellular e- acceptor) | 0.088 ± 0.022 | 0.268 ± 0.098 | [112] |
| Geobacter sulfurreducens (Bacterium) | Fumarate (intracellular e- acceptor) | 0.331 ± 0.094 | 1.96 ± 0.37 | [112] |
| Rat Liver (Mammal) | Cytoplasm | ~ 0.006 (Eh = -241 mV) | ~ 0.63 (Eh = -393 mV) | [111] |
| Rat Liver (Mammal) | Mitochondria | ~ 0.0003 (Eh = -318 mV) | ~ 0.16 (Eh = -415 mV) | [111] |
Table 2: Adenylate Profiles in Microbial Systems under Different Conditions
| Organism | Growth Condition | Adenylate Energy Charge (AEC) | ATP/ADP Ratio | Citation |
|---|---|---|---|---|
| Geobacter sulfurreducens | Suspended cultures (Fumarate or Fe(III)-citrate) | ~ 0.47 | Not Specified | [112] |
| Geobacter sulfurreducens | Biofilm on Anodes | ~ 0.47 | Significantly higher than suspended cultures | [112] |
Experimental Protocols for Determining Redox Poise
Accurate measurement of redox metabolites is technically challenging due to their rapid turnover and susceptibility to oxidation during sample processing. The following protocols outline standardized methods for determining these key ratios.
Protocol for Quantitative Extraction and Spectrophotometric Assay of NAD(P)H/NAD(P)+
This method, adapted from studies on Geobacter sulfurreducens and E. coli, involves rapid quenching of metabolism, differential extraction of oxidized and reduced forms, and enzymatic cycling assays for quantification [112].
- Culture Harvesting and Quenching: For microbial cultures, rapidly collect cells by fast filtration (for aerobic cultures) or immediate centrifugation under anaerobic conditions. For mammalian cells, quickly aspirate media and proceed to extraction. The key is to quench metabolic activity in less than a second to preserve the in vivo ratios.
- Differential Extraction:
- Reduced Forms (NADH & NADPH): Lyse the cell pellet in an alkaline buffer (e.g., 0.2 M NaOH) and heat at 60°C for 5-10 minutes. This destroys the oxidized forms while stabilizing the reduced ones. Neutralize the extract before assay.
- Oxidized Forms (NAD+ & NADP+): Lyse a separate cell pellet in a hot acidic buffer (e.g., 0.2 M HCl) and heat at 60°C for the same duration. This destroys the reduced forms while stabilizing the oxidized ones. Neutralize before assay.
- Enzymatic Cycling Assay:
- For NADH/NAD+: The concentration is determined by coupling the lactate dehydrogenase (LDH) reaction to a colorimetric or fluorescent signal. The assay mixture contains lactate, LDH, and a tetrazolium salt (e.g., MTT). NAD+ in the sample is reduced to NADH by LDH, and the generated NADH reduces MTT to a colored formazan product, which is measured spectrophotometrically. The ratio is calculated from the concentrations of the reduced and oxidized forms [112].
- For NADPH/NADP+: A similar principle is used, typically with glucose-6-phosphate dehydrogenase (G6PDH) as the coupling enzyme.
Protocol for Live-Cell Imaging with Genetically Encoded Biosensors
Genetically encoded sensors allow for non-destructive, real-time monitoring of redox dynamics with high spatiotemporal resolution in living cells [113] [114].
- Sensor Selection and Expression:
- For NADPH/NADP+ Redox Status: Use the NERNST biosensor. NERNST is a fusion of rice NADPH-thioredoxin reductase C (NTRC) with a redox-sensitive green fluorescent protein (roGFP2). It selectively transduces the NADP(H) redox status into a fluorescent readout [113].
- For NADH levels: Use the Frex or Peredox sensors. These are fusions of a bacterial Rex protein (which undergoes conformational change upon NADH binding) with circularly permuted fluorescent proteins [114].
- Cell Transformation/Transfection: Introduce the plasmid encoding the biosensor into the target cells (bacterial or mammalian) using standard methods (e.g., electroporation, chemical transformation, or lipofection).
- Ratiometric Fluorescence Imaging:
- Transfer the expressing cells to an imaging chamber (e.g., a confocal microscope or a microplate reader).
- For NERNST, acquire fluorescence emission at ~510 nm while alternately exciting at two wavelengths: 390 nm (oxidized roGFP2 peak) and 490 nm (reduced roGFP2 peak). The ratio of emissions (I390/I490) is directly related to the oxidation degree of the probe and, thus, the NADP(H) redox status [113].
- For Frex, excite at ~480 nm and measure emission at ~514 nm; an increase in fluorescence corresponds to an increase in free NADH [114].
- Data Calibration and Analysis:
- At the end of the experiment, calibrate the sensor response by treating cells with 10 mM Dithiothreitol (DTT) to fully reduce the sensor (Rmin) and then with 10 mM H2O2 to fully oxidize it (Rmax). The oxidation degree (OxD) is calculated as: OxD = (R - Rmin) / (Rmax - Rmin) [113].
Visualization of Redox Pathway Organization
The following diagrams illustrate the core organization of redox metabolism and the experimental workflow for its assessment, highlighting key differences and commonalities between systems.
Core Redox Metabolism and Regulation
Experimental Workflow for Redox Poise Determination
The Scientist's Toolkit: Essential Research Reagents
This section details key reagents and tools used in modern redox biology research, as featured in the cited experiments and methodologies.
Table 3: Key Reagent Solutions for Redox Poise Research
| Reagent / Tool Name | Type / Category | Primary Function in Research | Example Application |
|---|---|---|---|
| NERNST Biosensor | Genetically Encoded Fluorescent Biosensor | Ratiometric, non-destructive estimation of NADP(H) redox status (ENADP(H)) in live cells. | Monitoring NADP(H) dynamics during bacterial growth, environmental stress in plants, and metabolic challenges in mammalian cells [113]. |
| Frex/Peredox Sensors | Genetically Encoded Fluorescent Biosensor | Specific detection of free NADH levels (Frex) or the NADH:NAD+ ratio (Peredox) in live cells. | Real-time assessment of the metabolic profile of living cells, study of redox alterations in malignant cells [114]. |
| RX1 Probe | Small Molecule Chemical Probe | Selective measurement of thioredoxin reductase (TrxR) activity, enabling assessment of redox flux in live cells. | Correlating the activity of a major antioxidant enzyme with the metabolic state of the cell in diseases like cancer and diabetes [115]. |
| TrxRFP1 / roGFP2-based probes | Genetically Encoded Fluorescent Biosensor | Measurement of the redox poise of specific systems, such as the thioredoxin or glutathione pathways. | Determining the steady-state oxidation state of thiol-based antioxidant systems in various cellular compartments [115]. |
| Enzymatic Assay Kits (e.g., LDH, G6PDH) | Biochemical Reagent | Enable spectrophotometric or fluorometric quantification of specific metabolites (e.g., NADH, NADPH) in cell lysates via coupled reactions. | Used in extraction-based protocols to determine absolute concentrations and ratios of NAD(H) and NADP(H) [112]. |
| Dithiothreitol (DTT) | Reducing Agent | Strong chemical reductant used to fully reduce disulfide bonds in proteins and biosensors. | In-situ calibration of biosensors like NERNST to define the minimum ratio (Rmin) for fully reduced state [113]. |
| Hydrogen Peroxide (H₂O₂) | Oxidizing Agent | Exogenous source of oxidative stress; used to fully oxidize redox-sensitive probes. | In-situ calibration of biosensors like NERNST to define the maximum ratio (Rmax) for fully oxidized state [113]. |
Pathological Ratio Shifts in Barth Syndrome and Neurodegenerative Disease
The quantitative assessment of specific biochemical ratios has emerged as a powerful diagnostic and research tool across pathological conditions. This guide compares the alterations in ATP/ADP, NADPH/NADP+, and related metabolic ratios between Barth syndrome and neurodegenerative diseases, two distinct categories of disorders with underlying mitochondrial dysfunction. Barth syndrome, an X-linked disorder caused by mutations in the TAFAZZIN gene, demonstrates characteristic cardiolipin abnormalities that disrupt mitochondrial energy production [116] [117]. Neurodegenerative diseases such as Alzheimer's disease (AD) and Parkinson's disease (PD) exhibit progressive metabolic alterations that precede clinical symptoms by decades [118] [119]. Understanding these ratio shifts provides crucial insights into disease mechanisms and potential therapeutic targets.
The comparative analysis of these ratios reveals both unique and shared pathological features across diseases. In Barth syndrome, the monolysocardiolipin/cardiolipin (MLCL:CL) ratio serves as a pathognomonic diagnostic biomarker [116] [120], while neurodegenerative conditions demonstrate progressive shifts in NAD+/NADH ratios and related redox pairs that correlate with disease progression [55] [121]. This guide systematically compares the experimental methodologies, quantitative findings, and clinical implications of these ratio alterations to facilitate cross-disciplinary research approaches.
Experimental Protocols for Ratio Quantification
Cardiolipin Analysis in Barth Syndrome
Sample Preparation: The MLCL:CL ratio assay requires specific sample handling protocols. For clinical diagnosis, a 5 mL blood sample is collected in appropriate anticoagulant tubes, though the test can also be performed on patient-derived fibroblast cell lysates or dried blood spots [120]. Fibroblasts are cultured under standard conditions, harvested at confluence, and lysed in specialized buffers that preserve cardiolipin integrity. Lipid extraction is typically performed using modified Bligh-Dyer techniques with chloroform-methanol solvents [122].
Mass Spectrometry Analysis: Liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) represents the gold standard for cardiolipin speciation and quantification [122] [120]. The methodology employs multiple reaction monitoring (MRM) to identify specific cardiolipin species based on mass-to-charge ratios and fragmentation patterns. The MLCL:CL ratio is calculated by comparing the peak areas of monolysocardiolipin to mature tetralinoleyl-cardiolipin species. The assay demonstrates high specificity and sensitivity for Barth syndrome, with results typically available within several weeks when processed through specialized laboratories like the Laboratory of Genetic Metabolic Diseases at Amsterdam University Medical Center [120].
Quality Control: Internal standards including deuterated cardiolipin analogs are essential for quantification accuracy. Simultaneous analysis of control samples from healthy individuals ensures appropriate assay calibration. The diagnostic cutoff for MLCL:CL ratio in Barth syndrome is well-established, with affected individuals typically showing ratios dramatically elevated above the normal range of 0-0.3 [117].
NAD+/NADH and ATP/ADP Quantification in Neurodegenerative Research
Sample Collection for Neurodegenerative Studies: Cerebrospinal fluid (CSF) collection via lumbar puncture after an overnight fast represents the primary methodology for human biomarker analysis in neurodegenerative diseases [119]. CSF samples are immediately aliquoted into polypropylene cryotubes, maintained on dry ice, and transferred to -80°C freezers for long-term storage to prevent analyte degradation. For tissue-based studies, post-mortem brain samples require rapid processing and specialized preservation protocols.
Enzymatic Cycling Assays: Sensitive enzymatic cycling assays provide precise quantification of redox pairs including NAD+/NADH and NADP+/NADPH [2]. These assays exploit substrate-specific enzymes coupled to colorimetric or fluorometric readouts. For NAD+ quantification, samples are treated with alcohol dehydrogenase and ethanol, converting NAD+ to NADH which reacts with specific tetrazolium salts to generate colored formazan products measurable at 565 nm [2]. Parallel assays under acidic and basic conditions allow separate quantification of oxidized and reduced forms.
Advanced Imaging Techniques: Fluorescence lifetime imaging microscopy (FLIM) enables non-invasive measurement of NADH and FAD (flavin adenine dinucleotide) in living cells and tissues [118]. This technique exploits the distinct fluorescence lifetimes of free versus protein-bound NADH, providing insights into metabolic states without requiring cell disruption. The optical redox ratio (NADH/FAD) derived from FLIM data serves as a sensitive indicator of mitochondrial function in neuronal tissues [118].
Table 1: Core Methodologies for Metabolic Ratio Analysis
| Analysis Target | Primary Methodology | Sample Types | Key Technical Considerations |
|---|---|---|---|
| MLCL:CL Ratio | LC-MS/MS | Blood, fibroblasts, dried blood spots | Requires specialized lipid extraction; internal standards essential |
| NAD+/NADH Ratio | Enzymatic cycling assays | CSF, tissue homogenates, cell lysates | Acid/base treatment separates oxidized/reduced forms |
| ATP/ADP Ratio | Luciferase-based assays | Fresh tissue, cultured cells | Requires rapid processing to preserve labile phosphates |
| NADPH/NADP+ Ratio | Enzymatic cycling assays | CSF, tissue homogenates | Parallel assays with G6PDH and glutathione reductase |
| Optical Redox Ratio | FLIM | Living cells, tissue sections | Measures NADH/FAD fluorescence lifetimes |
Comparative Analysis of Pathological Ratios
Barth Syndrome: Cardiolipin Abnormalities and Metabolic Consequences
Barth syndrome demonstrates a characteristic and pathognomonic alteration in the MLCL:CL ratio due to defective cardiolipin remodeling caused by TAFAZZIN mutations [116] [117]. The MLCL:CL ratio in affected individuals typically ranges from 70 to 253 in fibroblasts, dramatically elevated from the normal range of 0-0.3 [117]. This specific biochemical signature results from the accumulation of monolysocardiolipin (MLCL) and decreased mature tetralinoleyl-cardiolipin, disrupting mitochondrial membrane integrity and function.
The abnormal MLCL:CL ratio directly impacts electron transport chain efficiency, particularly affecting complexes I, III, and IV, which require cardiolipin for proper assembly and function [122]. This disruption manifests as reduced oxidative phosphorylation capacity, with secondary effects on ATP production and cellular energy balance. Additionally, approximately 70% of Barth syndrome patients demonstrate 3-methylglutaconic aciduria, with urinary 3-methylglutaconic acid (3-MGC) levels elevated 5- to 20-fold above normal ranges [116]. This organic aciduria reflects broader mitochondrial dysfunction and represents an additional diagnostic ratio utilized in clinical assessment.
Supporting laboratory findings include lactic acidosis, hypocholesterolemia (total cholesterol <110 mg/dL), and neutropenia (absolute neutrophil count <1,500 cells/µL) [116]. These systemic manifestations underscore the far-reaching consequences of the primary cardiolipin abnormality and its impact on overall metabolic homeostasis.
Table 2: Characteristic Ratio Alterations in Barth Syndrome
| Biochemical Parameter | Normal Range | Barth Syndrome Range | Clinical Significance |
|---|---|---|---|
| MLCL:CL Ratio (fibroblasts) | 0-0.3 | 70-253 | Pathognomonic diagnostic biomarker |
| Urinary 3-Methylglutaconic Acid | 3.7±1.8 μg/mg Cr (adults) | 44.6±25 μg/mg Cr | Supportive diagnostic marker |
| Plasma 3-Methylglutaconic Acid | 162±68 nmol/L | 1,088±435 nmol/L | Indicator of mitochondrial dysfunction |
| Total Cholesterol | >110 mg/dL | <110 mg/dL | Common finding (hypocholesterolemia) |
| Absolute Neutrophil Count | >1,500 cells/µL | <1,500 cells/µL | Infection risk indicator |
Neurodegenerative Diseases: Redox Imbalances and Energetic Deficits
Neurodegenerative diseases demonstrate progressive alterations in NAD+/NADH ratios and related metabolic parameters that evolve throughout disease course. Longitudinal studies in Alzheimer's disease reveal that cerebrospinal fluid biomarkers begin changing 15-20 years before clinical symptom onset, with amyloid beta (Aβ) abnormalities detectable approximately 17.1 years pre-onset and phosphorylated tau changes appearing about 15.8 years pre-onset [119]. These proteinopathies precede measurable neuronal damage, indicated by neurofilament light chain (NfL) elevations approximately 11.6 years before diagnosis.
The brain's extreme energy demands render it particularly vulnerable to redox imbalances. Under normal conditions, the brain constitutes approximately 2% of body mass yet consumes 20% of total oxygen and 25% of glucose [118]. Neurons and glial cells maintain distinct metabolic profiles, with neurons preferentially utilizing oxidative metabolism while astrocytes employ higher glycolytic activity [118]. In neurodegenerative conditions, this delicate balance is disrupted, leading to progressive NAD+ deficiency and altered NAD+/NADH ratios that impair cellular resilience.
Research indicates that NADH reductive stress, characterized by NADH accumulation, drives metabolic reprogramming in neurodegenerative contexts [121]. This reductive stress typically originates from mitochondrial dysfunction, nutrient overload, or enzymatic malfunctions, creating a vicious cycle of impaired electron transport chain function and increased reactive oxygen species production. The resulting energy deficit manifests as reduced ATP/ADP ratios, particularly in vulnerable neuronal populations.
Table 3: Metabolic Ratio Timeline in Alzheimer's Disease Progression
| Biomarker | Change Point Before Symptoms | Direction of Change | Associated Process |
|---|---|---|---|
| CSF Aβ42/Aβ40 Ratio | 17.1 years | Decreased | Amyloid plaque formation |
| CSF p-tau181 | 15.8 years | Increased | Tau pathology |
| Neurofilament Light Chain | 11.6 years | Increased | Axonal damage |
| Whole Brain White Matter Volume | 11.6 years | Decreased | White matter degeneration |
| Total Ventricular Volume | 9.7 years | Increased | Brain atrophy |
Pathway Visualization and Molecular Interactions
Cardiolipin Remodeling Pathway in Barth Syndrome
Cardiolipin Remodeling Defect in Barth Syndrome
NAD+ Metabolism in Neurodegenerative Contexts
NAD+ Metabolism in Neurodegeneration
Research Reagent Solutions Toolkit
Table 4: Essential Research Reagents for Metabolic Ratio Studies
| Reagent/Category | Specific Examples | Research Application | Technical Considerations |
|---|---|---|---|
| Mass Spectrometry Standards | Deuterated cardiolipin analogs (d4-CL), Internal NAD+ standards | Quantitative normalization for LC-MS/MS assays | Ensure minimal isotopic interference; match extraction efficiency |
| Enzymatic Assay Kits | NAD/NADH-Glo Assay, ATP Bioluminescence Assay Kit | High-throughput screening of metabolic ratios | Linear range validation required for different sample types |
| Metabolic Inhibitors | G6PDi-1 (PPP inhibitor), Rotenone (Complex I inhibitor) | Pathway perturbation studies | Dose-response characterization essential for specificity |
| Fluorescent Probes | 10-N-Nonyl Acridine Orange (NAO), NADH autofluorescence | Live-cell imaging of metabolic states | Potential phototoxicity; validation against biochemical methods |
| Cell Culture Models | Patient-derived fibroblasts, iPSC-derived neurons | Disease-specific mechanistic studies | Confirm preservation of pathological phenotypes in culture |
| Antibodies for Detection | Anti-cardiolipin antibodies, Anti-NADK antibodies | Protein localization and expression analysis | Specificity validation via knockout controls recommended |
Comparative Discussion and Research Implications
The pathological ratio shifts in Barth syndrome and neurodegenerative diseases reveal both divergent and convergent metabolic disruptions. Barth syndrome demonstrates a highly specific biochemical signature centered on the MLCL:CL ratio, directly resulting from a single genetic defect in cardiolipin remodeling [116] [117]. In contrast, neurodegenerative diseases exhibit progressive alterations in NAD+/NADH ratios that represent downstream consequences of multifactorial processes including protein aggregation, oxidative stress, and inflammatory responses [118] [55].
From a diagnostic perspective, Barth syndrome benefits from a definitive biochemical test (MLCL:CL ratio) that provides clear diagnostic confirmation [120]. Neurodegenerative diseases lack such pathognomonic single ratios, instead relying on biomarker panels that evolve over extended timelines [119]. The different temporal patterns are striking: while Barth syndrome manifestations typically present in infancy with acute metabolic decompensation, neurodegenerative diseases demonstrate gradual ratio alterations over decades preceding clinical symptoms.
Therapeutic strategies targeting these ratio disturbances similarly diverge. Barth syndrome management focuses on symptomatic treatment of cardiac and hematological manifestations, with ongoing research investigating cardiolipin-targeted approaches [116]. In neurodegenerative contexts, NAD+ precursors and mitochondrial stabilizers represent promising investigative avenues aimed at restoring redox balance [55] [121]. Future research directions include developing small molecules that enhance cardiolipin remodeling efficiency and compounds that boost NAD+ bioavailability while mitigating reductive stress.
The comparative analysis of these ratio disturbances highlights the importance of context-specific interpretation. The MLCL:CL ratio provides exceptional diagnostic specificity for Barth syndrome but lacks relevance in neurodegenerative contexts. Conversely, NAD+/NADH ratio alterations represent a common final pathway in numerous neurological conditions but offer limited diagnostic specificity. These distinctions underscore the necessity of selecting appropriate ratio biomarkers based on clinical context and research objectives.
This comparison guide provides a foundation for cross-disciplinary research approaches, facilitating the transfer of methodological insights between seemingly distinct pathological conditions. The continued refinement of ratio quantification methodologies and expansion of reference databases will enhance both diagnostic precision and therapeutic development across the spectrum of metabolic disorders.
Conclusion
This analysis establishes that ATP/ADP and NADPH/NADP+ ratios serve as integrative biomarkers reflecting the complex interplay between cellular energy status, redox balance, and metabolic pathway flux. The consistent finding across studies is that these ratios are highly dynamic and context-dependent, responding to nutritional status, oxidative stress, genetic background, and disease pathology. Future research should prioritize developing standardized methodologies for ratio quantification across biological compartments and establishing validated reference ranges for specific tissues and pathological conditions. For biomedical and clinical research, targeting the enzymatic regulators of these ratios—such as NAD kinase or electron transport chain components—represents a promising therapeutic frontier for metabolic diseases, cancer, and neurodegenerative disorders. The integration of metabolic ratio analysis with genomic and proteomic data will enable more precise metabolic phenotyping for personalized medicine approaches.
References
- 1. Adenylate kinase [https://en.wikipedia.org/wiki/Adenylate_kinase]
- 2. Oxidative Stress Induces the Phosphorylation of NAD+ to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12553597/]
- 3. Cofactor (biochemistry) [https://en.wikipedia.org/wiki/Cofactor_(biochemistry)]
- 4. Conformational dynamics of adenylate kinase in crystals - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10883716/]
- 5. The energy landscape of adenylate kinase during catalysis [https://pmc.ncbi.nlm.nih.gov/articles/PMC4318763/]
- 6. A survey of synthetic nicotinamide cofactors in enzymatic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4866995/]
- 7. Engineering a nicotinamide mononucleotide redox cofactor ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7546441/]
- 8. Biomimetic cofactors and methods for their recycling [https://www.sciencedirect.com/science/article/abs/pii/S136759311830142X]
- 9. NAD(H) and NADP(H) Redox Couples and Cellular Energy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5737637/]
- 10. NADH reductive stress drives metabolic reprogramming [https://pubmed.ncbi.nlm.nih.gov/40769852/]
- 11. Reductive stress: The key pathway in metabolic disorders ... [https://www.sciencedirect.com/science/article/pii/S2090123225000311]
- 12. A Pilot Study Using Patient Fibroblasts and a Mouse Model [https://www.mdpi.com/2218-273X/15/1/38]
- 13. Metabolic Reprogramming Shapes the Progression and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12374702/]
- 14. Review P/O ratios of mitochondrial oxidative phosphorylation [https://www.sciencedirect.com/science/article/pii/S0005272804002701]
- 15. Insights into Metabolic Reprogramming in Tumor Evolution ... [https://www.mdpi.com/2072-6694/16/20/3513]
- 16. Architecting the metabolic reprogramming survival risk ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-024-05138-2]
- 17. Cell cycle progression is regulated by intertwined redox ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4459109/]
- 18. NAD(H) and NADP(H) in plants and mammals - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC12178909/]
- 19. Estimating physical conditions supporting gradients of ATP ... [https://www.sciencedirect.com/science/article/pii/S0006349525003765]
- 20. Semisynthetic biosensors for mapping cellular ... [https://elifesciences.org/articles/32638]
- 21. Compositional data analysis enables statistical rigor in ... [https://www.nature.com/articles/s41467-025-56249-3]
- 22. Comparative Analysis of Biochemical and Cellular Assay ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12429910/]
- 23. Redox regulation: mechanisms, biology and therapeutic ... [https://www.nature.com/articles/s41392-024-02095-6]
- 24. Homeostatic regulation of NAD(H) and NADP(H) in cells [https://www.sciencedirect.com/science/article/pii/S2352304223004294]
- 25. NADPH homeostasis in cancer: functions, mechanisms ... [https://www.nature.com/articles/s41392-020-00326-0]
- 26. In Vivo Changes of the Oxidation-Reduction State of NADP ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC167085/]
- 27. Redox Homeostasis and Prospects for Therapeutic Targeting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8355971/]
- 28. The Importance of Energy Balance in Improving ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3075755/]
- 29. Evaluating the contribution of plant metabolic pathways in ... [https://link.springer.com/article/10.1007/s11120-024-01106-5]
- 30. Citric acid cycle [https://en.wikipedia.org/wiki/Citric_acid_cycle]
- 31. Mitochondrial TCA cycle metabolites control physiology ... [https://www.nature.com/articles/s41467-019-13668-3]
- 32. Systematic Comparison of C3 and C4 Plants Based on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3521184/]
- 33. In planta study of photosynthesis and photorespiration using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7320160/]
- 34. The advantages and the limitations of LC-MS/MS and Elisa ... [https://www.biotrial.com/bioanalysis-lc-ms-ms-vs-elisa/]
- 35. Liquid chromatography-tandem mass spectrometry in ... [https://www.sciencedirect.com/science/article/abs/pii/S0009898124020989]
- 36. Utility, promise, and limitations of liquid chromatography‐mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8597589/]
- 37. Comparison of HPLC-RI, LC/MS-MS and enzymatic assays ... [https://www.sciencedirect.com/science/article/abs/pii/S0308814617307136]
- 38. Meta-analysis of NAD(P)(H) quantification results exhibits ... [https://www.nature.com/articles/s41598-023-29607-8]
- 39. Comparison of HPLC-RI, LC/MS-MS and enzymatic assays ... [https://pubmed.ncbi.nlm.nih.gov/28530589/]
- 40. Assays for NAD+-Dependent Reactions and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6409209/]
- 41. Mass-spectrometry based metabolomics - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC12105183/]
- 42. Metabolite Ratios as Quality Indicators for Pre-Analytical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8465943/]
- 43. An Overview of Pre-Analytical Factors Impacting ... [https://www.mdpi.com/2218-1989/14/9/474]
- 44. Pre-Analytical Sample Quality: Metabolite Ratios as an ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0121495]
- 45. In planta study of photosynthesis and photorespiration ... [https://www.nature.com/articles/s41467-020-17056-0]
- 46. The Use of Microdosing for In vivo Phenotyping ... [https://link.springer.com/article/10.1007/s13318-024-00896-2]
- 47. The impact of the exposome on cytochrome P450 ... [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1639646/full]
- 48. The impact of the exposome on cytochrome P450-mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12554611/]
- 49. Cytochromes P450 and P-Glycoprotein Phenotypic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9693601/]
- 50. Empirical Drug Dosage Validates Pharmacogenomic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12619694/]
- 51. The genetic landscape of major drug metabolizing ... [https://www.nature.com/articles/s41397-022-00288-2]
- 52. Direct real-time quantification of mitochondrial oxidative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5129772/]
- 53. Investigating mitochondrial redox state using NADH and ... [https://www.sciencedirect.com/science/article/pii/S0891584916303926]
- 54. The Crucial Role of NAD+ in Mitochondrial Metabolic ... [https://www.sciencedirect.com/org/science/article/pii/S032795452500060X]
- 55. The role of NAD+ metabolism and its modulation ... [https://www.nature.com/articles/s44324-025-00067-0]
- 56. Mitochondrial, metabolic and bioenergetic adaptations ... [https://www.nature.com/articles/s41419-025-07596-y]
- 57. Isotopically nonstationary metabolic flux analysis (INST-MFA) [https://www.sciencedirect.com/science/article/abs/pii/S0958166917302549]
- 58. Isotopically nonstationary metabolic flux analysis (INST-MFA) [https://medschool.vanderbilt.edu/mpb/publication/isotopically-nonstationary-metabolic-flux-analysis-inst-mfa-putting-theory-into-practice/]
- 59. Systematic comparison of local approaches for isotopically nonstationary metabolic flux analysis - PMC [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10280729/]
- 60. Model Validation and Selection in Metabolic Flux Analysis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10922127/]
- 61. Isotopically nonstationary MFA (INST-MFA) of autotrophic ... [https://pubmed.ncbi.nlm.nih.gov/24222417/]
- 62. Metabolic flux and flux balance analyses indicate the ... [https://www.sciencedirect.com/science/article/pii/S1096717625001211]
- 63. Flux sampling is a powerful tool to study metabolism under ... [https://www.nature.com/articles/s41540-019-0109-0]
- 64. Metabolic Flux Ratio Analysis of Genetic and Environmental ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC94132/]
- 65. Measurements of the ATP/ADP and NADPH/NADP+ Ratios [https://bio-protocol.org/exchange/minidetail?id=9273383&type=30]
- 66. Rethinking 13C-metabolic flux analysis – The Bayesian ... [https://www.sciencedirect.com/science/article/pii/S1096717624000508]
- 67. Cellular ATP levels alone do not reliably reflect overall ... [https://www.oaepublish.com/articles/jtgg.2025.02]
- 68. Brain NAD Is Associated With ATP Energy Production and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7772416/]
- 69. Bioenergetic biomarkers as predictive indicators and their ... [https://www.nature.com/articles/s41398-025-03367-7]
- 70. Metabolic implications of non-electrogenic ATP/ADP ... [https://www.sciencedirect.com/science/article/pii/S0005272821000438]
- 71. ATP requirements for growth reveal the bioenergetic impact of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7617979/]
- 72. NAD+ metabolism: pathophysiologic mechanisms and ... [https://www.nature.com/articles/s41392-020-00311-7]
- 73. Fuel-induced amplification of insulin secretion in mouse ... [https://link.springer.com/article/10.1007/s00125-007-0849-z]
- 74. Centrifugation-induced release of ATP from red blood cells [https://pmc.ncbi.nlm.nih.gov/articles/PMC6124747/]
- 75. Effect of High Temperature on Photosynthesis in Beans (II. ... [https://pubmed.ncbi.nlm.nih.gov/12226443/]
- 76. A bacterial effector protein targets plant ferredoxin-NADP+ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12591422/]
- 77. Transition to 37°C reveals importance of NADPH in ... [https://insight.jci.org/articles/view/126376]
- 78. A family of NADPH/NADP+ biosensors reveals in vivo ... [https://www.nature.com/articles/s41467-024-55302-x]
- 79. Whole-cell modeling in yeast predicts compartment ... [https://www.nature.com/articles/s41467-022-28467-6]
- 80. Compartment-specific metabolome labeling enables the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8536584/]
- 81. Absorbance vs. Luminescence Assays [https://byonoy.com/journal/resources-absorbance-vs-luminescence-assays/]
- 82. Comparison of Bioluminescent Kinase Assays Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3096547/]
- 83. Comparison of Colorimetric and Fluorometric ... [https://www.mdpi.com/2227-9040/10/12/542]
- 84. A comparison of two colorimetric assays, based upon ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3819989/]
- 85. Comparison to Other Quantitation Methods [https://www.merckmillipore.com/DE/de/life-science-research/protein-detection-quantification/direct-detect-spectrometer/Comparison-to-Other-Methods/H6Sb.qB.06QAAAFPqLJFgZOm,nav]
- 86. Bioluminescence readout lateral flow immunoassay using ... [https://pubs.rsc.org/en/content/articlehtml/2025/an/d5an00030k]
- 87. A Novel Smartphone-Based Bioluminescence Color ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11932295/]
- 88. ATP vs ADP: Key Differences and Functions in the Body [https://www.creative-proteomics.com/resource/atp-vs-adp-differences-functions-body.htm]
- 89. Regulation of Muscle Glycogen Metabolism during Exercise [https://pmc.ncbi.nlm.nih.gov/articles/PMC5872716/]
- 90. Metabolic adaptation to weight loss: implications for the athlete [https://jissn.biomedcentral.com/articles/10.1186/1550-2783-11-7]
- 91. Differences between the Resting Metabolic Profile of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10383823/]
- 92. Influence of ATP turnover and metabolite changes on IMP ... [https://pubmed.ncbi.nlm.nih.gov/2399963/]
- 93. Machine Learning-Driven Metabolic Syndrome Prediction [https://pmc.ncbi.nlm.nih.gov/articles/PMC11675332/]
- 94. Independent Validation and Assay Standardization of ... [https://www.sciencedirect.com/science/article/pii/S001650852200823X]
- 95. External Validation of the Prognostic Prediction Model for 4 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8257261/]
- 96. DNA methylation-based predictors of metabolic traits in ... [https://www.sciencedirect.com/science/article/pii/S000292972400421X]
- 97. A Comparative Analysis of Data Normalization Approaches ... [https://www.mdpi.com/2079-3197/12/7/137]
- 98. Preclinical modeling of metabolic syndrome to study the ... [https://www.nature.com/articles/s41598-024-71202-y]
- 99. Optimization and validation of the international metabolic ... [https://www.nature.com/articles/s41408-025-01338-1]
- 100. Genetic Factors in Drug Metabolism | AAFP [https://www.aafp.org/pubs/afp/issues/2008/0601/p1553.html]
- 101. Increased risk of hospitalization for ultrarapid of... metabolizers [https://pmc.ncbi.nlm.nih.gov/articles/PMC5317339/]
- 102. Table of Pharmacogenetic Associations | FDA [https://www.fda.gov/medical-devices/precision-medicine/table-pharmacogenetic-associations]
- 103. Metabolic flux analysis and the NAD(P)H/NAD(P)+ ratios in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5776761/]
- 104. Assimilatory Force and Regulation of Photosynthetic ... [https://link.springer.com/chapter/10.1007/978-1-4684-8571-4_45]
- 105. NADP+/NADPH ratio - Bacteria Escherichia coli [https://bionumbers.hms.harvard.edu/bionumber.aspx?s=n&v=6&id=105023]
- 106. In vivo changes of the oxidation-reduction state of NADP ... [https://pubmed.ncbi.nlm.nih.gov/12857827/]
- 107. ATP homeostasis and signaling in plants - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11009363/]
- 108. NAD(H) and NADP(H) in plants and mammals - ScienceDirect [https://www.sciencedirect.com/science/article/pii/S1674205225001637]
- 109. Nicotinamide adenine dinucleotide phosphate [https://en.wikipedia.org/wiki/Nicotinamide_adenine_dinucleotide_phosphate]
- 110. Increased demand for NAD+ relative to ATP drives aerobic ... [https://www.sciencedirect.com/science/article/pii/S1097276520309047]
- 111. Redox organization of living systems - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11313653/]
- 112. Changes in phosphorylation of adenosine phosphate and ... [https://www.sciencedirect.com/science/article/abs/pii/S1567539415000274]
- 113. NERNST: a genetically-encoded ratiometric non- ... [https://www.nature.com/articles/s41467-023-38739-4]
- 114. Real-Time Assessment of the Metabolic Profile of Living ... [https://www.sciencedirect.com/science/article/abs/pii/B9780124166189000182]
- 115. Flux versus poise: Measuring the dynamic cellular activity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9253492/]
- 116. Barth Syndrome - GeneReviews® - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK247162/]
- 117. New clinical and molecular insights on Barth syndrome [https://ojrd.biomedcentral.com/articles/10.1186/1750-1172-8-27]
- 118. Altered substrate metabolism in neurodegenerative disease [https://pmc.ncbi.nlm.nih.gov/articles/PMC8555332/]
- 119. Change points for dynamic biomarkers in the Alzheimer's ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12311614/]
- 120. Barth Syndrome Diagnostic Laboratory Testing [https://www.barthsyndrome.org/barthsyndrome/diagnose/methods-for-obtaining-a-barth-syndrome-diagnosis.html]
- 121. NADH reductive stress drives metabolic reprogramming [https://www.sciencedirect.com/science/article/pii/S0962892425001576]
- 122. The Role of Cardiolipin in Mitochondrial Function and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11012098/]