Benchmarking Microbial Chassis for Polyketide Production: A 2025 Guide for Synthetic Biology and Drug Discovery
The efficient heterologous production of polyketides is pivotal for drug discovery and development, yet the selection and optimization of a chassis strain remain significant challenges.
Benchmarking Microbial Chassis for Polyketide Production: A 2025 Guide for Synthetic Biology and Drug Discovery
Abstract
The efficient heterologous production of polyketides is pivotal for drug discovery and development, yet the selection and optimization of a chassis strain remain significant challenges. This article provides a contemporary, comprehensive guide for researchers and scientists, synthesizing the latest advances in chassis benchmarking. We explore the foundational principles of chassis selection, detail cutting-edge metabolic engineering methodologies, present systematic troubleshooting and optimization strategies, and establish a rigorous framework for the comparative validation of host performance. By integrating insights from recent high-impact studies on Streptomyces, E. coli, and yeast platforms, this review serves as an essential resource for streamlining the development of high-yield microbial cell factories for diverse polyketides.
The Polyketide Production Landscape: Why Your Chassis Choice Matters
Selecting an optimal microbial host is a critical first step in the efficient discovery and production of polyketides, a class of natural products with widespread pharmacological applications. This guide objectively compares the performance of various chassis strains, supported by recent experimental data, to provide a framework for researchers in metabolic engineering and drug development.
Polyketides are synthesized by polyketide synthases (PKSs), which are categorized into three types based on their domain structure and mechanism. Type I PKSs are large, modular assembly lines where each module catalyzes one elongation step, commonly found in bacteria and fungi and responsible for producing complex macrolides like erythromycin [1]. Type II PKSs are complexes of discrete, monofunctional proteins that operate iteratively to produce aromatic compounds, such as actinorhodin and tetracyclines [2] [1]. Type III PKSs are homodimeric enzymes that use acyl-CoA substrates directly and are typically involved in producing simpler aromatic compounds in plants [3].
An ideal chassis must efficiently express these often-large biosynthetic gene clusters (BGCs), provide ample precursor supply, and demonstrate compatibility with the target polyketide's structural class. The host's genetic stability, manipulation ease, and fermentation characteristics are also crucial practical considerations [2].
Comparative Performance of Common Polyketide Chassis
The table below summarizes key performance metrics for commonly used and emerging chassis strains, based on recent heterologous expression studies.
Table 1: Comparative Performance of Chassis Strains for Polyketide Production
| Chassis Strain | PKS Type Compatibility | Reported Production Titer (Example) | Genetic Tractability | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| Streptomyces aureofaciens Chassis2.0 | Type II (Primary), Type I | Oxytetracycline: 370% increase vs. commercial strain [2] | Moderate | High precursor supply; Native high-yield T2PKs producer; Efficient for tri-, tetra-, and penta-ring T2PKs [2] | Limited to Streptomyces; Requires cluster knockout [2] |
| Escherichia coli | Type I, Type III | Limited success with Type II PKSs [2] | High | Fast growth; Extensive genetic tools; Well-characterized physiology [3] | Challenges with soluble expression of minimal PKS for Type II compounds; Lack of native post-modification enzymes [2] |
| Saccharomyces cerevisiae | Type III, Type I (Fungal) | Various flavonoids and stilbenes [3] | High | Eukaryotic protein processing; Compartmentalization; Tolerant to secondary metabolites [3] | Suboptimal efficiency for some pathways; Long fermentation cycles; Limited precursor pool [2] [3] |
| Streptomyces albus J1074 | Type II, Type I | Oxytetracycline: Not detected in heterologous expression [2] | High | Well-established model system; Efficient genetic manipulation [2] | Often requires extensive metabolic engineering for high titer; Low native precursor flux [2] |
| Yarrowia lipolytica | Type III | Emerging host for plant polyketides [3] | Moderate (Improving) | High acetyl-CoA flux; Oleaginous [3] | Limited genetic tools compared to model hosts [3] |
Experimental Workflow for Chassis Evaluation and Engineering
The following diagram outlines a generalizable experimental protocol for evaluating and engineering a chassis strain, synthesizing methodologies from recent studies.
In-Depth Analysis: Streptomyces Chassis2.0 as a Case Study
A recent breakthrough demonstrates the power of rational chassis selection and engineering. The industrial chlortetracycline producer Streptomyces aureofaciens J1-022 was identified as a promising host due to its inherent high flux through type II polyketide pathways [2].
Table 2: Key Reagents and Research Solutions for Chassis Engineering
| Reagent / Solution | Function in Research | Specific Example / Application |
|---|---|---|
| ExoCET Method | Direct cloning of large biosynthetic gene clusters (BGCs) into shuttle vectors [2]. | Used to construct p15A_oxy plasmid containing the complete oxytetracycline BGC from S. rimosus [2]. |
| High-Yield Industrial Strain | Provides a native background with optimized precursor supply and tolerance for polyketides [2]. | S. aureofaciens J1-022, a high-yield chlortetracycline producer, served as the base for Chassis2.0 [2]. |
| In-Frame Deletion | Removes native BGCs to eliminate competition for essential precursors like malonyl-CoA [2]. | Creation of a "pigmented-faded" host (S. aureofaciens Chassis2.0) by deleting two endogenous T2PKs clusters [2]. |
| HPLC-MS Analysis | Detects, identifies, and quantifies polyketide products from fermentation broths [2]. | Used to confirm the production of oxytetracycline, actinorhodin, and the novel compound TLN-1 in Chassis2.0 [2]. |
Key Experimental Findings:
- Eliminating Precursor Competition: The engineered Chassis2.0, with two endogenous T2PK gene clusters knocked out, showed a 370% increase in oxytetracycline production compared to a commercial strain, highlighting the importance of redirecting metabolic flux [2].
- Broad Substrate Compatibility: Chassis2.0 successfully produced diverse polyketides, including the tri-ring actinorhodin, flavokermesic acid, and a novel pentangular polyketide (TLN-1), by directly activating an unidentified BGC [2].
- Superior Performance over Model Strains: In contrast, conventional model chassis like S. albus J1074 and S. lividans TK24 failed to produce any detectable oxytetracycline when transformed with the same BGC, underscoring the limitation of model systems without extensive engineering [2].
Strategic Recommendations for Chassis Selection
Based on the comparative data, researchers can use the following framework for selecting a chassis:
- For Type II Aromatic Polyketides: Engineered industrial Streptomyces strains (e.g., Chassis2.0) are superior due to their native compatibility and high precursor supply [2].
- For Type I Modular Polyketides: E. coli and S. cerevisiae remain strong candidates, but success may depend on the specific PKS and require extensive engineering for soluble expression and post-translational modifications [1] [3].
- For Plant-Derived Type III Polyketides: S. cerevisiae is often ideal due to its eukaryotic machinery and ability to functionally express plant P450 enzymes for downstream tailoring [3].
The field is moving towards developing specialized chassis for specific polyketide classes rather than seeking a universal host. Future efforts will likely focus on further engineering precursor pathways and regulatory networks in these specialized hosts to unlock the full potential of polyketide biodiscovery.
The genus Streptomyces is a cornerstone of modern biotechnology, renowned for its robust capacity to produce a vast array of medically relevant natural products, including antibiotics, immunosuppressants, and anticancer agents [4]. However, a significant challenge persists in the field: the majority of natural product biosynthetic gene clusters (BGCs) in native strains are either silent under standard laboratory conditions or produce compounds at yields too low for practical application [4]. Heterologous expression—the process of transferring BGCs into a genetically tractable "chassis" strain—has emerged as a powerful strategy to overcome these limitations. This approach allows researchers to awaken silent clusters and optimize production. Yet, the success of this strategy is heavily dependent on the choice of host, as the complex genetic circuits and precursor requirements of natural product biosynthesis necessitate a diverse panel of heterologous hosts [4] [2]. No single host can universally express all BGCs efficiently, creating a pressing need for new, high-performing chassis strains tailored for specific classes of compounds, such as polyketides [5]. This guide provides an objective comparison of the latest engineered Streptomyces strains, benchmarking their performance as dedicated platforms for polyketide production.
Comparative Analysis of Engineered Streptomyces Chassis Strains
Rigorous benchmarking is essential for selecting an appropriate chassis strain. The table below summarizes the key characteristics and performance data of recently developed Streptomyces hosts, providing a direct comparison for researchers.
Table: Performance Comparison of Streptomyces Chassis Strains for Polyketide Production
| Chassis Strain | Parental Strain | Key Genetic Modifications | Polyketide Types Tested | Key Performance Findings | Reported Yields (Comparative) |
|---|---|---|---|---|---|
| Streptomyces sp. A4420 CH [4] | Streptomyces sp. A4420 | Deletion of 9 native polyketide BGCs [4] | Type I and II PKS; diverse chemical scaffolds [4] | Produced all 4 tested benchmark metabolites; outperformed parental strain and other standard hosts in every condition [4] | Consistent high-yield producer across all tested BGCs [4] |
| Chassis2.0 [2] | S. aureofaciens J1-022 (industrial CTC producer) | In-frame deletion of two endogenous T2PK gene clusters [2] | Type II PKS (tetracyclines, tri-ring, penta-ring) [2] | 370% increase in oxytetracycline production; efficient synthesis of tri-ring and novel penta-ring T2PKs [2] | High-yield production of OTC, ACT, FK, and novel TLN-1 [2] |
| S. coelicolor M1152 [4] | S. coelicolor M145 | Deletion of four native BGCs; introduction of rpoB mutation [4] | Type I and II PKS [4] | Failed to produce oxytetracycline; requires extensive engineering for some T2PKs [4] [2] | Limited efficiency for heterologous T2PKs (e.g., OTC not detected) [2] |
| S. lividans TK24 [4] | S. lividans 66 | Removal of SLP2 and SLP3 plasmids [4] | Type I and II PKS [4] | Failed to produce oxytetracycline; a widely adopted host but with variable efficiency [4] [2] | Limited efficiency for heterologous T2PKs (e.g., OTC not detected) [2] |
The data reveal a clear trend: strains engineered from industrial producers or metabolically gifted wild types, such as the A4420 CH strain and Chassis2.0, demonstrate superior performance and broader compatibility compared to traditional model hosts like S. coelicolor M1152 and S. lividans TK24. The A4420 CH strain's key advantage is its remarkable consistency and ability to successfully express all tested polyketide BGCs, a feat not achieved by any other host in its benchmarking study [4]. Conversely, Chassis2.0 shows exceptional prowess specifically for type II aromatic polyketides, enabling not only overproduction but also the direct activation of cryptic BGCs for novel compound discovery [2].
Experimental Protocols for Benchmarking Chassis Strains
To ensure the reproducibility of chassis strain evaluations, researchers must adhere to standardized experimental workflows. The following protocols detail the key methodologies used to generate the comparative data.
Genome Sequencing and Cluster Identification
A critical first step in chassis development is the comprehensive genomic characterization of a candidate strain. The standard protocol involves:
- Genomic DNA (gDNA) Extraction: High-quality, high-molecular-weight gDNA is isolated from the candidate strain.
- Hybrid Genome Sequencing: A combination of long-read (e.g., Oxford Nanopore) and short-read (e.g., Illumina) sequencing technologies is employed to achieve a complete and accurate genome assembly [4].
- BGC Identification and Analysis: The assembled genome is analyzed using specialized bioinformatics tools like antiSMASH to identify and annotate all native biosynthetic gene clusters, particularly those for polyketides [4] [6]. This map guides the strategic deletion of competing pathways to create a metabolically simplified host.
Heterologous Expression and Metabolite Analysis
The core of chassis benchmarking involves testing its ability to produce compounds from heterologously expressed BGCs.
- BGC Cloning and Vector Construction: Target BGCs are cloned from donor strains, often using advanced techniques like ExoCET, into E. coli-Streptomyces shuttle vectors [2].
- Conjugative Transfer: The constructed vector is introduced into the chassis strain via conjugation from E. coli. Successful exconjugants are selected and validated by PCR [2].
- Fermentation and Metabolite Profiling: Engineered strains are cultivated in appropriate liquid media. After a defined fermentation period, culture broth is extracted, and metabolites are analyzed using Liquid Chromatography-Mass Spectrometry (LC-MS). The key is to compare the production profile of the chassis to that of the parental strain and other established hosts under identical conditions [4] [2].
Figure 1: Experimental workflow for chassis development and benchmarking
The Scientific Toolkit: Essential Reagents for Chassis Development
The engineering and evaluation of Streptomyces chassis strains rely on a suite of specialized reagents and tools. The following table outlines the core components of the scientific toolkit.
Table: Essential Research Reagents and Tools for Streptomyces Chassis Engineering
| Reagent / Tool | Function / Description | Application in Chassis Work |
|---|---|---|
| antiSMASH Software [7] [6] | A bioinformatics platform for the genome-wide identification, annotation, and analysis of biosynthetic gene clusters. | Critical first step for mapping native BGCs to be deleted and for analyzing heterologous BGCs for expression. |
| ExoCET Technology [2] | (Exonuclease in vitro Combined with RecET) A cloning method that enables direct capture and manipulation of large DNA fragments with high efficiency. | Used for seamless cloning of large, intact polyketide BGCs into expression vectors for heterologous expression [2]. |
| E. coli-Streptomyces Shuttle Vector [2] | A plasmid capable of replication in both E. coli (for cloning) and Streptomyces (for expression). | Serves as the vehicle for introducing heterologous BGCs into the chassis strain. |
| LC-MS / HPLC-ELSD [4] [7] | (Liquid Chromatography-Mass Spectrometry / Evaporative Light Scattering Detector) Analytical techniques for separating, detecting, and quantifying metabolites. | Essential for profiling secondary metabolites, confirming compound production, and comparing yields between different chassis strains. |
| ISP2 & TSBY Media [7] [8] | Standard culture media for the cultivation and fermentation of Streptomyces strains. | Used for routine culture maintenance and optimized production of secondary metabolites during benchmarking. |
The strategic development of specialized chassis strains like Streptomyces sp. A4420 CH and Chassis2.0 marks a significant leap forward in natural product discovery and biomanufacturing. The evidence demonstrates that moving beyond traditional model organisms to engineer hosts from industrially proven or metabolically versatile backgrounds is a powerful paradigm [4] [2]. The A4420 CH strain offers a robust, general-purpose platform for a wide range of polyketides, while Chassis2.0 provides a specialized and highly efficient system for type II aromatic polyketides. As genomics and synthetic biology tools continue to advance, the establishment of a diverse and well-characterized panel of heterologous hosts will be crucial for unlocking the vast potential of silent biosynthetic pathways [4] [5]. This will not only accelerate the discovery of novel therapeutics to combat antimicrobial resistance but also streamline the efficient production of known, high-value compounds. The future of the Streptomyces workhorse lies in its continued rational engineering into a versatile and powerful biofactory.
Polyketides represent a cornerstone of modern pharmacotherapy, providing the foundation for numerous antibiotics, anticancer agents, and immunosuppressants [9]. These complex natural products are synthesized by massive enzyme complexes known as polyketide synthases (PKSs), which in their native hosts—often slow-growing, genetically intractable actinomycetes—present significant challenges for large-scale production and engineering [9] [10]. The pursuit of an optimal heterologous host for polyketide biosynthesis has therefore become a central focus in metabolic engineering, with Escherichia coli emerging as a powerful contender despite its inherent limitations [11] [9].
This guide objectively benchmarks E. coli against alternative microbial platforms for polyketide production, with particular emphasis on the experimental strategies that have overcome its native limitations. We frame this comparison within the broader thesis that chassis selection must balance genetic tractability, precursor availability, and pathway compatibility to maximize polyketide titers and enable combinatorial biosynthesis of novel compounds [2].
Native Limitations ofE. colifor Polyketide Synthesis
Despite its well-established genetics and rapid growth, E. coli initially presented several formidable barriers to polyketide production. The bacterium naturally lacks the specialized metabolism of native polyketide producers, creating four primary challenges that required systematic addressing.
Key Challenges and Engineering Solutions
- Limited Precursor Availability: E. coli maintains tight regulation of malonyl-CoA, a central polyketide building block, primarily directing it toward fatty acid synthesis. This results in an insufficient pool for high-level polyketide production [12].
- Lack of Post-Translational Activation: Native E. coli lacks a phosphopantetheinyl transferase (PPTase) with broad specificity, leaving the acyl carrier protein (ACP) domains of heterologous PKSs inactive [9].
- Difficulty Expressing and Folding Large PKS Proteins: The megasynthases characteristic of type I PKSs often posed challenges for heterologous expression, solubility, and correct folding in E. coli [9] [10].
- Absence of Specialized Tailoring Enzymes: Many polyketides require glycosylation or other modifications by enzymes not natively present in E. coli, limiting production of fully elaborated bioactive molecules [13].
Comparative Performance Benchmarking:E. colivs. Alternative Chassis
To objectively evaluate E. coli as a polyketide production platform, we compare its performance against Streptomyces, the most common native host, using quantitative data from recent studies.
Table 1: Chassis Performance Comparison for Polyketide Production
| Feature | E. coli (Engineered) | Streptomyces Chassis | Experimental Evidence |
|---|---|---|---|
| Genetic Manipulation | Highly tractable; extensive toolbox [14] | Moderate; strain-dependent [2] | Direct genome editing and plasmid recombineering established in E. coli [14] |
| Growth Cycle | Rapid (hours) [9] | Slow (days) [2] | E. coli doubling time ~20 min; Streptomyces requires complex developmental cycle |
| Precursor Supply (Malonyl-CoA) | Engineered for enhanced supply [12] | Naturally abundant but regulated | E. coli engineered with orthogonal MatBC pathway significantly increases malonyl-CoA [12] |
| Type I PKS Production | Successful (e.g., 6-dEB, 20 mg/L) [9] | Native excellence | 6-deoxyerythronolide B (6-dEB) produced in engineered E. coli BAP1 [9] |
| Type II PKS Production | Recently achieved [10] | Native excellence | Soluble KS/CLF heterodimers expressed; anthraquinones synthesized [10] |
| Glycosylated Products | Achieved with pathway engineering (e.g., Erythromycin D) [13] | Native capability | Erythromycin D production increased 60-fold in engineered E. coli via TDP-sugar pathway optimization [13] |
| Model System Compatibility | Plug-and-play combinatorial biosynthesis demonstrated [10] | Limited HTP compatibility | E. coli pipeline used to produce new-to-nature compounds neomedicamycin and neochaetomycin [10] |
The data reveal a clear trade-off: while Streptomyces strains like S. aureofaciens Chassis2.0 demonstrate superior native compatibility and titers for complex molecules like oxytetracycline (370% increase over commercial strains) [2], E. coli offers unparalleled genetic accessibility and speed, enabling rapid prototyping and combinatorial biosynthesis.
Engineering Breakthroughs: Methodologies and Protocols
The transformation of E. coli into a viable polyketide producer required coordinated solutions across multiple biological scales, from individual enzymes to overall cellular physiology.
Key Experimental Strategies and Workflows
Strategy 1: Enhancing Malonyl-CoA Availability
A critical breakthrough involved rewiring central metabolism to increase the supply of malonyl-CoA, a key polyketide precursor. A 2025 study detailed a controlled strategy combining gene disruption, orthogonal pathway introduction, and adaptive laboratory evolution [12].
Experimental Protocol: Malonyl-CoA Enhancement in E. coli [12]
- Strain Construction: Knock out the bioH gene, creating a biotin auxotroph and disrupting the native malonyl-CoA biosynthetic pathway.
- Orthogonal Pathway Integration: Integrate the matC (malonate transporter) and matB (malonyl-CoA synthetase) genes from Rhizobium trifolii into the E. coli genome under a lacUV5 promoter.
- Precursor Supplementation: Grow engineered strains in medium supplemented with malonate (0-20 mM) to provide the direct substrate for the MatBC pathway.
- Titer Validation: Measure polyketide production (e.g., using flaviolin as a reporter) via absorbance at 340 nm and 520 nm.
- Strain Improvement: Perform adaptive laboratory evolution (ALE) by serially passaging strains in malonate-containing medium to select for mutations that further boost malonyl-CoA and polyketide levels.
Strategy 2: Plasmid Stabilization Without Antibiotics
A 2024 study addressed the industrial problem of plasmid instability in antibiotic-free cultures by developing a symbiotic plasmid-host system [15].
Experimental Protocol: Antibiotic-Free Plasmid Maintenance [15]
- Chromosomal Deletion: Knock out the essential folP gene (involved in folate biosynthesis) from the E. coli chromosome.
- Plasmid Engineering: Create a plasmid carrying both the folP gene and the target biosynthetic gene (e.g., aadL for phenylpyruvic acid production).
- Promoter Engineering: Tune the expression strength of folP using different constitutive promoters (e.g., J23100, J23101) to optimize plasmid copy number and product yield.
- Stability Testing: Culture the engineered strain for multiple serial passages in antibiotic-free medium, monitoring both plasmid retention and product titer.
- Bioconversion Assay: Use stabilized strains for whole-cell biotransformation, achieving 18.7 g/L phenylpyruvic acid within 14 hours.
Strategy 3: Heterologous Expression of Type II PKSs
The functional expression of type II PKSs in E. coli remained elusive for decades until a 2019 study established a plug-and-play production line [10].
Experimental Protocol: Type II PKS Reconstitution in E. coli [10]
- Bioinformatic Identification: Select candidate ketosynthase (KS) and chain-length factor (CLF) pairs from organisms like Photorhabdus luminescens with predicted E. coli compatibility.
- Co-expression with Chaperones: Express KS/CLF heterodimers in E. coli with molecular chaperones (GroEL/ES) to ensure proper folding and solubility.
- Pathway Reconstitution: Co-express minimal PKS components with phosphopantetheinyl transferase (Sfp) to activate acyl carrier proteins.
- Combinatorial Biosynthesis: Swap tailoring enzymes (e.g., ketoreductases, cyclases) from phylogenetically diverse sources (bacteria, fungi, plants) to create structural diversity.
- Compound Characterization: Identify novel polyketides (e.g., neomedicamycin) using LC-MS/MS and NMR spectroscopy.
The Scientist's Toolkit: Essential Research Reagents
Successful engineering of E. coli for polyketide production relies on specialized genetic tools and biological reagents.
Table 2: Key Research Reagents for Polyketide Engineering inE. coli
| Reagent / Tool | Function | Example Application |
|---|---|---|
| Sfp Phosphopantetheinyl Transferase | Activates ACP domains of PKSs by adding phosphopantetheine cofactor | Essential for functional expression of DEBS in E. coli BAP1 strain [9] |
| MatBC Malonate Assimilation System | Provides orthogonal pathway for malonyl-CoA synthesis from exogenous malonate | Increases intracellular malonyl-CoA pool; improves flaviolin production 70% [12] |
| λ-Red Recombinase System | Enables efficient chromosomal modifications and plasmid recombineering | Used for gene knockouts (e.g., bioH, folP) and pathway integration [15] [14] |
| BAP1 and K207-3 Engineered Strains | Specialist E. coli hosts with enhanced propionyl-CoA/methylmalonyl-CoA metabolism | Production of 6-dEB and erythromycin precursors [9] [13] |
| Triple-Selection Recombineering Cassette | Combines positive/negative selection with fluorescence for precise plasmid engineering | Enables robust plasmid manipulation at any copy number without antibiotic dependence [14] |
| ExoCET Cloning Technology | Facilitates direct cloning of large biosynthetic gene clusters | Used to construct E. coli-Streptomyces shuttle vectors with complete oxytetracycline BGC [2] |
The experimental evidence demonstrates that E. coli has successfully transitioned from a fundamentally incompatible host to a powerful platform for polyketide biosynthesis. Through systematic metabolic engineering, it now achieves production titers for certain molecules that rival or surpass those in engineered Streptomyces hosts, particularly for type I polyketides and an expanding repertoire of type II compounds [12] [10].
The benchmarking data reveal a clear decision framework for chassis selection: Streptomyces remains optimal for producing complex aromatic polyketides with minimal engineering, while E. coli excels in high-throughput combinatorial biosynthesis and rapid prototyping of novel compounds. Future directions will likely focus on refining dynamic control of precursor pathways, expanding the suite of compatible PKS systems, and further automating the engineering workflow to fully realize E. coli's potential as a plug-and-play platform for natural product discovery and production.
This guide provides an objective comparison of emerging and specialized microbial hosts for polyketide production, benchmarking their performance against conventional chassis to inform strategic selection for metabolic engineering and drug development research.
The pursuit of optimal microbial chassis for polyketide production is central to advancing pharmaceutical and biotechnology applications. This guide benchmarks the performance of several emerging hosts—including novel yeast and Streptomyces strains—against established alternatives. Quantitative comparisons focus on growth metrics, polyketide titers, and production efficiency. The evaluation reveals that specialized industrial Streptomyces strains, engineered for enhanced precursor supply and reduced metabolic competition, currently outperform other hosts in complex polyketide synthesis. Concurrently, non-conventional yeasts like Yarrowia lipolytica show exceptional promise for simpler polyketides, particularly when equipped with novel malonyl-CoA pathways. The data underscores a trend toward developing dedicated, metabolically streamlined chassis tailored for specific polyketide classes.
Host Performance Benchmarking
The table below summarizes key quantitative data for the featured chassis, enabling direct comparison of their performance and characteristics.
Table 1: Performance Benchmarking of Specialized Microbial Chassis
| Host Organism | Key Engineering Feature | Target Product(s) | Reported Titer / Performance | Key Advantage |
|---|---|---|---|---|
| Streptomyces aureofaciens Chassis2.0 [2] | Deletion of two endogenous T2PKs gene clusters | Oxytetracycline, Actinorhodin, Flavokermesic acid, TLN-1 | 370% increase in oxytetracycline vs. commercial strains; High-efficiency tri- and penta-ring T2PKs [2] | Superior chassis-product compatibility for diverse Type II Polyketides (T2PKs) [2] |
| Saccharomyces cerevisiae XP [16] | Novel isolate with native rapid growth | General chassis capability; L-lactic acid (as proof-of-concept) | Doubling time of 43.61 min (YPD2 medium), nearly 2x faster than S288C [16] | Innate robustness and fast growth in high sugar & low pH conditions [16] |
| Streptomyces sp. A4420 CH [17] | Deletion of 9 native polyketide BGCs | Four distinct heterologous polyketides | Successful production of all 4 tested polyketides, outperforming common Streptomyces hosts [17] | High metabolic capacity and superior sporulation/growth pattern [17] |
| Yarrowia lipolytica [18] | Engineered L-glutamate/L-aspartate to malonyl-CoA pathways | Triacetic acid lactone (TAL), 4-hydroxy-6-hydroxyethyl-2-pyrone (HHEP) | HHEP titer of 6.4 g/L in shaking flask [18] | Novel, thermodynamically favorable pathway for malonyl-CoA synthesis [18] |
| E. coli K207-3–MatBC [12] | Orthogonal malonyl-CoA pathway (matC & matB) integrated into genome | Flaviolin (M-CoA proxy), Pikromycin derivatives | 70% increase in flaviolin production with 20 mM malonate [12] | Controllable malonyl-CoA levels via malonate supplementation [12] |
Experimental Protocols for Host Evaluation
To ensure reproducibility and provide clarity on the data sources, this section details the key experimental methodologies cited in the performance benchmarks.
Protocol: Benchmarking Type II Polyketide Production in Streptomyces Chassis2.0
This protocol is derived from the development and validation of S. aureofaciens Chassis2.0 [2].
- 1. Chassis Construction:
- Strain Background: Begin with S. aureofaciens J1-022, a high-yield chlortetracycline producer.
- Gene Cluster Deletion: Execute an in-frame deletion of two endogenous T2PKs gene clusters to create a pigmented-faded host, designated Chassis2.0. This mitigates precursor competition and simplifies product detection [2].
- 2. Heterologous Expression:
- Vector Construction: Use ExoCET technology to clone the complete oxytetracycline (OTC) biosynthetic gene cluster (BGC) from S. rimosus ATCC 10970 into an E. coli-Streptomyces shuttle plasmid (e.g., p15A_oxy).
- Transformation: Introduce the constructed plasmid into the model chassis (e.g., S. albus J1074, S. lividans TK24) and the newly created Chassis2.0 via conjugation or protoplast transformation.
- Verification: Confirm successful integration of the OTC BGC through PCR verification (e.g., using primers for key OTC genes) [2].
- 3. Fermentation and Analysis:
- Culture Conditions: Grow strains in appropriate liquid media (exact composition not specified in source).
- Metabolite Extraction: Use standard solvent extraction (e.g., ethyl acetate) from culture broth.
- Product Quantification: Analyze extracts using High-Performance Liquid Chromatography (HPLC). Compare OTC yields from Chassis2.0 against yields from model chassis and commercial production strains [2].
Protocol: Enhancing Malonyl-CoA Supply in Yarrowia lipolytica
This protocol outlines the engineering strategy used to boost polyketide precursor supply in Y. lipolytica [18].
- 1. Pathway Engineering:
- Design: Retrosynthetically design two novel malonyl-CoA pathways using L-glutamate and L-aspartate as substrates. These pathways leverage oxidative deamination and decarboxylation reactions to generate malonate semialdehyde, which is then converted to malonyl-CoA by malonyl‐CoA reductase (MCR).
- Implementation: Clone genes encoding the key enzymes (e.g., IboH for the glutamate route, aspartate transaminase and MCR for the aspartate route) into expression vectors suitable for Y. lipolytica.
- 2. Strain Evaluation:
- Host Strain: Use Y. lipolytica Po1fK as the base strain.
- Transformation: Introduce the engineered pathway plasmids into the host.
- Screening: Use the production of the simple polyketide triacetic acid lactone (TAL) as a reporter to validate the functionality and efficiency of the novel pathways compared to the native acetyl-CoA carboxylase (ACC) route [18].
- 3. Fermentation and Metabolite Analysis:
- Culture: Ferment engineered strains in YPD-based medium in shaking flasks.
- Analysis: Quantify TAL and the newly discovered polyketide 4‐hydroxy‐6‐hydroxyethyl‐2‐pyrone (HHEP) using chromatographic methods (e.g., GC-MS or HPLC) to determine final titers [18].
Pathway and Workflow Visualization
The synthesis of polyketides relies on key precursors and specialized enzymatic machinery. The following diagrams illustrate the core metabolic pathways and engineering workflows discussed in this guide.
Engineered Malonyl-CoA Pathways in Yeast
This diagram visualizes the novel malonyl-CoA synthesis pathways engineered in Yarrowia lipolytica to boost polyketide production [18]. These pathways provide an efficient, ATP-independent alternative to the native route.
Streptomyces Chassis Development Workflow
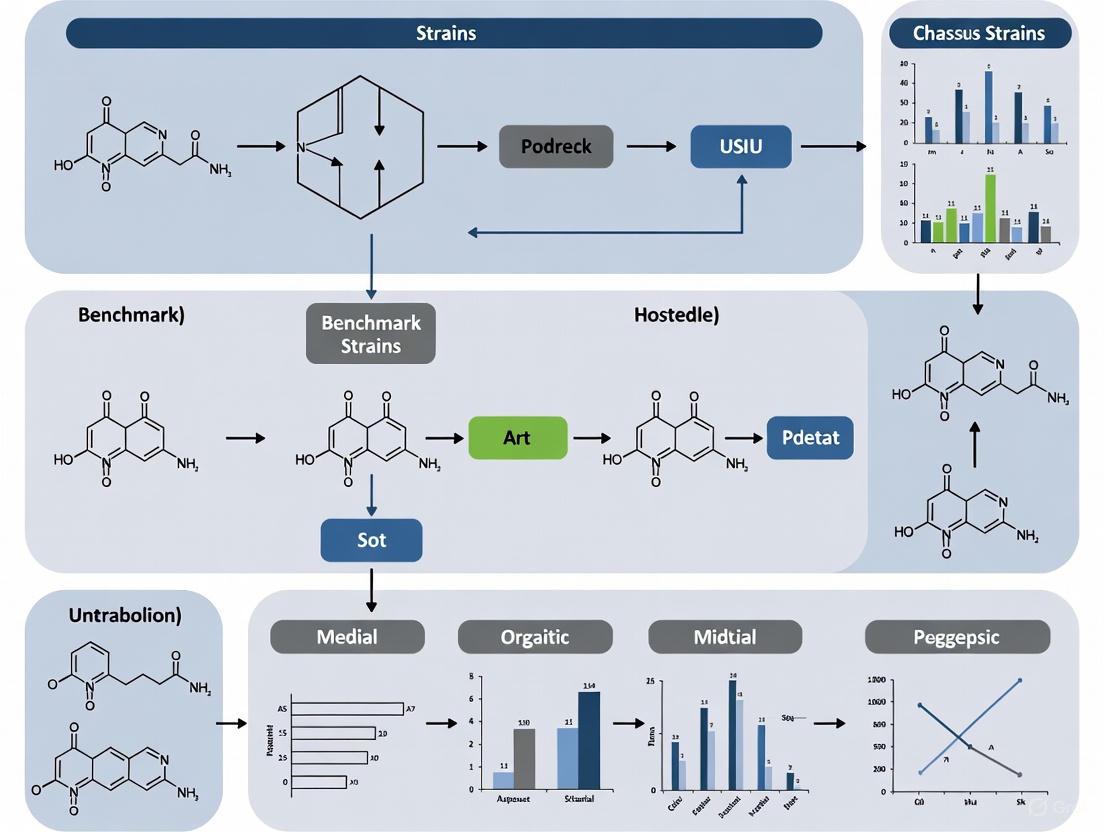
This flowchart outlines the strategic process for developing and validating a high-performance Streptomyces chassis for heterologous polyketide production, as demonstrated with Chassis2.0 and Streptomyces sp. A4420 CH [2] [17].
The Scientist's Toolkit: Essential Research Reagents
Successful engineering and evaluation of these specialized hosts require a suite of key reagents and tools. The following table details essential solutions used in the featured studies.
Table 2: Key Research Reagent Solutions for Chassis Engineering
| Research Reagent | Function in Chassis Development | Example Application / Note |
|---|---|---|
| ExoCET Technology [2] | Facilitates direct cloning and assembly of large biosynthetic gene clusters (BGCs) into shuttle vectors. | Used to construct the p15A_oxy plasmid containing the complete oxytetracycline BGC for heterologous expression [2]. |
| CRISPR/Cas9 System [16] | Enables precise, efficient genome editing for deleting competing gene clusters or introducing new genes. | A highly efficient tool for rapid multiplexed gene knockout in yeast and streptomyces chassis strains [16]. |
| Cre/loxP System [16] | Allows for recyclable marker selection and curated genomic rearrangements, enabling multiple rounds of engineering. | Useful for iterative genome editing in diploid industrial yeast strains, aiding in the construction of stable auxotrophs [16]. |
| Malonyl-CoA Biosensor [12] | Indirectly quantifies intracellular malonyl-CoA levels by linking them to the production of a colored compound. | The type III PKS RppA converts M-CoA to THN (which forms red flaviolin), providing a visual and quantifiable readout [12]. |
| Orthogonal MatBC Pathway [12] | Provides external control over intracellular malonyl-CoA levels via supplementation of malonate. | Integrated into the E. coli genome to create a tunable system for enhancing polyketide production without plasmid dependency [12]. |
The successful microbial production of polyketides, a class of pharmaceutically valuable natural products, hinges on two fundamental pillars: adequate precursor supply and optimal chassis-product compatibility. Precursor supply provides the essential building blocks for polyketide backbone assembly, primarily malonyl-CoA and methylmalonyl-CoA, while chassis-product compatibility ensures the host organism possesses the necessary enzymatic machinery and cellular environment for efficient biosynthesis and tolerance of the target compound. Without both elements functioning synergistically, efforts to achieve high-titer polyketide production face significant bottlenecks. This guide objectively compares the performance of major chassis strains, highlighting how different engineering strategies address these critical aspects to enhance polyketide production.
Chassis Strain Comparison: Performance Data and Analysis
The choice of host organism significantly influences the success of polyketide production projects. The table below summarizes key performance metrics for prominent engineered chassis strains, highlighting their advantages and limitations.
Table 1: Comparative Performance of Engineered Chassis Strains for Polyketide Production
| Chassis Strain | Polyketide Type | Key Engineering Strategy | Production Performance | Notable Advantages | Primary Limitations |
|---|---|---|---|---|---|
| Streptomyces aureofaciens Chassis2.0 [2] | Type II (Oxytetracycline, Actinorhodin) | In-frame deletion of two endogenous T2PKs gene clusters [2] | 370% increase in OTC vs. commercial strains; High-efficiency tri-ring T2PKs production [2] | High native precursor flux; Compatible with diverse T2PKs structures; Efficient discovery and overproduction [2] | Limited to Streptomyces specialists; Requires specialized genetic tools [2] |
| Streptomyces sp. A4420 CH [17] | Type I & II Polyketides (e.g., Benzoisochromanequinone, Glycosylated Macrolide) | Deletion of 9 native polyketide BGCs [17] | Produced all 4 tested metabolites; Outperformed parental strain and model hosts [17] | Rapid growth and high sporulation rate; High metabolic capacity; Superior BGC activation [17] | Newer host with less established toolset; Requires genomic simplification [17] |
| Engineered E. coli (K207-3–MatBC) [19] | Type III PKS (Flaviolin) | Orthogonal malonate assimilation pathway (matC transporter + matB ligase); Native pathway disruption [19] | 70% increase in flaviolin with 20mM malonate; Tunable M-CoA levels [19] | Universal genetic tools; Rapid growth; Controllable precursor supply; Wide substrate promiscuity potential [19] | Requires functional PKS expression; Lack of native PPTases; Metabolic burden from complex pathways [19] |
| Conventional Model Streptomyces (S. albus J1074, S. lividans TK24) [2] [17] | Type II (Oxytetracycline) | Introduction of heterologous OTC BGC [2] | No TCs accumulation without further engineering [2] | Well-established genetic tools; Extensive prior characterization [17] | Often requires additional engineering for efficient production; Suboptimal heterologous expression efficiency [2] |
Experimental Protocols for Chassis Evaluation and Engineering
Protocol: Developing a Versatile Streptomyces Chassis
The development of Streptomyces aureofaciens Chassis2.0 provides a methodology for creating specialized hosts [2].
- Host Selection: Identify a native high-producing strain with desirable physiological traits. S. aureofaciens J1-022 was selected as a high-yield chlortetracycline producer [2].
- Genomic Simplification: Execute in-frame deletions of endogenous biosynthetic gene clusters (BGCs) to eliminate competition for precursors. In Chassis2.0, two native T2PKs gene clusters were deleted [2].
- Heterologous BGC Introduction: Clone the target BGC into an appropriate E. coli-Streptomyces shuttle plasmid (e.g., using ExoCET technology). Verify construct integrity via PCR [2].
- Fermentation and Analysis: Cultivate engineered strains in suitable media, then detect and quantify polyketide production using HPLC and comparison to authentic standards [2].
Protocol: Engineering an Orthogonal Malonyl-CoA Supply in E. coli
This protocol enables controllable precursor enhancement in a versatile bacterial host [19].
- Pathway Disruption: Knock out the native malonyl-CoA biosynthetic pathway (e.g., by disabling bioH, essential for biotin biosynthesis and Ac-CoA carboxylase function) to create a malonate-dependent phenotype [19].
- Orthogonal Pathway Integration: Genomically integrate an orthogonal malonate assimilation pathway (e.g., matC malonate transporter and matB malonyl-CoA ligase from Rhizobium trifolii under inducible promoters) at a defined safe site (e.g., downstream of ompW) [19].
- PKS Expression: Co-transform the strain with plasmids encoding the target polyketide synthase and necessary activation enzymes (e.g., sfp for phosphopantetheinylation) [19].
- Precursor Tuning and Production: Culture engineered strains in media with varying malonate concentrations (e.g., 0-20 mM) to tune intracellular malonyl-CoA levels and monitor polyketide output [19].
Visualizing the Chassis Selection and Engineering Framework
The following diagram illustrates the critical decision points and engineering strategies for optimizing polyketide production, integrating both precursor supply and chassis compatibility considerations.
The Scientist's Toolkit: Essential Research Reagents
Successful chassis engineering requires specific genetic tools and reagents. The following table details key solutions mentioned in the cited research.
Table 2: Key Research Reagent Solutions for Polyketide Chassis Engineering
| Reagent / Tool Name | Function / Application | Experimental Context |
|---|---|---|
| ExoCET Technology [2] | Direct cloning and assembly of large biosynthetic gene clusters (BGCs) into shuttle vectors. | Used for constructing p15A_oxy plasmid containing the complete oxytetracycline BGC for heterologous expression [2]. |
| Orthogonal MatBC Pathway [19] | Provides controllable malonyl-CoA supply independent of the native metabolic pathway. | Genomically integrated matC (transporter) and matB (ligase) in E. coli to enable malonate-concentration-dependent tuning of M-CoA levels [19]. |
| p15A_oxy Plasmid [2] | E. coli-Streptomyces shuttle plasmid carrying the complete OTC BGC. | Used for heterologous expression of oxytetracycline in various Streptomyces chassis strains [2]. |
| AntiSMASH Software [17] | In silico identification and analysis of secondary metabolite BGCs in microbial genomes. | Used for genome mining to identify native polyketide BGCs for deletion in the construction of the Streptomyces sp. A4420 CH strain [17]. |
| CRISPRi/sRNA Libraries [20] | Systems metabolic engineering tool for targeted repression of gene expression to re-direct metabolic flux. | Cited as a tool for optimizing titer, rate, and yield (TRY) in engineered E. coli by channeling cellular resources toward product formation [20]. |
Building a Better Factory: Strain Engineering and Pathway Refactoring Strategies
The efficient production of valuable natural products in microbial hosts is often hindered by intrinsic cellular complexity. Metabolic streamlining through the deletion of competing endogenous gene clusters is a foundational strategy in synthetic biology to construct optimized chassis strains. By removing non-essential biosynthetic pathways that consume precursors and energy, researchers can redirect metabolic flux toward target compounds, reduce background interference, and improve host performance [21]. This approach is particularly valuable for polyketide production, where competing pathways directly impact the availability of essential malonyl-CoA and methylmalonyl-CoA building blocks. The process involves identifying dispensable genomic regions, often located in variable sub-telomeric areas of the chromosome, and employing advanced genome editing tools for their precise removal [22]. This guide provides a comparative analysis of streamlined chassis strains, details experimental protocols for cluster deletion, and offers resources to facilitate the adoption of these techniques in polyketide research and drug development.
Comparative Analysis of Streamlined Streptomyces Chassis Strains
Extensive research has been conducted to engineer optimized Streptomyces chassis strains by deleting endogenous biosynthetic gene clusters (BGCs). The table below summarizes key engineered strains, their modifications, and observed phenotypic improvements.
Table 1: Engineered Streptomyces Chassis Strains for Heterologous Expression
| Strain Name | Parental Strain | Gene Cluster Deletions | Key Genotypic Modifications | Resulting Phenotypic Improvements |
|---|---|---|---|---|
| S. lividans ΔYA9 [23] | S. lividans TK24 | 9 clusters (228.5 kb total) | Deletion of endogenous BGCs; introduction of additional φC31 attB sites | Simplified metabolic background; improved growth in liquid production medium; superior heterologous production of various natural products [23]. |
| Streptomyces sp. A4420 CH [4] | Streptomyces sp. A4420 | 9 native polyketide BGCs | Removal of competing polyketide synthases (PKS) | Consistent sporulation and growth; outperformed common hosts in expressing four distinct heterologous polyketide BGCs [4]. |
| Chassis2.0 [2] | S. aureofaciens J1-022 | Two endogenous T2PK clusters (in-frame deletion) | Creation of a pigmented-faded host | Enhanced production of oxytetracycline (370% increase) and efficient synthesis of tri-ring T2PKs; activation of a cryptic pentangular T2PK cluster [2]. |
| S. fungicidicus Mutants [24] | S. fungicidicus TXX3120 | NRPS/PKS clusters (up to 54.4 kb) | Single and cumulative deletions using optimized HR and CRISPR/Cas9 | Improved growth characteristics, including prolonged logarithmic phase and increased biomass [24]. |
| S. coelicolor M1152 [23] | S. coelicolor M145 | 4 clusters (act, red, cpk, cda) | Introduction of rpoB mutation | Well-characterized, widely used host for heterologous expression of natural products [23]. |
| S. albus Del14 [22] [23] | S. albus J1074 | 15 BGCs (503 kb total) | Large-scale genome reduction | Cluster-free mutant used as a chassis for drug discovery and improved production of microbial drugs [22] [23]. |
The selection of an appropriate parental strain is the first critical step in chassis development. As highlighted in a 2025 study, high-yielding industrial strains often possess innate advantages over conventional model strains. For instance, Streptomyces aureofaciens was selected as a chassis for Type II polyketides due to its robust native metabolism, shorter fermentation cycle, and better genetic tractability compared to other potential hosts [2]. The principle of product-chassis compatibility suggests that a strain already proficient in producing structurally similar compounds will likely provide a more favorable enzymatic and precursor environment for the target pathway [2].
The performance gains from metabolic streamlining are significant and multi-faceted. Deletions can lead to a reduction in metabolic burden, freeing up cellular resources. This often results in increased biomass and a prolonged logarithmic growth phase, as observed in engineered S. fungicidicus mutants [24]. From a practical standpoint, a simplified metabolic profile drastically reduces background production during heterologous expression, which facilitates the detection and purification of novel target compounds [4] [23]. The cumulative effect is frequently a major enhancement in the titer of the desired molecule, as demonstrated by the 370% increase in oxytetracycline production in Chassis2.0 [2].
Experimental Protocols for Gene Cluster Deletion
Identification of Dispensable Genomic Regions
Before undertaking deletion experiments, a systematic bioinformatic analysis is essential to identify dispensable gene clusters and avoid synthetic lethality.
- Comparative Genomics: Performing multiple genome alignments with closely related model strains (e.g., S. coelicolor, S. albus J1074) helps identify conserved core regions and variable sub-telomeric regions. The latter are often enriched with non-essential BGCs and are prime targets for large-scale deletion [22].
- Phylogenetic Analysis: Constructing a phylogenetic tree based on 16S rDNA sequences helps understand the evolutionary relationship of the target strain to other Streptomyces species, guiding the selection of appropriate reference genomes for comparison [22].
- Transcriptomics and Metabolomics: Integrating RNA-seq data and LC-MS metabolite profiling of the wild-type strain under production conditions helps pinpoint which BGCs are actively expressed. This allows for the prioritization of clusters that are functionally active and could interfere with heterologous production [23].
- In silico Metabolic Modeling: Using genome-scale metabolic models (e.g., iBsu1103 for Bacillus subtilis) can predict the phenotypic consequences of gene deletions and identify potential auxotrophies that may arise, guiding the design of supplementation strategies [25].
Core Deletion Methodologies
Two primary technological approaches are widely used for the deletion of gene clusters: optimized homologous recombination and CRISPR-based systems.
Table 2: Comparison of Gene Cluster Deletion Methods
| Method | Key Features | Typical Deletion Size | Advantages | Limitations/Challenges |
|---|---|---|---|---|
| Optimized Homologous Recombination (HR) [24] | Uses selection (e.g., antibiotic resistance) and counter-selection markers (e.g., upp). | Up to 54.4 kb and larger [24]. | Proven reliability for very large deletions; does not require specialized nucleases. | Time-consuming and labor-intensive; involves multiple screening steps for crossover events [24]. |
| CRISPR/Cas9 Systems [24] | Relies on Cas9-induced double-strand breaks and homology-directed repair (HDR). | Efficient for a wide range of sizes. | Greatly shortened editing cycle; enables highly efficient and precise screening [24]. | Efficiency can vary across different Streptomyces species due to differences in genetic backgrounds [24]. |
| CRISPR/cBEST (Base Editing) [24] | Fusion of nCas9 with cytidine deaminase; introduces point mutations (C to T) without double-strand breaks. | Used for introducing stop codons rather than large deletions. | Avoids double-strand breaks; high efficiency in some strains. | Not suitable for deleting entire gene clusters; application is for gene inactivation. |
| Endogenous Type I-E CRISPR System [26] | Repurposes the native CRISPR system present in many Streptomyces strains for transcriptional regulation. | Used for gene repression/activation, not deletion. | Functions in a wide range of phylogenetically distant strains; effective for activating cryptic BGCs. | Primarily a tool for gene regulation rather than deletion. |
The following diagram illustrates a generalized workflow for implementing these core methodologies to create a streamlined chassis.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of metabolic streamlining protocols requires a suite of specialized reagents and genetic tools. The following table details key solutions for experiments in Streptomyces.
Table 3: Key Research Reagent Solutions for Gene Cluster Deletion
| Reagent/Material | Function/Application | Specific Examples |
|---|---|---|
| Counter-Selection Markers | Enables efficient selection of double-crossover events in homologous recombination, critical for marker-less deletion. | upp gene (encodes uracil phosphoribosyltransferase); selection with 5-fluorouracil [24]. |
| CRISPR/cBEST System | A cytosine base editor for introducing stop codons into target genes to inactivate them without full cluster deletion. | pcBEST plasmid (rat APOBEC-1 cytidine deaminase, nCas9, UGI) [24]. |
| CRISPR/Cas9 System with Counter-Selection | Combines the precision of Cas9 with efficient screening via a counter-selection marker to shorten the gene editing cycle. | pCas9-upp plasmid [24]. |
| Type I-E CRISPR Activators | Repurposes endogenous CRISPR systems to activate cryptic BGCs for characterization prior to deletion decisions. | Dual plasmid system for Cascade module and crRNA expression [26]. |
| Bacterial Artificial Chromosomes (BACs) | Facilitates the cloning and heterologous expression of large biosynthetic gene clusters in chassis strains. | Used for heterologous expression of oxytetracycline BGC in chassis comparison studies [2]. |
| Engineered E. coli Donor Strains | Essential for intergeneric conjugation, the primary method for introducing DNA into many Streptomyces species. | E. coli ET12567/pUZ8002 (non-methylating, carries conjugation machinery) [24]. |
Metabolic streamlining through the deletion of competing endogenous gene clusters is a powerful and well-established strategy for crafting high-performance microbial chassis. As demonstrated by the comparative data, engineered strains like S. lividans ΔYA9, Streptomyces sp. A4420 CH, and Chassis2.0 consistently outperform their parental strains by offering simplified metabolic backgrounds, improved growth characteristics, and significantly enhanced production of heterologous polyketides and other valuable compounds. The choice of deletion strategy—whether robust homologous recombination or rapid CRISPR/Cas9 systems—depends on the specific project needs and the genetic tractability of the host. As genome editing tools continue to advance and our understanding of streptomycete metabolism deepens, the rational design of specialized chassis strains will become increasingly precise and efficient. This progress will undoubtedly accelerate the discovery and production of novel pharmaceuticals, pushing the boundaries of synthetic biology and drug development.
Polyketides constitute a large class of structurally diverse natural products with a vast array of biological and pharmacological activities, including antibacterial, antifungal, anticholesterol, antiparasitic, anticancer, and immunosuppressive properties [27]. These valuable compounds are assembled by polyketide synthases (PKSs) through successive rounds of decarboxylative Claisen condensations between acyl thioesters [27]. The building blocks for these reactions are acyl-CoA precursors, with malonyl-CoA and methylmalonyl-CoA representing the most fundamental extender units for polyketide chain elongation [27] [28]. Despite their critical importance, the native intracellular pools of these CoA-thioesters are typically limited in most microbial hosts as they are tightly regulated for essential cellular functions [12] [28]. This limitation creates a significant bottleneck for heterologous polyketide production, making precursor enhancement a cornerstone strategy in metabolic engineering efforts. Within the context of benchmarking chassis strains for polyketide production research, engineering the supply of malonyl-CoA and methylmalonyl-CoA emerges as a critical determinant of overall success, directly influencing titers, product profiles, and economic viability.
Chassis Strain Performance: A Comparative Analysis
Diverse microbial hosts have been engineered to enhance the supply of malonyl-CoA and methylmalonyl-CoA. The table below provides a comparative analysis of the performance of key chassis strains in polyketide precursor and product synthesis.
Table 1: Performance Comparison of Engineered Chassis Strains for Precursor and Polyketide Production
| Host Strain | Engineering Strategy | Key Precursor/Enzyme Targeted | Polyketide/Output | Reported Titer/Level | Reference |
|---|---|---|---|---|---|
| E. coli BAP1 (ΔygfH) | Deletion of propionyl-CoA succinate transferase (ygfH) | Methylmalonyl-CoA | 6-Deoxyerythronolide B (6dEB) | 527 mg/L (bioreactor) | [29] |
| E. coli K207-3–MatBC | Genomic integration of malonate assimilation pathway (matBC) | Malonyl-CoA/Methylmalonyl-CoA | Flaviolin (via RppA) | 70% increase (vs. control) | [12] |
| Streptomyces sp. A4420 CH | Deletion of 9 native polyketide gene clusters | Native precursor pool optimization | Four distinct polyketide BGCs | Production of all 4 tested polyketides | [4] |
| Pseudomonas putida (Engineered) | Knockout of genes in glycolysis, TCA, and fatty acid synthesis | Malonyl-CoA | Flaviolin / Poly(3-hydroxybutyrate) | Significant increase (vs. parent) | [30] |
| Schizochytrium sp. HX-308 | Overexpression of MAT from FAS and PKS pathways | Malonyl-ACP | DHA-rich lipids | 88.5 g/L lipids (5-L fermenter) | [31] |
The performance disparities among these chassis strains highlight their inherent metabolic differences and specialized applications. The engineered E. coli BAP1 and K207-3 strains demonstrate highly effective solutions for producing propionyl-CoA-derived polyketides like 6-deoxyerythronolide B and for achieving tunable precursor supply [29] [12]. In contrast, the Streptomyces sp. A4420 CH strain showcases superior compatibility and versatility for expressing diverse, complex polyketide pathways, a critical asset for natural product discovery [4]. The engineering of non-traditional hosts like Pseudomonas putida and the oleaginous yeast Schizochytrium sp. further expands the available toolkit, offering advantages in handling toxic compounds and channeling precursors toward specific lipid-related products [30] [31].
Experimental Deep Dive: Key Protocols and Workflows
Establishing an Orthogonal Malonyl-CoA Supply in E. coli
A seminal study demonstrated a controlled strategy to overcome the tight native regulation of malonyl-CoA in E. coli [12]. The methodology can be summarized in a defined workflow.
Step 1 – Native Pathway Disruption: The endogenous malonyl-CoA biosynthetic pathway, dependent on the biotin-requiring acetyl-CoA carboxylase (ACC), was inhibited by knocking out the bioH gene, which is essential for biotin synthesis. This creates a biotin auxotrophic strain whose growth and malonyl-CoA production become dependent on an alternative, engineered pathway [12].
Step 2 – Orthogonal Pathway Introduction: Genes for a malonate assimilation pathway from Rhizobium trifolii were integrated into the genome. This pathway consists of a malonate transporter (matC) and a malonyl-CoA ligase (matB). The matB enzyme directly converts exogenous malonate into malonyl-CoA in an ATP-dependent reaction, bypassing the native ACC complex [12].
Step 3 – Precursor Supplementation and Production: The engineered strain is cultivated in a medium supplemented with malonate. The intracellular concentration of malonyl-CoA becomes directly tunable by varying the malonate concentration in the culture medium. This elevated and controllable pool is then harnessed for polyketide biosynthesis, for instance, by expressing the type III PKS RppA to produce flaviolin or hybrid type I PKSs for more complex products [12].
Enhancing Methylmalonyl-CoA for 6-Deoxyerythronolide B (6dEB) in E. coli
The heterologous production of the erythromycin precursor 6dEB in E. coli requires a sufficient supply of both propionyl-CoA and (2S)-methylmalonyl-CoA. A key metabolic engineering study focused on modulating native E. coli metabolism to enhance this supply [29].
Host Strain and Base Pathway: The base strain was E. coli BAP1, a derivative of BL21(DE3). This strain already contained the deoxyerythronolide B synthase (DEBS) from Sacchropolyspora erythraea, the sfp gene from B. subtilis for phosphopantetheinylation, and a hybrid substrate pathway comprising the native E. coli propionyl-CoA synthetase (PrpE) and a heterologous propionyl-CoA carboxylase (PCC) from Streptomyces coelicolor for converting propionyl-CoA to methylmalonyl-CoA [29].
Metabolic Engineering of Competing Pathways: The researchers systematically deleted or overexpressed genes encoding enzymes that connect native metabolism to the heterologous precursor pools.
- Deletion of ygfH: The ygfH gene encodes a propionyl-CoA:succinate CoA transferase, which consumes propionyl-CoA. Its deletion prevented this drain, resulting in a dramatic increase in 6dEB titer from 206 mg/L to 527 mg/L in a bioreactor [29].
- Overexpression of ygfG: Conversely, overexpressing ygfG (encoding a methylmalonyl-CoA decarboxylase, which converts methylmalonyl-CoA back to propionyl-CoA) reduced 6dEB production fourfold, validating this node as a critical drain [29].
- Deletion of sbm: Deletion of sbm (methylmalonyl-CoA mutase) did not significantly affect titers, indicating this was not a major competing pathway under the tested conditions [29].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful engineering of acyl-CoA precursor supply relies on a suite of key reagents and genetic tools. The following table catalogues essential components for related metabolic engineering experiments.
Table 2: Key Research Reagent Solutions for Precursor Engineering
| Reagent / Tool | Function / Role | Example Application |
|---|---|---|
| MatB/MatC System | Malonate transporter (MatC) and malonyl-CoA synthetase (MatB) | Establishing an orthogonal, tunable malonyl-CoA supply pathway in E. coli [12]. |
| Propionyl-CoA Carboxylase (PCC) | Carboxylates propionyl-CoA to form (2S)-methylmalonyl-CoA. | Providing methylmalonyl-CoA extender units for 6dEB biosynthesis in E. coli [29]. |
| Acetyl-CoA Carboxylase (ACC) | Native enzyme complex that carboxylates acetyl-CoA to form malonyl-CoA. | A common target for overexpression to enhance the native malonyl-CoA pool [27] [28]. |
| Type III PKS RppA | Converts malonyl-CoA to 1,3,6,8-tetrahydroxynaphthalene (THN), which auto-oxidizes to red flaviolin. | Serves as a rapid, colorimetric biosensor for reporting intracellular malonyl-CoA availability [12] [30]. |
| Phosphopantetheinyl Transferase (e.g., Sfp) | Activates acyl carrier proteins (ACPs) of PKSs by attaching the 4'-phosphopantetheine moiety from CoA. | Essential for post-translational activation of heterologous PKSs in non-native hosts like E. coli [29] [12]. |
| λ-Red Recombinase System | Enables efficient, PCR-mediated gene deletion or insertion in the chromosome. | Used for precise knockout of competing metabolic genes (e.g., ygfH, sbm) [29]. |
Pathway Visualization and Logical Workflows
The biosynthesis of malonyl-CoA and methylmalonyl-CoA is deeply rooted in central metabolism. The following diagram illustrates the major metabolic routes and engineering targets for enhancing their supply.
This pathway map highlights the two primary strategies for precursor enhancement: augmenting synthesis (e.g., overexpressing ACC, introducing MatBC) and reducing competitive drains (e.g., knocking out fatty acid synthesis genes or methylmalonyl-CoA-consuming pathways like ygfH). The provision of propionyl-CoA, the precursor for methylmalonyl-CoA, can be engineered through various routes, including the expression of native synthetases (PrpE) or the catabolism of branched-chain amino acids [29] [28].
The efficient microbial production of high-value polyketides, a class of bioactive compounds with widespread pharmaceutical applications, is consistently constrained by the host's native metabolism. Polyketides, which include antibiotics, anticancer agents, and immunosuppressants, are synthesized from acyl-CoA precursors like malonyl-CoA (M-CoA) and methylmalonyl-CoA (mM-CoA) [19]. In conventional engineered strains, these precursors are also essential for central metabolism, particularly fatty acid biosynthesis, leading to competition, tight regulatory control, and limited precursor availability for polyketide synthesis [19]. This conflict between production and growth pathways fundamentally limits titers and yields.
Orthogonal biosynthesis presents a strategic solution to this problem. It involves the design and implementation of synthetic metabolic pathways that operate in parallel to, and with minimal interaction with, the host's native metabolic network [32]. The core principle is to create a "decoupled" system where the production of a target chemical is independent of biomass synthesis, allowing for independent optimization [32]. This approach is particularly powerful for polyketide production, as it enables researchers to bypass native regulatory mechanisms that tightly control key precursors. By introducing non-native, controllable pathways for substrate generation, orthogonal systems can enhance flux toward polyketides while minimizing the metabolic burden and undesirable interactions that plague traditional engineering strategies. This guide benchmarks the performance of orthogonal systems against alternative metabolic engineering approaches, providing a structured comparison for researchers selecting chassis strains and pathways for polyketide production.
Core Concept: Defining Orthogonality in Metabolic Pathways
An ideal orthogonal pathway for chemical production is characterized by two key structural features. First, it shares no enzymatic steps with the cellular pathways responsible for generating the precursors required for biomass. Second, it features a single, well-defined metabolite that acts as a branch point from which the product-forming and biomass-forming pathways diverge [32]. This structure minimizes the network-wide interactions that typically make metabolic networks robust and optimized for growth, but which constrain their capability as cell factories [32].
The degree of orthogonality can be quantified using an Orthogonality Score (OS), a metric that measures the overlap between the set of reactions required for biomass production and the set of reactions required for the target chemical's production [32]. A score closer to 1 indicates a highly orthogonal network where production is essentially a biotransformation separate from native metabolism, while a score closer to 0 signifies significant overlap and interaction [32]. Analyses reveal that natural metabolic pathways, such as the Embden-Meyerhof-Parnas (EMP) pathway for succinate production, often have low orthogonality scores (0.41-0.45), whereas synthetic pathways designed for the same purpose can achieve higher scores (e.g., 0.56), making them more suitable for engineered overproduction [32].
Benchmarking Orthogonal Systems Against Alternative Strategies
The implementation of orthogonal biosynthesis for polyketide precursors can be achieved through various molecular strategies. The table below provides a performance comparison of the primary approaches, benchmarking them against traditional engineering methods.
Table 1: Performance Comparison of Metabolic Engineering Strategies for Enhancing Polyketide Precursors
| Strategy | Key Principle | Reported Performance / Effect | Key Advantages | Key Limitations |
|---|---|---|---|---|
| Orthogonal MatBC Pathway [19] | Introduce heterologous malonate importer (matC) and malonyl-CoA ligase (matB) to bypass native regulation. | - 70% increase in flaviolin (M-CoA proxy) at 20mM malonate.- Tunable M-CoA levels via malonate supplementation.- Reduced promiscuous activity of PKSs. | - Direct, exogenous control over precursor pool.- Bypasses native feedback inhibition.- Can be made essential for growth in engineered auxotrophs. | - Requires external malonate supplementation.- Potential transporter toxicity. |
| Orthogonal Type I FAS [33] | Express heterologous type I Fatty Acid Synthase (FAS) that directly produces acyl-CoAs. | - Shift in fatty alcohol chain-length profile.- Production of methyl ketones confirmed in vivo activity. | - Direct acyl-CoA production avoids futile cycles.- Not subject to native E. coli FAS regulation.- No ATP cost for CoA activation. | - Lower methyl ketone titers than native FAS.- Soluble expression of large enzyme complexes can be challenging. |
| Traditional ACC Overexpression [19] | Overexpress native Acetyl-CoA Carboxylase (ACC) to enhance M-CoA synthesis from Ac-CoA. | Commonly used, but limited by native regulatory mechanisms and small free M-CoA pool. | - Utilizes endogenous precursor (Ac-CoA).- No external substrate required. | - Tightly regulated by fatty acyl-ACP feedback inhibition.- Increased metabolic burden. |
| Cerulenin Supplementation [19] | Inhibit native FAS with cerulenin to divert M-CoA toward polyketides. | Inefficient for increasing polyketide titers. | - Simple pharmacological intervention. | - Cerulenin also inhibits PKS ketosynthase domains.- Not a specific solution. |
Featured Experimental Protocol: Implementing an Orthogonal Malonyl-CoA Pathway
The following section details the key methodology for implementing and validating one of the most effective orthogonal systems for polyketide research: the MatBC malonyl-CoA pathway in E. coli [19].
Strain and Pathway construction
The orthogonal system was built in two E. coli chassis strains, K207-3 and BAP1, both equipped with the sfp gene from Bacillus subtilis for phosphopantetheinylation of PKSs [19].
- Genomic Integration: The genes matC (malonate transporter) and matB (malonyl-CoA ligase) from Rhizobium trifolii were placed under the control of a lacUV5 promoter and integrated into the bacterial genome via homologous recombination at the ompW intergenic region, a recognized "safe site" for stable expression [19]. This resulted in strains K207-3-MatBC and BAP1-MatBC.
- Creating a Controllable System: To achieve absolute control over the native M-CoA biosynthesis, the native pathway was disrupted by deleting bioH, a gene essential for biotin biosynthesis [19]. Since the native Acetyl-CoA Carboxylase (ACC) complex requires a biotin cofactor, this knockout renders the cells auxotrophic for biotin and incapable of synthesizing M-CoA via the native route. Introducing the MatBC pathway into this background creates a strain whose growth and M-CoA production become strictly dependent on exogenous malonate, providing a powerful growth-based selection for optimizing the orthogonal system [19].
Analysis and Validation Methods
The functionality and performance of the engineered orthogonal pathway were assessed using the following analytical methods:
- Flaviolin Production Assay: To indirectly quantify intracellular M-CoA levels available for polyketide synthesis, the type III PKS RppA (1,3,6,8-tetrahydroxynaphthalene synthase) was expressed from a plasmid (pBADT–RppA-NT) [19]. RppA condenses M-CoA into THN, which spontaneously oxidizes to the red-colored pigment flaviolin. The production of flaviolin was monitored over 72 hours in Lysogeny Broth (LB) medium supplemented with varying concentrations of malonate (0-20 mM), 10 µM Isopropyl β-d-1-thiogalactopyranoside (IPTG) to induce matBC expression, and 0.2% arabinose to induce rppA expression [19]. Flaviolin accumulation, measured by its characteristic absorbance at 340 nm and 520 nm, serves as a reliable proxy for intracellular M-CoA levels [19].
- Polyketide Titer Analysis: The ultimate validation involved testing the system with target polyketide synthases. For example, hybrid pikromycin synthase (Pik127) variants with engineered acyltransferase domains were expressed in the orthogonal strains. The production of the resulting M-CoA-derived and mM-CoA-derived polyketide products was analyzed and quantified using liquid chromatography-mass spectrometry (LC-MS) to demonstrate the system's ability to support the synthesis of complex polyketides and alter product ratios by controlling precursor supply [19].
Diagram: Workflow for Implementing and Validating an Orthogonal Malonyl-CoA Pathway
The Scientist's Toolkit: Essential Research Reagents
The following table catalogues key reagents, enzymes, and genetic components essential for constructing and evaluating orthogonal biosynthesis systems for polyketide production.
Table 2: Essential Research Reagents for Orthogonal Pathway Engineering
| Reagent / Component | Function / Role in Experimentation | Example Source / System |
|---|---|---|
| Malonyl-CoA Ligase (MatB) | Catalyzes the ATP-dependent ligation of malonate and CoA to form malonyl-CoA, forming the core reaction of the orthogonal pathway. | Rhizobium trifolii [19] |
| Malonate Transporter (MatC) | Enables efficient cellular uptake of extracellular malonate, the feedstock for the orthogonal pathway. | Rhizobium trifolii [19] |
| Type III PKS RppA | Serves as a reporter enzyme; converts malonyl-CoA into THN/flaviolin, allowing for indirect, colorimetric quantification of intracellular M-CoA levels. | Streptomyces species [19] |
| Phosphopantetheinyl Transferase (Sfp) | Essential for activating type I and type II PKSs by attaching a phosphopantetheine moiety to their acyl carrier protein (ACP) domains. | Bacillus subtilis [19] |
| Type I Fatty Acid Synthase (FAS1) | An orthogonal FAS that directly produces acyl-CoAs, bypassing the native type II FAS and its regulation. | Corynebacterium glutamicum (e.g., FAS1A) [33] |
| Cerulenin | A natural inhibitor of ketosynthase domains; used in traditional engineering to inhibit native FAS and divert M-CoA, but also inhibits PKSs. | Chemical reagent [19] |
Pathway Visualization: Native vs. Orthogonal Malonyl-CoA Synthesis
The following diagram illustrates the logical and metabolic relationships between the native E. coli pathway for malonyl-CoA synthesis and the engineered orthogonal MatBC pathway, highlighting the points of regulation and control.
Diagram: Native vs. Orthogonal Pathway for Malonyl-CoA Production
The implementation of orthogonal biosynthesis pathways represents a paradigm shift in metabolic engineering for polyketide production. Moving beyond the traditional approach of modifying and overloading native metabolism, orthogonality focuses on creating parallel, minimally interacting systems that offer superior control and efficiency. As benchmarked in this guide, systems like the MatBC pathway in E. coli provide a tangible performance advantage by enabling tunable precursor supply, bypassing native regulation, and simplifying strain optimization through strategies like adaptive laboratory evolution [19].
The future of orthogonal biosynthesis is closely linked to advances in synthetic biology and computational tools. The integration of artificial intelligence and machine learning is already improving the identification of pathway enzymes, predictive modeling of flux, and rational strain engineering [34] [35]. Furthermore, the development of versatile, plug-and-play toolkits for orthogonal gene expression, such as the TriO system, is democratizing the combinatorial testing of pathway designs, making sophisticated metabolic engineering more accessible to academic researchers [36]. As the field progresses, the application of these orthogonal principles to a wider range of chassis organisms and target compounds will undoubtedly accelerate the development of efficient microbial cell factories for the sustainable production of valuable polyketides and other natural products.
Table of Contents
- Introduction
- ExoCET Methodology
- Performance Comparison
- Application Case Studies
- Research Reagent Solutions
- Conclusion
The exponential growth of genomic sequence data has created an urgent need for advanced molecular tools that can efficiently clone and manipulate large DNA fragments. Traditional methods for cloning biosynthetic gene clusters (BGCs) often face significant limitations in efficiency, size capacity, and precision, creating bottlenecks in natural product research and metabolic engineering. Among the emerging solutions, Exonuclease Combined with RecET recombination (ExoCET) has established itself as a powerful direct cloning technology that effectively addresses these challenges. This guide provides a comprehensive technical comparison of ExoCET against alternative genomic integration techniques, with experimental data and protocols specifically framed within the context of benchmarking chassis strains for polyketide production research. For synthetic biologists and natural product researchers, understanding the capabilities and applications of these tools is crucial for accelerating the discovery and optimization of valuable compounds, including medically relevant polyketides and other secondary metabolites.
ExoCET Methodology
Core Principles and Mechanism
The ExoCET (Exonuclease Combined with RecET recombination) system represents a significant advancement in direct DNA cloning technology by synergistically combining in vitro exonuclease assembly with highly efficient RecET homologous recombination in vivo. This dual mechanism enables researchers to directly clone targeted regions from complex genomic DNA with nucleotide precision into operational plasmids, bypassing many limitations of conventional cloning methods [37].
The process begins with the preparation of a linearized vector and genomic DNA containing the target region. When these components are incubated together with T4 polymerase (T4pol) as the in vitro exonuclease, the enzyme generates single-stranded overhangs that facilitate the annealing of complementary ends between the vector and target DNA. This pre-annealing step is crucial as it creates a single DNA molecule, effectively breaking the bottleneck of low co-transformation efficiency that traditionally limited the cloning of large fragments [38] [37]. The pre-assembled linear DNA is then electroporated into E. coli cells expressing the RecET recombination system, which catalyzes efficient linear-linear homologous recombination to produce the final cloned product [37].
Detailed Experimental Protocol
Step 1: Vector Preparation
- Design and linearize a BAC/YAC vector (e.g., pBeloBAC11-cm-ccdB-hyg) containing appropriate homology arms (typically 120 bp) specific to the target genomic region. The vector should include selection markers (e.g., chloramphenicol resistance) and elements for subsequent genomic integration [38].
- Incorporate unique adapter sequences at the termini to program how different DNA segments will recombine during assembly. These adapters should have GC content ≥40%, lack mononucleotide repeats of ≥7 bases, and show no detectable identity to the host genome [39].
Step 2: Genomic DNA Isolation
- Extract high-molecular-weight genomic DNA from the source organism using standard protocols. For Streptomyces species, this may involve growing cultures in appropriate media (e.g., TSB broth), harvesting mycelia, and performing enzymatic lysis followed by phenol-chloroform extraction and isopropanol precipitation [4].
Step 3: In Vitro Exonuclease Assembly
- Combine approximately 200-500 ng of linearized vector with a 5:1 molar ratio of genomic DNA in a reaction buffer containing T4 DNA polymerase.
- Incubate at room temperature for 30 minutes to allow exonuclease processing and annealing of complementary ends.
- Stop the reaction by heating at 75°C for 20 minutes [37].
Step 4: Electroporation and RecET Recombination
- Electroporate the pre-assembled DNA into recombinogenic E. coli strains expressing RecET, such as GB05-dir harboring pSC101-BAD-ETgA-tet [38].
- Plate transformants on selective media (e.g., chloramphenicol for BAC selection) and incubate at 37°C for 24-48 hours.
Step 5: Clone Verification
- Screen colonies by colony PCR using primers specific to vector-segment junctions.
- Verify positive clones by restriction digest analysis and Sanger sequencing.
- For large-scale applications, clone verification can achieve success rates of 93% or higher [39].
Performance Comparison
Direct Comparison of Cloning Technologies
Table 1: Technical comparison of ExoCET with alternative DNA cloning and integration technologies
| Technology | Mechanism | Max Capacity | Efficiency | Precision | Key Applications |
|---|---|---|---|---|---|
| ExoCET | In vitro exonuclease + RecET recombination | >142 kb [38] | High (direct cloning without library construction) [37] | Nucleotide precision [37] | Direct cloning of BGCs from complex genomes [38] [37] |
| Traditional Homologous Recombination | Homology-directed repair in host cells | Limited only by transformation efficiency | Variable, requires optimization | Moderate (depends on homology arm design) | BAC construction, gene knockouts |
| TAR Cloning | Transformation-associated recombination in yeast | >300 kb | Moderate (requires yeast system) | High | Cloning of large gene clusters |
| CRISPR-Cas9 Assisted Cloning | Cas9 nuclease + homology-directed repair | Varies with system | High with optimized gRNAs | Single-base with proper design | Targeted cloning of specific genomic regions |
| CReATiNG | Cas9 excision + programmable assembly | 230 kb demonstrated [39] | High (93% success rate) [39] | Defined by gRNA selection | Synthetic chromosome construction, genome streamlining |
Quantitative Performance Data
Table 2: Experimental performance metrics of ExoCET in published studies
| Application | Target Size | Efficiency | Comparison to Alternatives | Reference |
|---|---|---|---|---|
| Herpesvirus genome cloning | 142 kb | Successful direct cloning without plaque purification | Bypasses attenuating mutations from serial passage [38] | [38] |
| Bacterial genome cloning | 106 kb | Efficient retrieval from prokaryotic genome (4×10^6 bp) | Superior to RecET alone for large fragments [37] | [37] |
| Eukaryotic genome cloning | 53 kb | Effective from complex genome (3×10^9 bp) | Enables bioprospecting from mammalian blood DNA [37] | [37] |
| OTC BGC cloning in Streptomyces | ~20 kb | Successful construction of p15A_oxy shuttle plasmid | Enabled heterologous expression in Streptomyces chassis [2] | [2] |
Figure 1: ExoCET experimental workflow and key advantages. The process enables direct cloning of large DNA fragments with nucleotide precision through a combination of in vitro exonuclease assembly and RecET recombination in E. coli.
Application Case Studies
Heterologous Expression of Polyketides in Streptomyces
The application of ExoCET cloning has proven particularly valuable in the heterologous expression of polyketides in engineered Streptomyces chassis strains. In one comprehensive study, researchers employed ExoCET to directly clone the complete oxytetracycline (OTC) biosynthetic gene cluster from Streptomyces rimosus ATCC 10970 into an E. coli-Streptomyces shuttle vector, creating p15A_oxy [2]. This construct was then introduced into various Streptomyces chassis strains, including S. albus J1074 and S. lividans TK24, though neither of these conventional chassis strains accumulated detectable TCs without additional metabolic engineering [2].
This case study highlights both the efficiency of ExoCET for BGC cloning and the critical importance of chassis selection in polyketide production. Further experiments demonstrated that high-yielding industrial strains like S. aureofaciens J1-022 showed significantly better performance as chassis for T2PKs (type II polyketides) production, achieving a 370% increase in OTC production compared to commercial production strains when using optimized chassis designs [2].
Herpesvirus Genome Engineering
Beyond natural products discovery, ExoCET has demonstrated remarkable utility in virology research. Scientists successfully cloned the complete 142-kb pseudorabies virus (PRV) genome directly into a bacterial artificial chromosome (BAC) in E. coli using ExoCET [38]. This approach strategically placed the BAC vector at the terminus of the linear viral genome, enabling subsequent excision using restriction endonucleases for delivery into mammalian cells.
The ExoCET approach provided significant advantages over conventional methods for constructing herpesvirus BACs, which typically rely on homologous recombination in mammalian cells. Specifically, it eliminated the need for multiple rounds of plaque purification and avoided the accumulation of attenuating mutations that commonly occur during serial passage of viruses in cell culture [38]. The resulting viral BAC was stable in E. coli and amenable to further genetic manipulation using Red recombination or site-specific recombination methods, highlighting the system's versatility for large DNA virus engineering.
Activation of Cryptic Biosynthetic Pathways
ExoCET has also enabled the activation and characterization of cryptic biosynthetic pathways in optimized chassis strains. In one notable example, researchers identified Streptomyces sp. A4420 as a promising chassis for polyketide production due to its rapid growth and high metabolic capacity [4]. After engineering a metabolically streamlined derivative (CH strain) by deleting nine native polyketide BGCs, the team used direct cloning methods to express four distinct polyketide BGCs in this optimized host.
Remarkably, the engineered Streptomyces sp. A4420 CH strain demonstrated the capability to produce all tested metabolites under every condition, outperforming both its parental strain and other commonly used heterologous hosts including S. coelicolor M1152, S. lividans TK24, S. albus J1074, and S. venezuelae NRRL B-65442 [4]. This success highlights how the combination of advanced cloning technologies like ExoCET with carefully engineered chassis strains can unlock the biosynthetic potential of cryptic gene clusters that would otherwise remain silent in their native genomic context.
Figure 2: Application of ExoCET cloning for activation of cryptic biosynthetic pathways. The workflow enables discovery of novel natural products and enhanced production of valuable compounds through heterologous expression in optimized chassis strains.
Research Reagent Solutions
Essential Tools and Materials
Table 3: Key research reagents and materials for implementing ExoCET and related genomic integration techniques
| Reagent/Material | Function | Examples/Specifications | Application Notes |
|---|---|---|---|
| RecET-Expressing E. coli Strains | Provides recombination machinery for direct cloning | GB05-dir (pSC101-BAD-ETgA-tet), GB08-red [38] | Inducible expression systems preferred for controlling recombination |
| BAC/YAC Vectors | Molecular vehicle for cloning large inserts | pBeloBAC11-cm-ccdB-hyg, pASC1 [38] [39] | Must include appropriate selection markers and integration sites |
| T4 DNA Polymerase | In vitro exonuclease for end processing | Commercial preparations with optimized buffer systems | Critical for generating complementary overhangs |
| Cas9-gRNA Systems | Targeted genomic cleavage for DNA extraction | pML104 (TDH3p-Cas9) with IVT gRNAs [39] | Enables precise excision of target genomic regions |
| Modular Integration Cassettes | Site-specific integration of cloned DNA | Cre-lox, Vika-vox, Dre-rox, phiBT1-attP systems [40] | Enables orthogonal recombination strategies in chassis strains |
| Engineered Streptomyces Chassis | Heterologous expression hosts | S. coelicolor A3(2)-2023, Streptomyces sp. A4420 CH [4] [40] | Pre-engineered hosts with deleted native BGCs reduce metabolic competition |
ExoCET represents a paradigm shift in DNA cloning technology, offering researchers an efficient and precise method for accessing large genomic regions directly from complex genomes. When integrated with optimized chassis strains and complementary genomic integration techniques, this powerful toolkit significantly accelerates the discovery and production of valuable natural products, particularly in the challenging field of polyketide research. The experimental data and protocols presented in this guide provide researchers with a comprehensive resource for implementing these advanced techniques in their own metabolic engineering and natural product discovery pipelines. As synthetic biology continues to advance, the synergy between sophisticated cloning technologies like ExoCET and systematically engineered chassis strains will undoubtedly unlock new possibilities for microbial natural product synthesis and engineering.
The efficient discovery and production of Type II polyketides (T2PKs), a class of compounds with diverse pharmacological activities, is heavily dependent on the selection of an optimal microbial chassis [41]. Conventional Streptomyces model chassis often require extensive metabolic engineering and still yield suboptimal production, creating a significant bottleneck in natural product development [41] [4]. This case study objectively evaluates the performance of an engineered chassis, Streptomyces aureofaciens Chassis2.0, against established alternative hosts for the production of oxytetracycline and tri-ring T2PKs. We present comparative experimental data framed within a broader thesis on systematic chassis benchmarking for polyketide research, providing researchers with quantitative metrics for host selection.
Experimental Benchmarking: Chassis2.0 vs. Conventional Hosts
Chassis2.0 Development and Engineering Rationale
The parental strain for Chassis2.0, S. aureofaciens J1-022, was initially selected as a high-yield producer of chlortetracycline [41] [42]. The principle of "product-chassis compatibility" guided this selection, hypothesizing that a native high-yield producer of a tetra-ring T2PK would possess favorable innate metabolism for related compounds [41]. To create a versatile host, researchers executed an in-frame deletion of two endogenous T2PK gene clusters, resulting in a pigmented-faded host designated Chassis2.0 [41] [42]. This engineering strategy aimed to mitigate precursor competition and reduce background interference, streamlining metabolism toward target heterologous products.
Quantitative Performance Comparison for Oxytetracycline Production
The oxytetracycline (OTC) biosynthetic gene cluster (BGC) from S. rimosus ATCC 10970 was heterologously expressed in Chassis2.0 and conventional chassis hosts for direct comparison [41].
Table 1: Heterologous Oxytetracycline Production in Various Streptomyces Chassis
| Chassis Strain | OTC Production | Relative Performance vs. Commercial Strains | Genetic Modifications Required |
|---|---|---|---|
| S. aureofaciens Chassis2.0 | High | 370% increase [41] | Deletion of 2 native T2PK clusters [41] |
| S. albus J1074 | Not detectable [41] | Not applicable | Introduction of OTC BGC only [41] |
| S. lividans TK24 | Not detectable [41] | Not applicable | Introduction of OTC BGC only [41] |
| S. rimosus HP126 (Hyperproducer) | 4,490 mg/L [43] | Baseline (Wild-type produced 70 mg/L) [43] | Classical mutagenesis; large genomic rearrangements [43] |
Experimental results demonstrated that Chassis2.0 achieved a 370% increase in OTC production relative to commercial production strains, a significant enhancement without further pathway engineering [41]. In contrast, conventional model chassis like S. albus J1074 and S. lividans TK24 failed to produce detectable amounts of OTC when transformed with the same BGC, highlighting the critical importance of host innate metabolism [41].
Performance Evaluation for Tri-Ring Type II Polyketides
Chassis2.0 was further tested for its capacity to produce tri-ring type T2PKs, including actinorhodin (ACT) and flavokermesic acid (FK) [41].
Table 2: Production of Tri-Ring T2PKs in Engineered Chassis Strains
| Chassis Strain | Genetic Background | Production of Tri-ring T2PKs (e.g., Actinorhodin) | Notable Capabilities |
|---|---|---|---|
| S. aureofaciens Chassis2.0 | High-yield CTC producer; 2 native clusters deleted [41] | High efficiency [41] | Also activated a pentangular T2PK BGC, producing TLN-1 [41] |
| Streptomyces sp. A4420 CH | 9 native polyketide BGCs deleted [4] | Capable (Part of 4-polyketide test panel) [4] | Produced all 4 tested distinct polyketides; outperformed parental & common hosts [4] |
| S. coelicolor M1152 | Engineered with rpoB mutation; 4 clusters deleted [4] | Varies | 20-40 fold yield increases for some natural products [4] |
The chassis demonstrated high-efficiency synthesis of these tri-ring compounds, confirming its versatility beyond tetracycline-like molecules [41]. Furthermore, researchers directly activated a previously unidentified biosynthetic gene cluster associated with pentangular T2PKs in Chassis2.0, leading to the production of a structurally distinct compound, TLN-1, at high production levels [41] [42]. This underscores the chassis's utility not only for overproduction but also for the discovery of novel natural products.
Detailed Experimental Protocols
Protocol 1: Construction of Chassis2.0 from S. aureofaciens J1-022
The development of Chassis2.0 involved a targeted genetic simplification to eliminate competition for biosynthetic precursors [41].
- Strain Selection: Begin with S. aureofaciens J1-022, a native high-yield chlortetracycline producer [41].
- Cluster Identification: Identify two native T2PK biosynthetic gene clusters within the host genome.
- In-Frame Deletion: Perform precise, in-frame deletions of the two identified gene clusters. This method avoids leaving behind antibiotic resistance markers, creating a cleaner host [41].
- Phenotypic Validation: Confirm successful deletion by observing a pigmented-faded phenotype in the resulting strain, designated Chassis2.0 [41].
- Fermentation Validation: Validate chassis performance in complex starch-containing media or defined minimal media (e.g., with mannitol as a carbon source) to assess growth and baseline production capabilities [43].
Protocol 2: Heterologous Expression of OTC BGC in Chassis2.0
This protocol details the process for introducing and expressing the oxytetracycline pathway in Chassis2.0 [41].
- BGC Cloning: Using genomic DNA from S. rimosus ATCC 10970 as a template, clone the complete OTC BGC. Verify the integrity of the cloned cluster by alignment with previously validated sequences [41].
- Vector Construction: Employ ExoCET technology to construct an E. coli-Streptomyces shuttle plasmid (e.g., p15A_oxy) containing the complete OTC BGC [41].
- Conjugation: Introduce the constructed plasmid into Chassis2.0 via conjugation from E. coli.
- Strain Verification: Confirm successful integration of the OTC BGC through PCR verification. Designate the final heterologous expression strain (e.g., Chassis2.0_otc) [41].
- Fermentation and Analysis: Culture the verified strain in a suitable production medium. Monitor OTC accumulation over time using analytical methods such as HPLC or LC-MS, typically showing production initiation upon phosphate limitation [43].
Workflow Visualization: From Chassis Development to Compound Production
The following diagram illustrates the logical workflow from chassis construction to the heterologous production of diverse T2PKs, as demonstrated in the case study.
The Scientist's Toolkit: Key Research Reagents and Materials
Table 3: Essential Reagents for Chassis Engineering and Heterologous Expression
| Reagent / Material | Function / Application | Example Source / Specification |
|---|---|---|
| Streptomyces aureofaciens J1-022 | Parental strain for developing Chassis2.0 [41] | Wild-type, high-yield chlortetracycline producer |
| S. rimosus ATCC 10970 gDNA | Template for cloning the oxytetracycline BGC [41] | Genomic DNA from wild-type OTC producer |
| ExoCET Technology | Cloning method for constructing E. coli-Streptomyces shuttle plasmids [41] | Used for assembling large BGCs in plasmid p15A_oxy |
| Complex Starch Medium | Production medium mimicking industrial conditions [43] | For high-titer fermentations |
| Mannitol Minimal Medium | Defined medium for controlled systems biology studies [43] | Facilitates interpretation of multi-omics data |
| E. coli-Streptomyces Shuttle Vector | Plasmid for transferring BGCs into chassis host [41] | e.g., p15A-based vectors |
| AntiSMASH Software | Bioinformatics tool for BGC identification in host genomes [4] | Critical for profiling native clusters to delete |
Discussion and Comparative Analysis
The experimental data positions S. aureofaciens Chassis2.0 as a highly competitive chassis, particularly for aromatic polyketides. Its standout feature is the combination of high yield and versatility, efficiently producing tetra-ring antibiotics (OTC), tri-ring pigments (ACT, FK), and enabling the discovery of penta-ring compounds (TLN-1) [41]. This performance is attributed to its robust native metabolism as an industrial producer and reduced precursor competition after engineering.
When benchmarked against other engineered hosts, Chassis2.0's 370% OTC yield increase over commercial strains is notable [41]. Other specialized chassis, like Streptomyces sp. A4420 CH (with 9 deleted BGCs), also demonstrate broad capabilities by successfully producing all four tested polyketide types in a comparative study [4]. Model engineered strains like S. coelicolor M1152 can yield 20-40 fold increases for some compounds but may fail entirely to activate others, such as the OTC BGC [41] [4]. This confirms a core thesis in chassis benchmarking: no single host is universally optimal, and a panel of specialized chassis is required for comprehensive natural product discovery and production [4].
The choice of chassis thus depends on the research goal. For maximizing the yield of a known tetracycline or related compound, Chassis2.0 is an excellent choice. For expressing a diverse library of unknown BGCs to discover new molecules, a panel including Chassis2.0, Streptomyces sp. A4420 CH, and minimized hosts like S. albus Del14 would provide the highest probability of success [41] [4].
Within metabolic engineering, a primary objective is the development of optimal microbial chassis for the production of high-value natural products. For polyketides—a class of compounds with extensive pharmaceutical applications—the intracellular level of malonyl-CoA is a critical determinant of final product titer. This case study objectively benchmarks the performance of contemporary engineering strategies for enhancing malonyl-CoA levels in E. coli, providing a comparative analysis of their effectiveness in boosting polyketide production. The data and methodologies outlined herein serve as a framework for selecting and implementing chassis engineering protocols.
Benchmarking Malonyl-CoA Enhancement Strategies
Four principal metabolic engineering strategies have been developed to overcome the native low pool of malonyl-CoA in E. coli. The performance of these strategies is benchmarked in the table below, with quantitative data on their efficacy in improving both malonyl-CoA levels and polyketide production.
Table 1: Benchmarking of Malonyl-CoA Enhancement Strategies in E. coli
| Engineering Strategy | Key Genetic Modifications | Impact on Malonyl-CoA | Polyketide Titer Improvement | Reported Advantages & Limitations |
|---|---|---|---|---|
| Orthogonal Pathway (MatBC) [12] | Disruption of native bioH; genomic integration of matC (transporter) and matB (ligase) under lacUV5 promoter. |
Tunable increase with malonate supplementation [12]. | ~70% increase in flaviolin production [12]. | Advantage: Precise external control, reduces promiscuous PKS activity.Limitation: Requires malonate supplementation. |
| Precursor & Enzyme Enhancement [44] | Overexpression of Acetyl-CoA Carboxylase (ACC); deletion of pta, ackA, adhE; overexpression of Acetyl-CoA Synthetase (Acs). |
15-fold increase [44]. | 4-fold higher phloroglucinol titer [44]. | Advantage: Works with standard carbon sources.Limitation: High metabolic burden; tight regulation is challenging. |
| Biosensor-Guided sRNA Knockdown [45] | Use of RppA-based biosensor; screening of sRNA library to identify knockdown targets enhancing malonyl-CoA. | Identified 14 gene targets for increased accumulation [45]. | Naringenin: 103.8 mg/L; Resveratrol: 51.8 mg/L [45]. | Advantage: High-throughput; identifies non-intuitive targets.Limitation: Requires biosensor implementation and library screening. |
| Computational Prediction (CiED) [46] | Gene deletions predicted by Cipher of Evolutionary Design (CiED) algorithm, coupled with ACC and CoA pathway overexpression. | Increased carbon flux toward malonyl-CoA predicted and validated [46]. | Naringenin: >660% increase (100 mg/L/OD); Eriodictyol: >420% increase (55 mg/L/OD) [46]. | Advantage: Systems-level, rational design.Limitation: Relies on accuracy of metabolic model. |
Experimental Protocols for Key Strategies
This protocol creates a strain whose malonyl-CoA pool and growth are externally controlled by malonate.
- Strain Background: Start with an E. coli BAP1 or K207-3 strain harboring the sfp gene for PKS activation.
- Disruption of Native Pathway: Knock out the bioH gene to create a biotin auxotroph and disable the native acetyl-CoA carboxylase (ACC)-dependent pathway.
- Integration of MatBC: Genomically integrate the matC (malonate transporter) and matB (malonyl-CoA synthetase) genes from Rhizobium trifolii into the intergenic region downstream of ompW using homologous recombination. Genes are placed under the control of a lacUV5 promoter.
- Culture and Production:
- Culture the engineered strain in a suitable medium (e.g., Lysogeny Broth).
- Induce the MatBC system with IPTG (e.g., 10 µM).
- Supplement the medium with malonate (0-20 mM) to directly control malonyl-CoA levels.
- Co-express the polyketide synthase of interest and quantify product formation.
This strategy augments the native biosynthetic pathway by increasing the flux from acetyl-CoA to malonyl-CoA.
- Plasmid Construction: Clone the genes encoding the four subunits of acetyl-CoA carboxylase (ACC) – accA, accB, accC, accD – into a high-copy expression vector (e.g., pRSFDuet-1).
- Competing Pathway Deletion: Delete genes involved in byproduct formation to redirect carbon flux. Key targets include:
ptaandackA: Acetate production pathway.adhE: Ethanol production pathway.
- Acetate Assimilation: Overexpress acetyl-CoA synthetase (acs) to convert exogenous or accumulated acetate back into acetyl-CoA.
- Culture and Production:
- Cultivate the engineered strain in a medium like LB supplemented with 2% glucose.
- Induce ACC overexpression.
- Monitor cell growth and polyketide production, noting potential growth defects due to metabolic burden.
This method uses a colorimetric biosensor to screen for gene knockdowns that increase malonyl-CoA.
- Biosensor Strain Construction: Transform the host E. coli strain with a plasmid containing the rppA gene from Streptomyces griseus (e.g., in pTacCDFS vector). RppA is a type III PKS that converts malonyl-CoA into red-colored flaviolin.
- sRNA Library Introduction: Introduce a genome-scale synthetic small regulatory RNA (sRNA) library into the biosensor strain. This library allows for targeted knockdown of gene expression.
- Colorimetric Screening: Plate the transformed cells and screen for colonies exhibiting a deeper red color compared to the control. The color intensity correlates with intracellular malonyl-CoA levels.
- Target Validation: Isolate the sRNA plasmids from the colored colonies and sequence them to identify the gene knockdown targets. These targets can be validated and implemented in a production strain.
Pathway Diagrams for Key Metabolic Engineering Strategies
The following diagrams illustrate the logical flow and key components of two primary engineering strategies benchmarked in this study.
The Scientist's Toolkit: Essential Research Reagents
The following table details key reagents, strains, and genetic tools essential for implementing the benchmarked malonyl-CoA engineering strategies.
Table 2: Essential Research Reagents for Malonyl-CoA Engineering in E. coli
| Reagent / Tool | Function / Role | Example Source / Identifier |
|---|---|---|
| MatBC System | Orthogonal pathway: Malonate import (MatC) and conversion to Malonyl-CoA (MatB). | Rhizobium trifolii genes [12]. |
| Acetyl-CoA Carboxylase (ACC) | Key enzyme complex converting Acetyl-CoA to Malonyl-CoA in the native pathway. | E. coli genes accA, accB, accC, accD [44]. |
| RppA Biosensor | Type III PKS that produces red flaviolin from Malonyl-CoA for colorimetric screening. | rppA gene from Streptomyces griseus [45]. |
| sRNA Library | Library for targeted knockdown of gene expression to identify beneficial mutations. | Genome-scale synthetic sRNA library for E. coli [45]. |
| Engineered E. coli Strains | Specialized chassis with features like PPTase activity for PKS activation. | BAP1, K207-3 [12]. |
| CiED Algorithm | Computational model to predict gene deletions for optimizing metabolic flux. | Cipher of Evolutionary Design computational tool [46]. |
This benchmarking guide demonstrates that the choice of malonyl-CoA engineering strategy is contingent on research goals and resource constraints. The Orthogonal MatBC pathway offers precise external control and is highly effective for focused polyketide production with malonate supplementation. In contrast, biosensor-guided screening and computational approaches provide powerful, high-throughput methods for discovering non-intuitive genetic modifications that rewire central metabolism. The classic strategy of precursor enhancement and ACC overexpression remains a potent, well-established method. Ultimately, the optimal chassis strain for polyketide production may integrate elements from multiple strategies, combining external control of malonyl-CoA with internal rewiring of metabolic networks to maximize titers of these valuable compounds.
Solving the Bottlenecks: A Troubleshooting Guide for Low Titer and Yield
In the strategic benchmarking of microbial chassis for polyketide production, diagnosing precursor limitation stands as a fundamental challenge determining success in heterologous expression. Polyketides constitute one of the largest families of clinically valuable natural products, including antibiotics, anticancer agents, and immunosuppressants, yet their titers in native producers often remain impractically low [2] [17]. Overcoming precursor limitations requires precise analytical methodologies to quantify key metabolic intermediates that fuel polyketide biosynthesis. The availability of universal five-carbon precursors isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) for terpenoid backbones, as well as short-chain acyl-CoAs for polyketide chains, directly constrains production yields [47] [2]. Effective monitoring enables researchers to identify specific metabolic bottlenecks, guide targeted engineering interventions, and select optimal chassis strains based on quantitative precursor availability rather than phenotypic observations alone.
This guide provides a systematic comparison of analytical platforms and computational tools for monitoring precursor metabolites, presenting experimental data from polyketide production studies to benchmark performance across methodologies. By integrating these approaches within a structured experimental framework, metabolic engineers can transform the black box of precursor metabolism into an engineered pipeline optimized for yield.
Analytical Platforms for Targeted Metabolite Quantification
Performance Comparison of Major Analytical Techniques
Table 1: Comparison of Analytical Platforms for Targeted Metabolite Quantification
| Platform | Metabolite Coverage | Sensitivity | Quantitation Capability | Best Applications in Precursor Monitoring | Key Limitations |
|---|---|---|---|---|---|
| LC-MS/MS (Triple Quadrupole) | Targeted (dozens to hundreds) | High (pM-nM) | Excellent with internal standards | Absolute quantification of known precursors (acyl-CoAs, IPP, DMAPP) | Requires method development per metabolite; limited to targeted analytes |
| LC-QTOF-MS | Wide (hundreds to thousands) | Moderate (nM-μM) | Good with reference standards | Simultaneous targeted quantification and untargeted discovery | Lower sensitivity vs. triple quadrupole for targeted analysis |
| GC-MS | Volatile/semi-volatile metabolites | High (pM-nM) | Excellent with derivatization | Central carbon metabolites (organic acids, sugars) | Requires chemical derivatization; not ideal for labile compounds |
| NMR | Broad (all detectable compounds) | Low (μM-mM) | Absolute without standards | Structural elucidation; non-destructive analysis | Poor sensitivity; limited for low-abundance precursors |
Liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) using triple quadrupole instruments operating in multiple reaction monitoring (MRM) mode represents the gold standard for sensitive, specific quantification of known precursor metabolites. This approach enables researchers to monitor specific precursor-to-product ion transitions with high specificity, achieving sensitivity in the pico- to nanomolar range [48] [49]. When applied to monitoring acyl-CoAs in engineered Streptomyces strains, LC-MS/MS has demonstrated the capability to quantify these critical polyketide precursors at intracellular concentrations as low as 0.1-5 nmol/gDCW [50].
Liquid chromatography-quadrupole time-of-flight mass spectrometry (LC-QTOF-MS) provides a complementary approach, particularly valuable during strain development when the full spectrum of metabolic changes remains unknown. A recently developed LC-QTOF-MS method with data-independent acquisition (MSE) enables simultaneous targeted quantification of specific precursors and untargeted profiling of broader metabolic consequences, achieving accuracy of 84-131% and precision of 1-17% RSD for urinary metabolites, performance metrics that translate well to microbial systems [49]. This dual capability makes LC-QTOF-MS particularly valuable for connecting precursor availability to unexpected metabolic consequences of genetic modifications.
Experimental Protocol: Quenching and Extraction for Accurate Precursor Measurement
Proper sample preparation is critical for obtaining accurate intracellular metabolite measurements, as precursors like ATP and acyl-CoAs can turnover in seconds if metabolism is not effectively quenched [50].
Recommended Protocol:
- Rapid Quenching: For suspension cultures, employ fast filtration followed by immediate immersion in cold (-40°C) acidic acetonitrile:methanol:water (40:40:20, v/v/v) with 0.1 M formic acid. For adherent cultures, rapidly aspirate media and directly add quenching solvent [50].
- Extraction: Add extraction solvent (typically -20°C 80% methanol or acetonitrile:methanol:water) to quenched cells in a 5:1 solvent-to-biomass ratio. Vortex vigorously for 30 seconds and incubate at -20°C for 1 hour with periodic mixing.
- Pellet Removal: Centrifuge at 16,000 × g for 15 minutes at -9°C to remove protein and cell debris.
- Sample Processing: Transfer supernatant to a new tube and evaporate under nitrogen gas. Reconstitute in MS-compatible solvent for analysis.
- Standard Addition: Use stable isotope-labeled internal standards (e.g., 13C-labeled metabolites) when possible to correct for matrix effects and extraction efficiency variations [50].
Acidic quenching solvents have demonstrated superiority in preserving the integrity of energy charge metabolites, with experiments showing that non-acidic solvents allow interconversion of 3-phosphoglycerate to phosphoenolpyruvate and ATP to ADP during processing, while acidic conditions (0.1 M formic acid) prevent these artifacts [50].
Figure 1: Integrated workflow for precursor metabolite analysis from sample preparation through analytical measurement to specific applications in polyketide precursor monitoring.
Computational Approaches for Predicting Metabolic Capacity
Machine Learning for Metabolite Prediction from Genomic Data
Machine learning approaches have emerged as powerful tools for predicting metabolic potential directly from sequencing data, offering advantages when large-scale metabolomic profiling remains impractical. In comparative evaluations across six independent studies comprising over 900 microbiome-metabolome paired samples, machine learning approaches outperformed reference-based methods for predicting metabolite occurrences, achieving the highest F1-scores [51]. These models can predict the likelihood of specific precursor metabolite production based solely on genomic features, providing valuable insights during chassis selection before experimental characterization.
The Evolutionary Heterogeneous Decision Tree (EvoHDTree) algorithm has demonstrated particular promise in metabolomic applications, achieving F1-scores of 0.714-0.985 for metabolite-based classification in biomedical applications [52]. When applied to strain benchmarking, such approaches can prioritize chassis strains with innate genetic capacity for precursor biosynthesis, complementing experimental measurements.
Reference-Based Metabolic Potential Prediction
Reference-based pipelines including MIMOSA and Mangosteen leverage curated databases like KEGG and BioCyc to connect genomic features to metabolic potential through known biochemical transformations [51]. These methods identify which precursor biosynthesis pathways exist in a chassis organism and can predict the taxonomic origins of specific metabolites within complex communities. However, their dependence on database completeness represents a significant limitation, particularly for novel or poorly characterized biosynthetic pathways common in polyketide biosynthesis [51].
Table 2: Computational Tools for Metabolic Potential Prediction
| Tool | Approach | Input Data | Output | Performance Metrics | Applications in Chassis Selection |
|---|---|---|---|---|---|
| EvoHDTree | Machine learning | Metabolomic or genomic features | Metabolite classification | F1-score: 0.714-0.985 [52] | Prioritizing strains with high precursor potential |
| MelonnPan | Machine learning | Paired microbiome-metabolome data | Metabolite abundance prediction | Outperformed reference-based methods [51] | Predicting precursor production from genomic data |
| MIMOSA | Reference-based (KEGG) | Metagenomic data | Metabolic potential | Database-dependent accuracy [51] | Identifying precursor pathways in chassis genomes |
| Mangosteen | Reference-based (KEGG/BioCyc) | Metagenomic data | Compound prediction | Limited by database coverage [51] | Mapping precursor biosynthesis capacity |
Case Studies: Monitoring Precursors in Polyketide-Producing Chassis
Streptomyces Chassis Engineering for Enhanced Precursor Supply
Strategic chassis engineering requires meticulous precursor monitoring to validate metabolic rewiring. In the development of Streptomyces aureofaciens Chassis2.0 for type II polyketide production, researchers executed precise deletions of competing endogenous gene clusters to redirect precursor flux toward heterologous pathways [2]. This engineered strain demonstrated a 370% increase in oxytetracycline production compared to conventional production strains, achievement directly attributable to improved precursor availability rather than merely transcriptional activation [2].
Similarly, in the engineering of Streptomyces sp. A4420 as a polyketide-focused chassis, deletion of nine native polyketide biosynthetic gene clusters created a metabolically simplified host with enhanced precursor supply for heterologous expression [17]. When evaluated against established hosts including S. coelicolor M1152, S. lividans TK24, and S. albus J1074, the engineered A4420 CH strain uniquely produced all four tested polyketide compounds across all conditions, outperforming both its parental strain and conventional hosts [17]. This performance advantage stemmed from intrinsic differences in precursor metabolism and availability between chassis options.
Analytical Benchmarking Across Platforms
Comparative metabolomic studies provide valuable insights into platform selection for specific precursor monitoring applications. When comparing UHPLC-HRMS and FTIR spectroscopy for metabolic profiling, UHPLC-HRMS demonstrated 8-17% higher accuracies (≥83%) for classification tasks involving homogeneous sample groups [53]. However, FTIR spectroscopy outperformed mass spectrometry-based approaches when analyzing unbalanced populations, achieving 83% accuracy where UHPLC-HRMS failed to generate viable models [53]. This finding highlights the importance of matching analytical platform to experimental design in precursor limitation studies.
Essential Research Reagent Solutions
Table 3: Key Research Reagents for Precursor Metabolite Analysis
| Reagent/Category | Specific Examples | Function in Precursor Monitoring | Application Notes |
|---|---|---|---|
| Stable Isotope Labels | 13C-glucose, 15N-ammonium, 2H-water | Tracing precursor incorporation into products | Enables flux analysis; reveals rate-limiting steps |
| Internal Standards | 13C/15N-labeled amino acids, acyl-CoAs | Quantification normalization | Corrects for matrix effects and recovery variations |
| Quenching Solvents | Acidic acetonitrile:methanol:water (40:40:20) | Immediate metabolic arrest | Preserves labile metabolites like ATP and acyl-CoAs |
| Extraction Solvents | 80% methanol, acetonitrile:methanol:water | Metabolite liberation from cells | Optimized for polar metabolite recovery |
| Derivatization Reagents | MSTFA (for GC-MS), dansyl chloride | Chemical modification for detection | Enhances volatility (GC-MS) or detectability (LC-MS) |
| Authentic Standards | IPP, DMAPP, acetyl-CoA, malonyl-CoA | Method calibration and quantification | Essential for absolute concentration determination |
Stable isotope labels represent particularly powerful reagents for diagnosing precursor limitations, as they enable researchers to trace the incorporation of simple carbon sources into complex polyketide scaffolds. By feeding 13C-labeled glucose or acetate to production strains and monitoring labeling patterns in intracellular precursor pools and final products, metabolic engineers can identify which precursors become limiting and which pathways contribute most significantly to product biosynthesis [50].
Diagnosing precursor limitation in polyketide production chassis requires an integrated analytical strategy rather than reliance on a single methodology. The most comprehensive approach combines targeted LC-MS/MS quantification of known precursor metabolites with stable isotope tracing to elucidate metabolic flux patterns and machine learning prediction of metabolic potential from genomic data. This multi-layered strategy enables researchers to not only identify current limitations but predict how genetic modifications will impact precursor supply.
Experimental evidence from Streptomyces chassis development demonstrates that strategic precursor monitoring can dramatically improve polyketide titers, with documented cases of several-fold production increases resulting from engineered precursor enhancements [2] [17]. As synthetic biology advances toward more complex polyketide structures, the precision of precursor monitoring will increasingly determine success in heterologous production. The tools and methodologies compared in this guide provide a foundation for evidence-based chassis selection and engineering, transforming precursor limitation diagnosis from a constraint to a design parameter in polyketide metabolic engineering.
Addressing Enzyme Solubility and Activity in Non-Native Hosts
A foundational goal in metabolic engineering is the successful transfer of biosynthetic pathways into heterologous hosts for the efficient production of valuable compounds. For polyketides—a class of pharmaceutically important secondary metabolites—this process is frequently hampered by one critical bottleneck: the poor solubility and inadequate activity of enzymes when expressed in non-native chassis strains [54] [55]. Enzyme insolubility can lead to protein aggregation, inclusion body formation, and a complete loss of function, thereby derailing entire biosynthetic pathways [55]. This guide provides a comparative analysis of current strategies to overcome these challenges, offering experimental data and methodologies to aid researchers in selecting and benchmarking the most appropriate chassis for their polyketide production goals.
Comparative Analysis of Chassis Strains for Polyketide Production
The choice of host organism is a primary determinant of success in heterologous expression. The table below compares the performance of three key chassis strains for polyketide production, highlighting their inherent advantages and limitations.
Table 1: Performance Comparison of Chassis Strains for Polyketide Production
| Chassis Strain | Reported Titer for Model Polyketide | Key Advantages | Major Limitations |
|---|---|---|---|
| Escherichia coli | Low (e.g., challenges with minimal PKS solubility) [2] | • Rapid growth & high-density cultivation• Well-established genetic tools & parts [56] | • Frequent insoluble expression of large enzyme complexes [2] [55]• Lack of native post-translational modifications |
| Streptomyces albus / lividans (Model Strains) | Low to Moderate (e.g., 0.2 mg/L to 127 mg/L) [2] | • Native producer of polyketides• Compatible with complex PKS machinery | • Often requires extensive metabolic engineering for high yields [2]• Suboptimal efficiency for diverse T2PKs |
| Streptomyces aureofaciens Chassis2.0 (Engineered) | High (e.g., ~3.8 g/L Chlortetracycline; 370% increase in Oxytetracycline) [2] | • High native precursor flux• Superior product-chassis compatibility [2]• Efficient for tri-, tetra-, and penta-ring T2PKs | • Genetic manipulation can be more complex than in E. coli• Longer fermentation cycles than bacterial hosts |
Key Insights from Comparative Data
- E. coli remains a popular starting point due to its unparalleled ease of use, but its application is often limited to simpler pathways. One of the most significant hurdles is the insoluble expression of minimal polyketide synthase (PKS) complexes, which are core to polyketide biosynthesis [2].
- Model Streptomyces strains, while more genetically tractable, often show unsatisfactory efficiency for heterologous expression of type II polyketides (T2PKs), typically requiring multiple rounds of metabolic engineering to achieve commercially viable titers [2].
- Engineered Industrial Strains present a promising solution. The engineered Streptomyces aureofaciens Chassis2.0, developed by deleting competing endogenous gene clusters, demonstrates the principle of product-chassis compatibility [2]. It successfully achieved high-yield production of diverse T2PKs, including oxytetracycline, actinorhodin, and the newly discovered pentangular polyketide TLN-1, without further pathway optimization [2].
Core Experimental Protocols for Assessing Host Compatibility
To systematically address enzyme solubility and activity, researchers employ a suite of experimental protocols. The following workflows are critical for benchmarking chassis performance.
Protocol 1: In Silico Solubility Prediction and Pathway Assembly (ProPASS)
This computational workflow prioritizes enzyme selection based on predicted solubility before experimental construction [54].
Detailed Methodology:
- Pathway Construction: Identify possible biosynthetic pathways from a host's native metabolites to the target polyketide using tools like ProPath, which explores abstract metabolic networks [54].
- Database Curation: Create a solubility database, such as ProSol DB, by calculating solubility propensity scores for enzymes from repositories like UniProtKB/Swiss-Prot. The tool ccSOL omics can be used, which employs neural networks to estimate solubility confidence in E. coli based on Fourier transform coefficients of protein sequences [54].
- Probabilistic Assembly: For each reaction in the candidate pathways, select enzyme sequences (associated with specific EC numbers) with the highest predicted solubility scores in the target host (e.g., E. coli) [54].
- Experimental Validation: Construct the top-ranked genetic implementations in the chosen host and measure protein expression (e.g., via SDS-PAGE/Western Blot) and solubility (e.g., via fractionation and analysis of soluble vs. insoluble fractions) [54].
Protocol 2: Deep Mutational Scanning for Activity-Stability Tradeoffs (EP-Seq)
Enzyme Proximity Sequencing (EP-Seq) is a powerful method for simultaneously analyzing thousands of enzyme variants to dissect the relationship between sequence, stability, and activity [57].
Detailed Methodology:
- Library Construction: Create a site-saturation mutagenesis library for the enzyme of interest, ensuring each variant is tagged with a Unique Molecular Identifier (UMI) [57].
- Stability/Expression Profiling:
- Display the variant library on the yeast surface.
- Stain expressed proteins with fluorescent antibodies against a tag (e.g., His-tag).
- Use Fluorescence-Activated Cell Sorting (FACS) to sort cells into bins based on fluorescence intensity, which serves as a proxy for protein expression level and folding stability.
- Sequence the UMIs in each bin to calculate an expression fitness score for each variant [57].
- Activity Profiling:
- For oxidoreductases, incubate the displayed library with the enzyme's substrate (e.g., D-amino acids for a D-amino acid oxidase). The reaction produces H~2~O~2~ as a byproduct.
- In a cascade reaction, use Horseradish Peroxidase (HRP) to convert H~2~O~2~ and a tyramide-based probe into a phenoxyl radical, which labels the yeast cell wall in proximity to the active enzyme.
- Sort cells based on this activity-dependent fluorescence and sequence the UMIs to calculate an activity fitness score for each variant [57].
- Data Integration: Cross-reference expression and activity scores to identify mutations that enhance catalytic activity without compromising stability, or to understand evolutionary constraints [57].
The logical relationship and workflow of the EP-Seq method can be visualized as follows:
Research Reagent Solutions: A Scientist's Toolkit
A range of molecular tools and reagents is essential for implementing the aforementioned protocols. The table below lists key solutions for tackling solubility and activity challenges.
Table 2: Essential Research Reagent Solutions for Heterologous Expression
| Reagent / Tool | Primary Function | Key Application in Research |
|---|---|---|
| Solubility Prediction Software (ccSOL omics) | Predicts solubility propensity of proteins in E. coli [54]. | Pre-screening enzyme variants and selecting optimal sequences for pathway assembly (ProPASS) [54]. |
| Fusion Tags (SUMO, MBP, Trx) | Enhances solubility of fused target proteins; aids in purification [56]. | Overcoming insolubility of difficult-to-express proteins; one-step affinity purification [56]. |
| Molecular Chaperone Plasmids | Co-expression of chaperones (e.g., GroEL/GroES) to assist proper protein folding in vivo [54]. | Reducing aggregation and increasing soluble yield of recombinant enzymes in E. coli [54]. |
| Specialized E. coli Strains (e.g., BL21(DE3), ArcticExpress) | Optimized for protein expression; contain features like tRNA for rare codons or cold-adapted chaperones. | Improving folding and solubility of heterologous proteins, especially those with complex structures or codon bias [55]. |
| Yeast Surface Display (YSD) | Display of protein libraries on yeast cell surface for high-throughput screening [57]. | Coupling genotype to phenotype for deep mutational scanning (EP-Seq) to assess stability and activity [57]. |
The journey to efficient polyketide production in heterologous hosts is complex and hinges on resolving enzyme solubility and activity. No single chassis is universally superior; the optimal choice is dictated by the specific project requirements.
- For rapid prototyping of simple pathways, E. coli, augmented with fusion tags and chaperones, offers speed and ease of use.
- For the expression of complex Type II PKS pathways without extensive engineering, an engineered industrial host like Streptomyces aureofaciens Chassis2.0 demonstrates clear advantages in yield and compatibility [2].
- For the fundamental improvement of a specific, problematic enzyme, high-throughput methods like EP-Seq provide unprecedented insights into stability-activity tradeoffs, enabling data-driven enzyme engineering [57].
A systematic approach that leverages in silico prediction, advanced screening technologies, and strategic chassis engineering is paramount to overcoming the persistent challenges of enzyme solubility and activity, thereby accelerating the discovery and production of valuable polyketide-based therapeutics.
Overcoming Genetic Instability and Poor Fermentation Characteristics
The economic viability of industrial-scale microbial fermentation is critically dependent on the stability and robustness of the production chassis. Genetic instability—the irreversible loss of production capacity due to mutations—and poor fermentation characteristics such as slow growth and complex nutrient requirements represent significant bottlenecks in bioprocess scale-up, particularly for high-value compounds like polyketides [58] [59]. These challenges often remain undetected during laboratory-scale development but manifest severely in industrial bioreactors where populations undergo 60-80 generations, allowing non-producing mutants to overtake the culture [58]. This comprehensive guide benchmarks contemporary microbial chassis and characterizes their performance stability for polyketide production, providing researchers with experimental data and methodologies for selecting and engineering robust production platforms.
Comparative Analysis of Microbial Chassis for Polyketide Production
Performance Benchmarking of Industrial Chassis
The table below summarizes the fermentation characteristics and genetic stability profiles of major microbial hosts used for polyketide synthesis, highlighting their advantages and limitations for industrial applications.
Table 1: Comparative analysis of microbial chassis for polyketide production
| Chassis Strain | Product/Class | Titer/Yield | Genetic Stability | Fermentation Characteristics | Key Advantages | Major Limitations |
|---|---|---|---|---|---|---|
| Streptomyces aureofaciens Chassis2.0 [2] | Type II Polyketides (Oxytetracycline, Actinorhodin) | 370% increase in OTC vs. commercial strains; High tri-ring T2PK efficiency | Maintained production over serial passages; Precursor competition mitigated via gene cluster deletion | Short fermentation cycle; Good sporulation; Easy genetic manipulation | High chassis-product compatibility; Efficient precursor channeling | Limited to actinomycetes-specific products |
| Escherichia coli (Engineered) [58] | Mevalonic Acid | Complete production loss after ~70 generations | Severe genetic instability; Diverse mutation modes (IS transposition, SNPs) | Rapid growth; High metabolic burden from heterologous expression | Well-established genetic tools; High growth rate | High metabolic burden; Unsolved instability in large-scale fermentation |
| Saccharomyces cerevisiae (Engineered) [60] [59] | Dicarboxylic Acids; Fluorescent Proteins | Stable succinate/fumarate production; Output decline over 100 generations (varies by integration site) | Model-predicted evolutionary stability; Copy number and locus-dependent stability | Industrially familiar; Compatible with waste streams | Robust industrial history; Eukaryotic enzyme expression | Variable output based on genomic integration site |
| Streptomyces sp. FR-008 [61] | Candicidin (Macrolide) | High native candicidin production | Streamlined genome (7.26 Mb); Stable after PKS cluster deletion | Fast-growing; Simplified conjugation | Smallest Streptomyces genome sequenced; Efficient DNA uptake | Limited product range without engineering |
Quantitative Stability Assessment Across Microbial Systems
The table below presents experimental stability metrics for different chassis, highlighting production decline rates and key influencing factors identified through controlled studies.
Table 2: Quantitative stability assessment of engineered production chassis
| Chassis Strain | Production Metric | Generations Assessed | Performance Decline | Key Instability Factors Identified |
|---|---|---|---|---|
| E. coli (Mevalonic Acid) [58] | Mevalonic Acid Titer | 80 | Complete loss by generation 70 | Escape rate: 2.5 × 10⁻⁸/generation; Production load: 30% |
| S. cerevisiae (Multi-copy Reporter) [59] | Fluorescent Protein Output | 100 | 20-60% decrease (site-dependent) | Copy number; Genomic integration locus; Metabolic burden |
| S. cerevisiae (Dicarboxylic Acids) [60] | Succinate/Fumarate/Malate | Evolved strains | Improved production after ALE | Evolutionary robustness; Model-predicted gene targets |
| S. aureofaciens Chassis2.0 [2] | Heterologous T2PKs | Production confirmed | No reported decline | Endogenous cluster deletion; Precursor availability |
Experimental Protocols for Stability Assessment
Serial Passage Fermentation Simulation
Principle: Industrial fermentations involving 60-80 cell generations from master cell bank to production scale enable spontaneous non-producing mutants to emerge and overtake populations. This protocol experimentally simulates these conditions to evaluate strain stability [58].
Procedure:
- Inoculum Preparation: Start from a single colony of the production strain in a seed culture medium.
- Serial Transfers: Perform sequential batch transfers every 8 hours (approximately 8-9 generations per transfer) for a total of 9 transfers (~80 generations total).
- Population Sampling: Collect and preserve population samples at each transfer point for subsequent analysis.
- Growth Rate Monitoring: Measure optical density (OD600) at regular intervals to calculate population growth rates.
- Product Titer Quantification: Analyze product concentration in culture supernatants using appropriate analytical methods (HPLC, GC-MS, fluorescence).
- Data Correlation: Statistically correlate growth rates with production titers to identify fitness-production trade-offs.
Key Parameters:
- Production Load: Calculated as (1 - (μproducer/μcontrol)) × 100%, where μ represents growth rate
- Escape Rate: Determined through model fitting of production decline data
Essential Gene Complementation for Plasmid Stabilization
Principle: Plasmid-based expression systems often require antibiotic selection, which is infeasible industrially. Essential gene complementation stabilizes plasmids without antibiotics by making host cell survival dependent on plasmid maintenance [62].
Procedure:
- Chromosomal Deletion: Delete an essential gene (infA, ssb, proC, dapD) from the host chromosome.
- Plasmid Engineering: Clone the deleted essential gene onto the expression plasmid.
- Continuous Cultivation: Perform phosphate-limited continuous fermentation at various dilution rates (0.033 h⁻¹, 0.1 h⁻¹) and temperatures (30°C, 37°C).
- Plasmid Retention Monitoring: Sample populations regularly and assess plasmid retention via selective plating and PCR.
- Structural Stability Assessment: Sequence plasmids from endpoint populations to identify mutations.
Evaluation Metrics:
- Segregational Stability: Plasmid retention rate over generations
- Structural Stability: Genetic integrity of expression cassettes
- Specific Productivity: Product formed per cell per unit time
Visualization of Genetic Instability Mechanisms and Experimental Workflows
Mechanisms of Genetically Driven Production Decline in Fermentations
Experimental Workflow for Chassis Stability Assessment
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key research reagents and methodologies for stability engineering
| Reagent/Method | Function | Application Example | Performance Notes |
|---|---|---|---|
| ExoCET Direct Cloning [2] | Large gene cluster assembly | Heterologous expression of complete oxytetracycline BGC | Maintains pathway integrity for functional expression |
| Essential Gene Complementation [62] | Plasmid stabilization without antibiotics | infA, ssb, dapD complementation in continuous fermentation | Maintains 100% plasmid retention for >500 hours |
| Ultra-Deep Population Sequencing [58] | Detection of rare genetic variants | Identification of multiple IS transposition events in E. coli | Reveals heterogeneity at >1000× coverage |
| Genome-Scale Metabolic Modeling [60] | Prediction of evolutionarily stable knockouts | ZWF1, SER3, SER33 deletion chassis for dicarboxylic acids | Couples growth to product formation |
| Promoter Library Screening [61] | Tunable expression optimization | GusA reporter assay in Streptomyces sp. FR-008 | Provides 60-860% expression range relative to ermE-p |
| Iodo-TMT Redox Proteomics [63] | Cellular redox status assessment | Quantification of oxidative stress in high vs. low toxin Karenia brevis | Measures redox stoichiometry of cysteine residues |
Strategic chassis selection and engineering are paramount for overcoming genetic instability and poor fermentation characteristics in industrial bioprocesses. The experimental data and comparative analysis presented demonstrate that streptomycetes chassis, particularly engineered variants like S. aureofaciens Chassis2.0, offer superior stability and productivity for complex natural products like polyketides [2]. In contrast, conventional hosts like E. coli exhibit significant limitations despite their genetic tractability [58]. The implementation of robust stability assessment protocols—including serial passage simulations, essential gene complementation systems, and deep sequencing of population dynamics—enables researchers to identify and mitigate instability mechanisms before scale-up. By integrating these approaches with modern engineering strategies like genome minimization [61] and model-guided design [60], researchers can develop microbial cell factories that maintain high productivity throughout industrial fermentation cycles, ultimately bridging the critical gap between laboratory promise and commercial implementation.
A primary challenge in metabolic engineering is that forcing microbes to overproduce valuable compounds often creates metabolic imbalances. Engineering pathways compete with cellular metabolism for essential resources, which can lead to metabolic burden, accumulation of toxic intermediates, and reduced cell growth, ultimately limiting final product titers, rates, and yields (TRY) [64]. Traditional "static" engineering strategies, such as gene knockouts or constitutive gene expression, are often insufficient to address these dynamic challenges because they cannot respond to changing conditions within the fermentation [65].
Dynamic metabolic engineering addresses this by creating genetically encoded control systems that allow cells to autonomously adjust their metabolic flux in response to their internal or external state [64]. This is analogous to "just-in-time transcription" found in natural systems [66]. By dynamically re-routing flux, these systems can manage trade-offs between cell growth and product formation, prevent the buildup of toxic intermediates, and enhance overall process robustness, making them particularly valuable for complex pathways like those producing polyketides and other toxic compounds [66] [65].
Comparing Major Dynamic Regulation Strategies
Dynamic control systems can be broadly categorized into pathway-dependent and pathway-independent strategies. The table below compares three key approaches applied in modern metabolic engineering.
Table 1: Comparison of Dynamic Regulation Strategies for Metabolic Engineering
| Strategy | Core Mechanism | Key Features | Demonstrated Applications & Performance |
|---|---|---|---|
| Quorum Sensing (QS) Systems [66] [67] | Uses self-produced signaling molecules (e.g., AHL) to trigger gene expression changes at high cell density. | • Pathway-independent• Broadly applicable across hosts and pathways• Links regulation to population density | E. coli: 5.5-fold increase in myo-inositol; glucaric acid from unmeasurable to >0.8 g/L [66].B. subtilis: Increased D-pantothenic acid to 14.97 g/L in fed-batch fermentation [67]. |
| Orthogonal Pathway Control [12] | Replaces native, tightly regulated pathways with an externally tunable, non-native pathway. | • Precise, external control over metabolite pools• Can be coupled with adaptive evolution• Reduces promiscuous enzyme activity | E. coli for Polyketides: Disrupted native malonyl-CoA synthesis; introduced MatBC malonate assimilation pathway. Enabled tunable M-CoA levels via malonate supplementation, enhancing polyketide and fatty acid titers [12]. |
| QS-Integrated CRISPRi [67] | Combines QS cell-density sensing with the programmability of CRISPR interference for gene knockdown. | • Fully autonomous & programmable• High precision in transcriptional regulation• Type I systems show lower cellular toxicity than Cas9 | B. subtilis: Dynamic regulation of TCA (citZ) or glycolysis (pfk) boosted D-pantothenic acid and riboflavin (2.49-fold increase) production [67]. |
Quorum Sensing for Autonomous Flux Control
The Esa QS system from Pantoea stewartii has been successfully implemented in E. coli to create a pathway-independent genetic control module [66]. The system functions as a genetic inverter: the transcriptional regulator EsaR activates a promoter (PesaS) in the absence of a threshold level of the quorum sensing molecule AHL. As the cell population grows, AHL synthesized by EsaI accumulates, eventually repressing the PesaS promoter [66].
Table 2: Key Components of the Esa QS System for Dynamic Regulation
| Component | Type | Function in the Circuit |
|---|---|---|
| EsaI | Enzyme (AHL synthase) | Produces the acyl-homoserine lactone (AHL) signal. Its expression level determines the timing of the metabolic switch. |
| EsaR | Transcriptional regulator | Binds and activates the PesaS promoter in the absence of AHL. Accumulated AHL causes it to dissociate, turning off transcription. |
| PesaS | Promoter | Drives expression of the target metabolic gene (e.g., pfkA); is active at low cell density and switched off at high cell density. |
| SsrA Degradation Tag | Peptide tag | Appended to the target protein (e.g., PfkA) to ensure rapid degradation once transcription is halted, enabling swift flux control. |
Orthogonal Pathway Control for Precise Precursor Provision
A 2025 study on polyketide production in E. coli addressed the bottleneck of limited malonyl-CoA (M-CoA) availability, a crucial precursor [12]. The researchers engineered an orthogonal system for M-CoA production by:
- Disrupting native regulation: The native M-CoA biosynthetic pathway was disrupted by deleting bioH, a gene essential for the function of the acetyl-CoA carboxylase (ACC) complex. This made the host auxotrophic for M-CoA.
- Introducing an orthogonal pathway: Genes for a malonate transporter (matC) and a malonyl-CoA ligase (matB) were integrated into the genome.
- External tuning: M-CoA levels could then be precisely controlled by the external addition of malonate, which rescues growth and provides the M-CoA precursor pool. This strategy not only increased M-CoA and polyketide titers but also reduced the promiscuous activity of PKSs toward undesired acyl-CoA substrates [12].
Integrated QS-CRISPRi for Programmable Autonomous Control
A cutting-edge tool developed in 2025 combines the autonomy of QS with the programmability of CRISPR. The QS-controlled type I CRISPRi (QICi) toolkit was designed for Bacillus subtilis [67]. This system uses the native PhrQ-RapQ-ComA QS circuit to control the expression of a type I CRISPR array. At high cell density, the QS signal activates the expression of CRISPR RNAs (crRNAs), which then guide a CRISPR complex to repress target genes [67]. This system is less toxic than the commonly used Cas9-based systems and provides a convenient and effective strategy for balancing complex pathways.
Experimental Protocols for Key Dynamic Regulation Strategies
Protocol: Implementing a QS-Based Metabolic Valve in E. coli
This protocol is adapted from studies that used the Esa QS system to dynamically regulate glycolytic flux [66].
Objective: To dynamically downregulate a native essential gene (e.g., pfkA) to redirect flux toward a heterologous production pathway.
Key Reagents:
- Strain: E. coli MG1655 with Δzwf to block the pentose phosphate pathway.
- Circuit Parts: Genomically integrated esaRI70V under a constitutive promoter; a library of promoter-RBS combinations driving esaI; the PesaS promoter controlling the target gene (e.g., pfkA) with a C-terminal SsrA degradation tag.
Procedure:
- Circuit Characterization: Create a library of strains (e.g., the LXX series) with varying expression levels of EsaI to generate a spectrum of switching times. Use a reporter like GFP under PesaS to characterize the cell density (OD600) at which the circuit switches off for each variant.
- Modeling and Prediction: Build a kinetic model incorporating enzyme kinetics and AHL production dynamics to simulate the effect of different Pfk-1 knockdown times on product titer and identify the theoretical optimal switching point.
- Strain Construction: Replace the native promoter of the target gene (pfkA) with the PesaS promoter in the host genome and append the SsrA tag. Use the characterized EsaI expression variants to control the timing of Pfk-1 knockdown.
- Fermentation and Validation: Cultivate engineered strains in a defined medium with glucose. Monitor cell growth, substrate consumption, and product formation (e.g., myo-inositol). Compare the performance of strains with different switching times against a static control to identify the optimal dynamic regulation profile.
Protocol: Engineering Orthogonal Malonyl-CoA Control
This protocol is based on the 2025 method for enhancing polyketide production in E. coli [12].
Objective: To decouple malonyl-CoA (M-CoA) supply from native regulation and enable external control over its intracellular concentration.
Key Reagents:
- Strains: PKS-compatible E. coli strains (e.g., K207-3, BAP1) with sfp for PKS activation.
- Genes: matC (malonate transporter) and matB (malonyl-CoA ligase) from Rhizobium trifolii.
Procedure:
- Disrupt Native Pathway: Delete the bioH gene in the host genome to disrupt biotin biosynthesis, rendering the native Ac-CoA carboxylase (ACC) complex non-functional and creating a biotin/M-CoA auxotroph.
- Integrate Orthogonal Pathway: Integrate the matBC genes, under an inducible (e.g., lacUV5) or constitutive promoter, into a defined genomic "safe site" (e.g., downstream of ompW).
- Validate Control and Functionality:
- Growth Rescue Test: Confirm that the engineered strain only grows in the presence of malonate in minimal medium.
- M-CoA/PKS Activity Assay: Transform the strain with a plasmid encoding a reporter PKS (e.g., RppA synthase that produces flaviolin). Cultivate strains with varying malonate concentrations (e.g., 0-20 mM) and measure product formation (e.g., flaviolin absorbance at 340/520 nm) to demonstrate tunable M-CoA levels.
- Polyketide Production: Introduce the target PKS pathway into the engineered strain. Perform fed-batch fermentations with optimized malonate feeding to maximize polyketide titer.
Pathway Diagrams and Experimental Workflows
The following diagrams illustrate the logical relationships and workflows for the dynamic regulation strategies discussed.
Quorum Sensing Mechanism for Dynamic Regulation
Orthogonal Malonyl-CoA Pathway Engineering
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Dynamic Metabolic Engineering Experiments
| Reagent / Tool | Category | Primary Function | Example Sources / Parts |
|---|---|---|---|
| Esa Quorum Sensing System | Genetic Circuit | Provides cell-density-dependent, autonomous control of gene expression. | EsaI, EsaR, PesaS promoter from Pantoea stewartii [66]. |
| MatBC Malonate Assimilation System | Orthogonal Pathway | Provides external control over intracellular malonyl-CoA levels. | matC (transporter) and matB (ligase) from Rhizobium trifolii [12]. |
| Type I CRISPRi System | Programmable Repressor | Enables precise gene knockdown; often less toxic than Cas9. | Endogenous type I system in B. subtilis or other hosts with cas3 deletion [67]. |
| SsrA Degradation Tag | Protein Tag | Ensures rapid degradation of target proteins, enabling swift metabolic flux changes. | Peptide tag (e.g., AADENYALAA or "LAA") [66]. |
| Metabolite Biosensors | Sensor | Links product or intermediate concentration to a measurable output (e.g., fluorescence) for screening. | Transcription factors or riboswitches responsive to specific metabolites [64]. |
| Promoter and RBS Libraries | Tuning Toolbox | Allows fine-tuning of gene expression levels to optimize circuit performance. | Pre-characterized promoter and RBS sequences with varying strengths [66] [64]. |
Adaptive Laboratory Evolution (ALE) for Unlocking Higher Production
In the pursuit of optimal microbial chassis for polyketide production, researchers are increasingly turning to Adaptive Laboratory Evolution (ALE) as a powerful strategy to overcome inherent physiological limitations. ALE is an experimental methodology that promotes the accumulation of beneficial mutations in microbial populations through long-term cultivation under selective pressure, leading to enhanced fitness and often unlocking novel production capabilities [68]. For polyketide biosynthesis—a domain of immense pharmaceutical importance due to compounds with antibacterial, anticancer, and immunosuppressant activities—ALE has emerged as a crucial tool for enhancing precursor supply, improving stress tolerance, and ultimately increasing titers of these valuable natural products [68] [12]. This guide provides a comparative analysis of ALE implementations across different microbial hosts, offering researchers benchmark data and methodologies for harnessing evolution to create superior polyketide production chassis.
Core ALE Techniques and Their Applications
ALE encompasses several distinct experimental approaches, each with specific advantages for particular applications in polyketide research. Understanding these methodologies is essential for selecting the appropriate strategy for a given production challenge.
Table 1: Comparison of Major ALE Methodologies
| ALE Method | Key Advantages | Limitations | Polyketide Production Applications |
|---|---|---|---|
| Serial Transfer | Easy to automate; suitable for high-throughput experiments; enables parallel evolution lines [68] | Difficult with aggregating cells; discontinuous growth; limited temporal control of conditions [68] | E. coli long-term evolution [68]; Co-culture evolution [68]; Antibiotic resistance studies [68] |
| Colony Transfer | Introduces single-cell bottlenecks; applicable to aggregating cells; enables evolutionary dynamics visualization [68] | Low-throughput; limited automation; discontinuous growth; difficult environmental control [68] | Mutation accumulation studies [68]; Antibiotic production enhancement in Streptomyces [68] |
| Continuous Culture | Constant growth rate control; stable population densities; continuous nutrient supply; precise environmental control [68] | Limited parallel replication; potential for biofilm formation on bioreactors [68] | Morbidostat for antibiotic resistance [68]; E. coli sugar synthesis from CO2 [68]; Turbidostat-based yeast evolution [68] |
Experimental Workflow for ALE in Polyketide Research
A generalized ALE workflow for enhancing polyketide production involves multiple stages from initial design to final strain characterization. The process typically requires several weeks to months depending on the generational time of the microorganism and the selection pressure applied.
ALE Implementation Across Microbial Chassis for Polyketide Production
Escherichia coli as a Polyketide Production Chassis
E. coli represents one of the most widely employed hosts for ALE studies due to its rapid growth, well-characterized genetics, and metabolic flexibility [69]. Recent work has demonstrated the power of ALE for enhancing malonyl-CoA availability—a critical precursor for polyketide biosynthesis—in engineered E. coli strains.
Table 2: ALE Applications in E. coli for Polyketide Production
| Engineering Target | ALE Strategy | Key Outcomes | Reference |
|---|---|---|---|
| Malonyl-CoA Enhancement | Disrupted native M-CoA pathway + orthogonal MatBC malonate assimilation; ALE under malonate dependency [12] | Identified mutations further boosting M-CoA and polyketide production; Enhanced understanding of M-CoA regulation [12] | Nature Chemical Biology (2025) [12] |
| Type II Polyketide Synthesis | Soluble expression of oviedomycin BGC proteins; Substrate channeling; Efflux transporter engineering [70] | 120 mg/L oviedomycin from glucose in 24 hours; Efficient heterologous production of type II polyketides [70] | Metabolic Engineering (2025) [70] |
| Genetic Circuit Optimization | Promoter engineering and RBS optimization of minimal PKS from Photorhabdus luminescens [71] | 181 mg/L SEK4 and 392 mg/L SEK4b production; Additional 25% yield increase via metabolic reprogramming [71] | ChemRxiv (2025) [71] |
The metabolic engineering strategy for enhancing malonyl-CoA in E. coli involves creating a dependency on an orthogonal pathway, then applying ALE to optimize this non-native system:
Streptomyces as a Specialized Polyketide Producer
Streptomyces species represent native producers of many polyketides, offering inherent compatibility with complex PKS machinery but presenting challenges in genetic manipulation and process optimization.
Table 3: Streptomyces Chassis Development for Type II Polyketides
| Strain/Strategy | Engineering Approach | Production Outcomes | Advantages/Limitations |
|---|---|---|---|
| S. aureofaciens Chassis2.0 | In-frame deletion of two endogenous T2PKs gene clusters; Precursor competition mitigation [2] | 370% oxytetracycline increase vs. commercial strains; Efficient tri-ring T2PKs production; Activated pentangular T2PKs BGC [2] | Advantages: High native precursor supply; Excellent PKS compatibility; Industrial-scale proven [2] Limitations: Longer fermentation cycles; Complex genetic manipulation [2] |
| S. coelicolor Heterologous Expression | Expression of oviedomycin BGC; Metabolic modeling to enhance malonyl-CoA and NADPH [70] | Improved oviedomycin production via enhanced precursor supply [70] | Advantages: Well-established genetic tools; Model organism Limitations: Often requires extensive engineering for high titers [2] |
Non-Traditional Hosts: Aurantiochytrium for PKS-Derived DHA
Beyond conventional bacterial hosts, marine protists like Aurantiochytrium offer unique advantages for producing polyketide-derived compounds, particularly the valuable omega-3 fatty acid DHA (docosahexaenoic acid).
Table 4: ALE in Aurantiochytrium for Enhanced DHA Production
| ALE Strategy | Evolution Conditions | Performance Metrics | Molecular Insights |
|---|---|---|---|
| Staged Acidic ALE | Multi-factor ALE: low pH (citric acid), low temperature (16°C), high dissolved oxygen [72] | 171.4% DHA increase vs. wild-type; 106.3% biomass increase; 243.8% total fatty acid yield increase [72] | Upregulated glycolysis and PKS pathway enzymes; Enhanced TCA cycle and PPP; Differential ATP/NADPH supply mechanisms [72] |
The multi-factor ALE approach implemented in Aurantiochytrium demonstrates how combined stressors can synergistically enhance production phenotypes:
Experimental Protocols for Key ALE Implementations
ALE for Malonyl-CoA Enhancement in E. coli
Objective: Increase intracellular malonyl-CoA levels to enhance polyketide production [12].
Methodology:
- Strain Construction:
- Disrupt native malonyl-CoA biosynthesis by deleting bioH (essential for biotin biosynthesis and ACC complex function)
- Integrate orthogonal MatBC malonate assimilation pathway (malonate transporter matC and malonyl-CoA ligase matB) into ompW intergenic region
- Transform with polyketide production plasmid (e.g., pBADT-RppA-NT for flaviolin production)
ALE Process:
- Conduct serial batch cultures in minimal medium with malonate as sole precursor for malonyl-CoA
- Maintain malonate dependency throughout evolution process
- Transfer cultures at regular intervals (typically 1-10% inoculum) to fresh medium
- Monitor flaviolin production spectrophotometrically (A340 and A520) as proxy for malonyl-CoA availability
Endpoint Analysis:
- Isolate individual clones from evolved populations
- Sequence genomes to identify causative mutations
- Characterize malonyl-CoA levels and polyketide production in bioreactors
Multi-Factor ALE in Aurantiochytrium for DHA Enhancement
Objective: Enhance acid tolerance and DHA production under fermentation conditions [72].
Methodology:
- Baseline Characterization:
- Cultivate wild-type Aurantiochytrium sp. PKU#Mn16 in M4 medium (glucose 20 g/L, peptone 1.5 g/L, yeast extract 1 g/L, sea salt 33 g/L)
- Establish growth and DHA production profiles under standard conditions (28°C, 170 rpm, pH 6.5)
Staged ALE Regimen:
- Implement orthogonal design combining temperature (16°C, 28°C), shaking speed (170 rpm, 230 rpm), and acid types (citric, acetic, hydrochloric)
- Initiate ALE at mild stress levels, gradually increasing selection pressure
- Conduct serial transfers maintaining 10% inoculum size
- Monitor biomass (dry cell weight), total fatty acids, and DHA content at each transfer
Systems Biology Analysis:
- Perform comparative transcriptomics of evolved vs. wild-type strains at early (metabolic peak) and late (metabolic decline) fermentation stages
- Analyze expression changes in glycolysis, PKS pathway, TCA cycle, and pentose phosphate pathway
- Validate metabolic rewiring through enzyme activity assays and metabolite profiling
The Scientist's Toolkit: Essential Research Reagents
Table 5: Key Research Reagents for ALE Polyketide Studies
| Reagent/Category | Specific Examples | Function/Application | Reference |
|---|---|---|---|
| Malonyl-CoA Pathway Engineering | MatBC system (matC transporter, matB ligase from Rhizobium trifolii); bioH deletion | Creates orthogonal malonyl-CoA biosynthesis pathway; Enables precursor control [12] | Nature Chemical Biology (2025) [12] |
| PKS Expression Systems | ovm BGC from S. antibioticus; pik PKS from S. venezuelae; RppA synthase | Provides polyketide-producing machinery; Enables flavonoid and complex polyketide production [12] [70] | Metabolic Engineering (2025) [70] |
| Solubility Enhancement Tools | Molecular chaperones (GroEL/GroES); Rare tRNAs; Fusion tags (MBP, GST) | Improves soluble expression of PKS proteins in heterologous hosts [70] | Metabolic Engineering (2025) [70] |
| Efflux Transporters | AcrAB-TolC system; MFS family transporters | Enhances product secretion; Reduces intracellular inhibition; Improves host tolerance [70] | Metabolic Engineering (2025) [70] |
| Analytical Standards | Flaviolin; SEK4; SEK4b; Oviedomycin; DHA standards | Enables accurate quantification of polyketide products and precursors [12] [72] [70] | Multiple Sources |
ALE has established itself as an indispensable strategy in the metabolic engineer's toolkit for unlocking higher polyketide production across diverse microbial chassis. The comparative data presented in this guide demonstrates that while E. coli offers exceptional genetic tractability and rapid evolution cycles, specialized producers like Streptomyces provide inherent PKS compatibility, and non-conventional hosts like Aurantiochytrium present unique opportunities for specific compound classes. The most successful implementations combine targeted genetic engineering with ALE, creating synthetic metabolic dependencies that drive evolutionary optimization toward desired phenotypes. As ALE methodologies continue to advance—particularly through integration with multi-omics analyses and machine learning—this approach will undoubtedly play an increasingly central role in developing next-generation microbial cell factories for pharmaceutical and industrial polyketide production.
In the pursuit of engineering efficient microbial cell factories for polyketide production, a critical challenge lies in overcoming the inherent limitations of native metabolic pathways. These pathways often prioritize cellular growth over the synthesis of desired, high-value compounds, leading to low yields and inefficient production processes. Within this benchmarking context, a sophisticated "two-layer strategy" has emerged as a powerful systematic approach for reprogramming cellular machinery. This methodology involves first modulating the specific biosynthetic pathway to enhance the production of a target molecule, followed by a second layer of engineering aimed at boosting the host's overall capacity to accumulate the final product. A prominent example of this strategy's success is its application in the oleaginous yeast Yarrowia lipolytica for the high-level production of palmitoleic acid (POA), a valuable polyketide-derived fatty acid with applications in nutraceuticals, cosmetics, and biofuel precursors [73]. This article provides a comparative guide on the implementation and performance of this two-layer strategy, benchmarking its efficacy against conventional single-layer metabolic engineering approaches.
The Two-Layer Engineering Framework: Principles and Workflow
The two-layer strategy is a structured metabolic engineering approach that systematically addresses the dual challenges of product specificity and overall titer. The foundational principle involves a clear functional separation of the engineering objectives:
- Layer 1: Pathway Modulation: The primary objective of this layer is to reshape the host's native metabolic network to favor the synthesis of the target compound. This is achieved by redirecting metabolic flux away from competing pathways and enhancing the activity of bottleneck reactions in the desired pathway. Key techniques include the expression of heterologous enzymes with higher specificity, the dynamic regulation of competitive pathways, and the knockout of genes responsible for product degradation or diversion.
- Layer 2: Capacity Enhancement: Once the pathway is optimized, the second layer focuses on amplifying the host cell's overall production capacity. This involves engineering the central metabolism to increase the supply of essential precursors, enhancing the cell's energy and cofactor regeneration systems, and preventing the degradation of the final product. The goal is to turn the cell into a high-yield factory for the target compound.
The experimental workflow for implementing this strategy is sequential and iterative, as visualized in the diagram below.
Case Study: High-Level Production of Palmitoleic Acid inYarrowia lipolytica
The application of this two-layer strategy in the oleaginous yeast Y. lipolytica for palmitoleic acid (POA) production serves as a benchmark for its effectiveness. In wild-type strains, POA typically constitutes only 2-3% of total fatty acids, as its direct precursor, palmitic acid (C16:0), is preferentially elongated to stearic acid (C18:0) [73]. The following sections detail the experimental protocols and the resulting performance data.
Layer 1: Modulation of the Fatty Acid Profile
The objective of the first layer was to reprogram the fatty acid synthesis machinery to favor the conversion of C16:0 to POA over its elongation to C18:0.
- Experimental Protocol 1: Screening Heterologous Desaturases
- Methodology: Researchers screened and expressed heterologous fatty acid desaturases from other organisms, such as CeFat5 from Caenorhabditis elegans, which demonstrated higher substrate specificity for C16:0 compared to the native yeast desaturase [73].
- Rationale: Introducing a more specific "gatekeeper" enzyme ensures that a larger proportion of the C16:0 pool is converted into the desired product, POA, rather than other fatty acids.
- Experimental Protocol 2: Dynamic Regulation of Fatty Acid Elongase
- Methodology: A key innovation was the implementation of dynamic regulation using a copper-responsive promoter to control the expression of the fatty acid elongase gene (YlELO1). Upon addition of copper ions to the fermentation broth, the expression of the elongase was downregulated [73].
- Rationale: This dynamic control strategically diverts the C16:0 pool away from the C18:0 elongation pathway and towards the newly enhanced POA synthesis pathway. This method is superior to a complete gene knockout, as it avoids potential negative impacts on cell growth that require small amounts of C18:0.
- Experimental Protocol 3: Blocking β-Oxidation
- Methodology: The gene PEX10, essential for the import of proteins into peroxisomes (where β-oxidation occurs), was deleted (Δpex10) to effectively block the degradation of fatty acids, including POA [73].
- Rationale: Preventing the breakdown of the product ensures that synthesized POA accumulates within the cell.
Layer 2: Boosting POA-Rich Lipid Accumulation
With a reshaped fatty acid profile, the second layer of engineering focused on amplifying the yeast's capacity to produce and store large quantities of POA-rich lipids.
- Experimental Protocol 4: Overexpressing a POA-Specific Acyltransferase
- Methodology: A heterologous POA-specific triacylglycerol forming acyltransferase was overexpressed [73].
- Rationale: This enzyme efficiently channels the free POA into triacylglycerols (TAGs), the primary storage lipid in yeast. This "pull" strategy helps to drive the overall flux of the pathway towards the end product and facilitates intracellular storage.
- Experimental Protocol 5: Introducing a Non-Carboxylative Malonyl-CoA Pathway
- Methodology: An artificial, non-carboxylative malonyl-CoA production (NCM) pathway was introduced into the strain [73].
- Rationale: Malonyl-CoA is the essential two-carbon building block for fatty acid synthesis. Native production consumes ATP. The NCM pathway provides an alternative, potentially more efficient route to generate this key precursor, thereby increasing the total flux through the fatty acid synthesis pathway.
- Experimental Protocol 6: Engineering Storage and Efflux
- Methodology: Further engineering to prevent lipid degradation and potentially enhance lipid droplet formation was conducted, building upon the PEX10 knockout [73].
- Rationale: Stabilizing the lipid storage compartments minimizes product loss and allows for higher intracellular titers.
Performance Benchmarking and Quantitative Comparison
The cumulative impact of this systematic two-layer engineering strategy on POA production was profound. The table below summarizes the quantitative performance metrics of the engineered strains compared to the baseline.
Table 1: Performance Benchmark of Two-Layer Engineering for Palmitoleic Acid Production in Y. lipolytica
| Strain / Engineering Step | POA (% of Total Fatty Acids) | POA Titer (g/L) | Fold Improvement (vs. Initial) |
|---|---|---|---|
| Initial Strain (Wild-type) | ~1.34% [73] | Not Specified | 1x |
| After Layer 1 Modulations | Data not explicitly stated in source | Data not explicitly stated in source | Significant increase |
| After Layer 2 Enhancements | 50.62% [73] | 25.60 g/L [73] | 37.7x [73] |
This performance data highlights the synergistic effect of the two-layer approach. The final engineered strain not only achieved a POA purity of over 50% in its lipid profile but also reached a record titer of 25.60 g/L in a bioreactor, marking a 37.7-fold improvement over the initial strain's production capacity [73]. This titer and purity are significantly superior to traditional sources of POA, such as macadamia nuts (~22% POA) or sea buckthorn (~40% POA), demonstrating the viability of engineered microbes as a sustainable and efficient production platform [73].
The Scientist's Toolkit: Essential Reagents and Solutions
The successful implementation of the two-layer strategy relies on a suite of specialized research reagents and genetic tools. The following table details key solutions and their functions in the context of metabolic engineering for polyketide and lipid production.
Table 2: Research Reagent Solutions for Two-Layer Metabolic Engineering
| Research Reagent / Tool | Function / Explanation |
|---|---|
| Heterologous Desaturases (e.g., CeFat5) | Enzymes from other organisms introduced into the chassis strain to create a more specific or efficient reaction step that is lacking in the native host [73]. |
| Inducible / Dynamic Promoters (e.g., Copper-Responsive) | Genetic elements that allow for precise, external control of gene expression. Essential for dynamic regulation strategies to balance growth and production [73]. |
| CRISPR-Cas9 Gene Editing System | A versatile tool for performing precise gene knockouts (e.g., PEX10, KU70), knock-ins, and other genomic modifications with high efficiency [73]. |
| Synthetic Pathway Modules (e.g., NCM Pathway) | Artificially designed and constructed metabolic pathways, often heterologous, introduced to bypass native regulatory mechanisms or to provide a more efficient route to key precursors like malonyl-CoA [73]. |
| Acyltransferases (e.g., POA-specific TAG acyltransferase) | Enzymes that catalyze the attachment of fatty acids to glycerol backbones. Overexpression of specific acyltransferases can channel flux towards desired storage lipids [73]. |
| Comparative Transcriptomics | An analytical method to compare gene expression profiles between engineered and control strains, helping to elucidate underlying mechanisms of improvement and identify new engineering targets [73]. |
Pathway Visualization: From Acetyl-CoA to Product Accumulation
The complex metabolic network involved in polyketide and fatty acid synthesis, and the key engineering interventions, can be visualized in the following simplified pathway diagram. This illustrates the journey from central metabolites to the high-titer accumulation of a target compound like POA.
Discussion and Comparative Outlook
The two-layer strategy represents a paradigm shift from earlier metabolic engineering efforts, which often focused on a single rate-limiting step. The presented case study demonstrates that the synergistic integration of pathway-specific modulation and host-capacity engineering is a robust framework for benchmarking and optimizing chassis strains. When compared to single-layer approaches—such as only overexpressing a desaturase or only blocking β-oxidation—the two-layer strategy delivers exponentially superior results by addressing the entire production pipeline, from precursor to stored product.
This systematic approach is transferable to the broader field of polyketide production. The core principles of dynamically redirecting flux, boosting precursor supply, and preventing degradation are universally applicable. Future work in benchmarking chassis strains will likely involve more advanced dynamic control systems, machine learning-guided design of synthetic pathways, and the integration of multi-omics data to further refine both layers of this powerful engineering strategy.
Head-to-Head: A 2025 Benchmarking Analysis of Leading Production Chassis
In the development of microbial cell factories for polyketide synthesis, achieving commercial viability depends on meeting stringent performance benchmarks. The core metrics of titer, yield, and rate form the foundational triad for evaluating bioprocess efficiency, directly impacting economic feasibility and scalability [74]. For polyketides—a class of metabolites with widespread pharmaceutical applications—these metrics are particularly crucial due to the structural complexity and typically low native production levels of these compounds. The fermentation cycle further completes this benchmarking framework by defining the temporal dimension of production, influencing volumetric productivity and overall process costs [2]. This guide provides a comparative analysis of these key performance indicators across different chassis strains and cultivation systems, offering researchers standardized methodologies for objective strain evaluation within the context of polyketide production research.
Defining the Core Benchmarking Metrics
Titer
Titer quantifies the final concentration of a target compound accumulated in the fermentation broth, typically expressed as mass per unit volume (e.g., g L⁻¹ or mg L⁻¹). It represents the cumulative efficiency of the chassis strain and process in converting substrates into the desired polyketide product. High titer is a primary goal in strain engineering, as it directly impacts downstream purification costs and determines the vessel size required for a given production output. In polyketide production, titers can vary significantly based on the chassis organism, the complexity of the polyketide, and the optimization of the biosynthetic pathway.
Yield
Yield measures the conversion efficiency of substrate to product, expressed as mass of product per mass of substrate (g product g substrate⁻¹) or as a percentage of the theoretical maximum. This metric is critical for assessing the economic viability of a bioprocess, as substrate costs often constitute a major portion of production expenses. Yield reflects the chassis strain's metabolic efficiency and the success of engineering strategies in minimizing carbon flux toward competing pathways. For polyketides, yield optimization often involves enhancing precursor supply (e.g., malonyl-CoA, methylmalonyl-CoA) and reducing loss to biomass formation or byproducts [28].
Productivity Rate
Productivity Rate (or volumetric productivity) defines the amount of product formed per unit volume per unit time (e.g., g L⁻¹ h⁻¹). This metric integrates both titer and fermentation cycle time, providing a measure of process intensification and bioreactor utilization efficiency. High productivity is particularly important for large-scale manufacturing where bioreactor occupancy directly impacts capital and operational expenditures. For polyketides with long biosynthetic pathways, achieving high productivity requires balancing cell growth with product synthesis phases [74].
Fermentation Cycle
Fermentation Cycle encompasses the total time required to complete a production batch, including lag phase, growth phase, production phase, and harvest. Shorter fermentation cycles generally improve volumetric productivity and reduce contamination risks but must be balanced against the time required for adequate biomass accumulation and product synthesis. For Streptomyces strains used in polyketide production, fermentation cycles can vary from days to weeks, significantly impacting comparative performance between chassis strains [2].
Comparative Performance of Chassis Strains for Polyketide Production
Table 1: Benchmarking metrics across different chassis strains and polyketide products
| Chassis Strain | Polyketide Product | Titer (mg L⁻¹) | Yield (g g⁻¹) | Productivity (mg L⁻¹ h⁻¹) | Fermentation Cycle | Reference |
|---|---|---|---|---|---|---|
| E. coli K207-3 | Triketide Lactone (Pik167) | 791 | - | - | 6-10 days | [75] |
| E. coli K207-3 | Tetraketide Lactone (Pik1567) | 100 | - | - | 6-10 days | [75] |
| S. aureofaciens Chassis2.0 | Oxytetracycline | 28,100 (28.1 g L⁻¹) | - | - | ~50% shorter than S. rimosus | [2] |
| S. aureofaciens Chassis2.0 | β-Arbutin | 28,100 (28.1 g L⁻¹) | - | - | ~50% shorter than S. rimosus | [2] |
| Streptomyces sp. A4420 CH | Benzoisochromanequinone | Higher than control strains | - | - | - | [17] |
| Streptomyces sp. A4420 CH | Glycosylated Macrolide | Higher than control strains | - | - | - | [17] |
Table 2: Comparative analysis of chassis strain characteristics for polyketide production
| Chassis Strain | Genetic Manipulation Efficiency | Precursor Availability | Fermentation Cycle Duration | Heterologous Expression Efficiency | Key Advantages |
|---|---|---|---|---|---|
| E. coli K207-3 | High | Engineered for enhanced malonyl-CoA and methylmalonyl-CoA | 6-10 days | Moderate to high for type I PKS | Established platform with precursor engineering |
| S. aureofaciens Chassis2.0 | Moderate | Native high flux toward polyketide precursors | Shorter cycle than related strains | High for type II polyketides | Industrial heritage, optimized for aromatic polyketides |
| Streptomyces sp. A4420 CH | High (rapid sporulation/growth) | High metabolic capacity for polyketides | - | Superior for diverse polyketide BGCs | Clean background with 9 native PKS clusters deleted |
| S. albus J1074 | Moderate | Moderate | - | Variable | Well-characterized, commonly used |
| S. lividans TK24 | Moderate | Moderate | - | Variable | Accepts methylated DNA, low protease activity |
Experimental Protocols for Metric Determination
Standard Fermentation Protocol for Polyketide Production in E. coli
This protocol outlines the standard procedure for polyketide production in engineered E. coli strains such as K207-3, with specific modifications for precursor supplementation [75]:
- Strain Preparation: Transform E. coli K207-3 with PKS expression plasmids containing appropriate antibiotic resistance markers (e.g., kanamycin, streptomycin, or chloramphenicol).
- Pre-culture Development: Inoculate 5 mL LB media containing appropriate antibiotics in 10 mL culture tubes. Incubate overnight at 37°C with shaking at 240 rpm.
- Production Culture: Use pre-culture to inoculate polyketide production media (0.3 mL for 30 mL scale, 3 mL for 300 mL scale) in non-baffled Fernbach flasks. Production media contains: 5 g L⁻¹ yeast extract, 10 g L⁻¹ casein, 15 g L⁻¹ glycerol, 10 g L⁻¹ NaCl, 100 mM potassium phosphate (pH 7.6), with appropriate antibiotics.
- Growth Phase: Incubate cultures at 37°C with shaking at 240 rpm until OD₅₆₀ reaches 0.6.
- Production Phase Induction: Cool cultures to 19°C and induce with 0.1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Supplement with sodium propionate (20-100 mM final concentration) for methylmalonyl-CoA-dependent PKS systems.
- Product Accumulation Phase: Continue incubation at 19°C for 6-10 days with shaking at 240 rpm.
- Product Quantification: Monitor polyketide production by HPLC analysis of culture extracts. For analysis, mix 500 μL culture with equal volume acetone, centrifuge, and analyze supernatant.
Heterologous Expression Protocol for Type II Polyketides in Streptomyces
This protocol describes the process for expressing type II polyketide biosynthetic gene clusters (BGCs) in Streptomyces chassis strains, based on methodologies applied to S. aureofaciens Chassis2.0 and Streptomyces sp. A4420 CH [2] [17]:
BGC Cloning and Vector Construction:
- Isolate target BGC from donor strain using appropriate method (e.g., cosmic library, PCR amplification, or direct cloning).
- For S. aureofaciens, use ExoCET technology to construct E. coli-Streptomyces shuttle vectors containing the complete BGC.
- Verify BGC integrity through PCR and sequencing.
Strain Transformation:
- Introduce BGC-containing vectors into Streptomyces chassis strains via protoplast transformation or conjugation from E. coli.
- For conjugation, use non-methylating E. coli ET12567/pUZ8002 as donor strain.
- Select exconjugants with appropriate antibiotics (e.g., apramycin).
Fermentation and Product Analysis:
- Inoculate spores or mycelial fragments into liquid medium suitable for polyketide production (e.g., SFM, ISP2, or optimized production media).
- Incubate at appropriate temperature (typically 28-30°C) with shaking for 3-14 days depending on chassis strain and target compound.
- Extract culture broth with equal volume of organic solvent (e.g., ethyl acetate, butanol).
- Analyze extracts for polyketide production using HPLC, LC-MS, or other appropriate analytical methods.
Analytical Method for Polyketide Quantification
HPLC Analysis Protocol [75]:
- Equipment: Waters 1525 HPLC system with Microsorb-MV 300-5 C18 column (4.6 × 250 mm)
- Mobile Phase:
- Solvent A: Water with 0.1% formic acid
- Solvent B: Acetonitrile with 0.1% formic acid
- Gradient: 5-100% B over 15 minutes, maintain at 100% B for 3 minutes
- Flow Rate: 1 mL min⁻¹
- Detection: UV-Vis or MS detection appropriate for target polyketide
Interrelationship of Benchmarking Metrics
The diagram above illustrates how the four benchmarking metrics interrelate within a polyketide production system. Titer, yield, rate, and fermentation cycle are interconnected rather than independent parameters, with complex trade-offs often existing between them. For instance, strategies to maximize titer (e.g., extended fermentation) may negatively impact productivity rate by prolonging the fermentation cycle. Similarly, maximizing yield through controlled feeding strategies may initially reduce productivity rate despite improving substrate conversion efficiency. The chassis strain characteristics fundamentally influence all metrics, determining the upper bounds of performance. These integrated metrics ultimately dictate the economic viability and scalability of the bioprocess, with optimal strain selection requiring careful consideration of all four parameters in relation to the specific polyketide target and production goals.
Essential Research Reagent Solutions
Table 3: Key reagents and materials for polyketide production experiments
| Reagent/Material | Function/Application | Example Usage | Key Considerations |
|---|---|---|---|
| Propionate (Sodium Salt) | Precursor for methylmalonyl-CoA biosynthesis | Supplementation at 20-100 mM in E. coli K207-3 cultures | Concentration optimization required to balance precursor supply with potential toxicity |
| Malonate | Enhances malonyl-CoA pool via orthogonal MatBC pathway | Used in engineered E. coli with malonate transporter | Enables bypass of native regulatory mechanisms controlling malonyl-CoA levels |
| Cerulenin | Fatty acid synthase inhibitor | Diverts malonyl-CoA from fatty acid synthesis to polyketide production | Nonspecific inhibition may also affect PKS ketosynthase domains |
| Isopropyl β-D-1-thiogalactopyranoside (IPTG) | Inducer for T7 RNA polymerase | Standard induction at 0.1 mM for PKS expression in E. coli | Concentration and induction timing critical for balancing protein expression and metabolic burden |
| MatBC Enzyme System | Orthogonal malonyl-CoA generation | Genomic integration in engineered E. coli strains | Provides tunable control over intracellular malonyl-CoA levels via malonate supplementation |
| Sfp Phosphopantetheinyl Transferase | Activates ACP domains of PKSs | Genome-encoded in specialized E. coli strains (e.g., BAP1, K207-3) | Essential for converting apo-PKS to active holo-form |
| T7 Promoter System | Controls PKS gene expression | Varying promoter strength tunes polypeptide stoichiometry in multi-gene PKS | Mutations at -12 or -5 positions enable fine-tuning of expression balance |
| ExoCET Cloning System | Facilitates BGC assembly and vector construction | Used for heterologous expression in Streptomyces chassis | Enables precise manipulation of large polyketide gene clusters |
The benchmarking metrics of titer, yield, productivity rate, and fermentation cycle provide a comprehensive framework for evaluating chassis strains for polyketide production. Current data demonstrates that specialized chassis strains optimized for specific polyketide classes can achieve dramatically improved performance metrics compared to conventional laboratory strains. The experimental protocols and analytical methods outlined enable standardized comparison across different systems, while the reagent toolkit provides essential components for pathway engineering and precursor optimization. As synthetic biology and metabolic engineering continue to advance, these benchmarking metrics will remain essential for guiding the development of next-generation chassis strains with enhanced capabilities for polyketide biosynthesis. Future directions will likely focus on further balancing the trade-offs between these metrics through dynamic regulation strategies and systems-level metabolic engineering.
Within the field of microbial metabolic engineering, the selection of an appropriate host chassis is a critical determinant of success for the heterologous production of valuable natural products. This guide provides a objective comparison of chassis strains for the production of type II polyketides (T2PKs), a class of compounds with diverse pharmacological activities. The analysis focuses on the performance of Streptomyces aureofaciens J1-022—an industrial high-yield strain engineered into "Chassis2.0"—against conventional Streptomyces model strains. The content is framed within a broader thesis on benchmarking chassis strains, providing researchers and drug development professionals with experimental data and methodologies to inform strain selection for polyketide production research.
Table 1: Fundamental Characteristics of Streptomyces Chassis Strains
| Feature | S. aureofaciens J1-022 (Chassis2.0) | Conventional Model Strains (e.g., S. albus J1074, S. lividans TK24) |
|---|---|---|
| Origin/Type | High-yield industrial chlortetracycline producer [41] | Laboratory-adapted model strains [41] |
| Endogenous T2PKs | Two clusters deleted in Chassis2.0 to minimize precursor competition [41] | Contain native T2PK gene clusters [41] |
| Colony Morphology & Genetic Stability | Robust sporulation and stable morphology, suitable for reliable genetic manipulation [41] | Variable; some industrial high-producers (e.g., S. rimosus) show shriveled phenotypes and genetic instability [41] |
| Fermentation Cycle | Shorter cycle, reducing contamination risk and accelerating validation [41] | Longer cycle (up to twice as long as J1-022 for some strains), slowing R&D iterations [41] |
| Primary Application | Optimized as a versatile chassis for diverse T2PKs discovery and overproduction [41] | Used for basic genetic studies and initial heterologous expression trials [41] |
Performance Benchmarking: Quantitative Production Data
The efficacy of a chassis is ultimately judged by its ability to produce target compounds. The following table summarizes experimental production data for various type II polyketides in different hosts.
Table 2: Comparative Production Efficiency of Type II Polyketides
| Polyketide Type / Compound | S. aureofaciens Chassis2.0 | Conventional Model Strains | Notes & Experimental Conditions |
|---|---|---|---|
| Tetra-ring: Oxytetracycline (OTC) | ~370% increase relative to commercial production strains [41] | No detectable accumulation (S. albus J1074, S. lividans TK24) [41] | Heterologous expression of OTC BGC from S. rimosus ATCC 10970 [41] |
| Tri-ring: Actinorhodin (ACT) & Flavokermesic Acid (FK) | High-efficiency synthesis [41] | Typically requires metabolic engineering for activation (e.g., knockout, regulator overexpression) [41] [76] | Chassis2.0 demonstrates inherent compatibility with tri-ring structures [41] |
| Penta-ring: TLN-1 | Direct activation of an unidentified BGC, leading to high production levels [41] | Information not specified in search results | Highlights the chassis's capability for novel compound discovery [41] |
| General PK-II Core Structures | Information not specified in search results | Production of BIQ, ANG, TCM, and PEN cores shown in S. coelicolor M1152 [76] | S. coelicolor M1152 is an engineered derivative with four endogenous clusters deleted [76] |
Detailed Experimental Protocols
To ensure reproducibility and provide a clear understanding of the underlying data, this section outlines the key experimental methodologies cited in the performance comparison.
Heterologous Expression of Oxytetracycline (OTC) BGC
- Cloning Method: The complete OTC biosynthetic gene cluster (BGC) was cloned from the genomic DNA of S. rimosus ATCC 10970. The ExoCET technology was employed to construct the E. coli-Streptomyces shuttle plasmid
p15A_oxy, which carries the entire OTC BGC [41]. - Strain Transformation: The constructed plasmid was introduced into both the conventional model strains (S. albus J1074, S. lividans TK24) and the candidate chassis. Successful integration was confirmed via PCR verification, resulting in strains designated as
J1074_otc,TK24_otc, etc [41]. - Fermentation & Analysis: The heterologous expression strains were cultured under appropriate conditions. The accumulation of tetracyclines (TCs) in the culture medium was analyzed, likely using chromatographic methods such as High-Performance Liquid Chromatography (HPLC), which revealed no production in the model strains but significant titers in the engineered S. aureofaciens host [41].
Development of S. aureofaciens Chassis2.0
- Rationale: To mitigate competition for essential precursors (malonyl-CoA, etc.) between the host's native metabolic pathways and heterologously introduced BGCs [41].
- Engineering Strategy: An in-frame deletion of two endogenous T2PKs gene clusters was executed in the parent strain S. aureofaciens J1-022. This resulted in a "pigmented-faded" host, designated Chassis2.0, which is effectively a clean slate for the production of non-native polyketides [41].
General Workflow for Heterologous PK-II Production
The following diagram illustrates the standard workflow for engineering a chassis strain to produce heterologous type II polyketides, as exemplified by the development and use of Chassis2.0.
The Scientist's Toolkit: Key Research Reagents
Table 3: Essential Reagents and Tools for Chassis Engineering and Polyketide Production
| Research Reagent / Tool | Function & Application | Reference |
|---|---|---|
| ExoCET Technology | A method for direct cloning and assembly of large biosynthetic gene clusters (BGCs) into shuttle plasmids. | [41] |
| E. coli-Streptomyces Shuttle Plasmid (e.g., p15A_oxy) | A vector that can be propagated in E. coli for easy manipulation and then transferred into Streptomyces for heterologous expression. | [41] |
| pXZ4/pXZ5 Plasmid System | An expression vector for Streptomyces containing the natural, self-regulatory PK-II operon promoter PactI and its activator, optimized for heterologous expression. | [76] |
| S. coelicolor M1152 Host | An engineered model host with four native secondary metabolite gene clusters deleted, designed to reduce background interference. | [76] |
| antiSMASH | A bioinformatics tool for the genome-wide identification, annotation, and analysis of secondary metabolite BGCs. | [77] |
The comparative data indicate that Streptomyces aureofaciens Chassis2.0 presents a superior platform for the overproduction and discovery of type II polyketides compared to conventional model strains. Its principal advantages include a dramatically enhanced production efficiency for complex molecules like oxytetracycline, a native cellular machinery that is highly compatible with a wide range of T2PKs without requiring extensive engineering, and a robust physiological profile that streamlines the research and development process. For research programs focused on the efficient heterologous production of complex polyketides, Chassis2.0 represents a compelling and high-performing chassis choice.
Escherichia coli has emerged as a premier heterologous host for the production of polyketides, a class of natural products with significant pharmaceutical importance. Among the various engineered E. coli strains, K207-3 and BAP1 represent landmark achievements in metabolic engineering for polyketide biosynthesis. These strains address critical challenges in heterologous expression, including precursor supply and post-translational activation of polyketide synthases (PKSs). This guide provides an objective comparison of the performance characteristics of K207-3 and BAP1 strains, focusing on their applications in type I and III polyketide production. The analysis is framed within the broader context of benchmarking chassis strains for polyketide research, offering researchers evidence-based selection criteria for their experimental designs.
Strain Origins and Genetic Lineage
The development of both BAP1 and K207-3 stems from foundational work in the early 2000s that established E. coli as a viable host for complex polyketide biosynthesis [78] [79]. The pioneering BAP1 strain, derived from E. coli BL21(DE3), was engineered to support the heterologous production of 6-deoxyerythronolide B (6-dEB), the macrocyclic core of the antibiotic erythromycin [78] [80]. This achievement demonstrated that E. coli could be engineered to supply the necessary propionyl-CoA and (2S)-methylmalonyl-CoA precursors and properly modify heterologous PKS enzymes.
K207-3 represents a further optimized strain developed to enhance polyketide production capabilities. This B-derived E. coli strain incorporates several improvements over its predecessors, including removal of the propionate degradation pathway (∆prpRBCD) and introduction of a propionyl-CoA carboxylase (PCC) to direct assimilated propionate toward methylmalonyl-CoA synthesis [81]. K207-3 also contains the sfp gene from Bacillus subtilis, encoding a phosphopantetheinyl transferase necessary for PKS activation, and the native prpE gene under an inducible T7 promoter for enhanced propionyl-CoA synthesis [12]. Additionally, K207-3 contains genes encoding the propionyl-CoA carboxylase from Streptomyces coelicolor, enabling the conversion of propionyl-CoA to methylmalonyl-CoA (mM-CoA) [12].
Table 1: Key Genetic Modifications in BAP1 and K207-3
| Genetic Feature | BAP1 | K207-3 |
|---|---|---|
| Parental Strain | BL21(DE3) | E. coli B |
| Propionate Catabolism | prpRBCD deleted | prpRBCD deleted |
| Phosphopantetheinyl Transferase | sfp integrated | sfp integrated |
| Propionyl-CoA Synthesis | prpE overexpressed | prpE overexpressed |
| Methylmalonyl-CoA Synthesis | S. coelicolor PCC expressed | S. coelicolor PCC expressed |
| Propionyl-CoA Carboxylase | Introduced for mM-CoA synthesis | Introduced for mM-CoA synthesis |
| Additional Modifications | - | Enhanced genetic stability |
The strategic engineering of these strains enables them to overcome E. coli's natural limitations in PKS precursor availability, particularly for (2S)-methylmalonyl-CoA, which is not naturally produced in significant quantities in wild-type E. coli [78]. Both strains have been utilized extensively for heterologous expression of various PKSs, with K207-3 generally demonstrating superior performance in production titers for many polyketide targets [82] [83].
Performance Comparison and Benchmarking Data
Quantitative Production Metrics
Extensive benchmarking experiments have revealed significant differences in the performance capabilities of BAP1 and K207-3 strains for polyketide production. The most comprehensive data exists for 6-deoxyerythronolide B (6-dEB) production, where both strains have been systematically evaluated under various cultivation conditions.
Table 2: Performance Comparison for 6-dEB Production
| Strain | Production Context | Max Titer (mg/L) | Key Production Conditions | Citation |
|---|---|---|---|---|
| BAP1 | Initial development | 20 | Propionate supplementation | [79] |
| BAP1 | Fed-batch optimization | 80-100 | High-cell density cultivation | [82] |
| K207-3 | Fed-batch process | 700 | Initial bioreactor evaluation | [82] |
| K207-3 | Optimized fed-batch | 1,100 | Medium and fermentation optimization | [82] |
| K207-3 | Plasmid system with Wood-Werkman cycle | 0.81 | Glucose sole carbon source | [81] |
The remarkable 11-fold improvement in 6-dEB titer achieved with K207-3 under optimized fed-batch conditions highlights the critical importance of host strain selection in heterologous polyketide production [82]. This titer of 1.1 g/L represents one of the highest reported for any complex polyketide produced heterologously in E. coli and demonstrates the potential for industrial-scale production using engineered K207-3 strains.
Beyond 6-dEB production, both strains have been successfully employed for the biosynthesis of various other polyketides. BAP1 has been used for production of 1,3,6,8-tetrahydroxynaphthalene (THN) via the type III PKS RppA, with flaviolin production serving as an indicator of intracellular malonyl-CoA levels [12]. Recent engineering efforts have further enhanced K207-3's capabilities through the introduction of an orthogonal malonyl-CoA biosynthesis pathway involving malonate transporter (matC) and malonyl-CoA ligase (matB), enabling tunable control over malonyl-CoA levels via malonate supplementation [12].
Strain Selection Guidelines
The comparative performance data suggests specific guidelines for strain selection in polyketide research:
K207-3 is generally preferred for high-titer production of type I polyketides, particularly when using fed-batch cultivation processes [82] [83].
BAP1 remains valuable for initial pathway validation and proof-of-concept studies, especially when rapid strain engineering and testing are prioritized.
K207-3 demonstrates superior genetic stability for maintaining large PKS-encoding plasmids during extended cultivations, a critical factor for high-cell density processes [82].
Both strains can be further engineered with accessory pathways to enhance precursor supply, such as the MatBC malonate assimilation system for improving malonyl-CoA availability [12].
Experimental Protocols and Methodologies
Standardized Production Protocol for 6-dEB
The highest reported titers for 6-dEB production in K207-3 were achieved using a optimized fed-batch bioprocess [82]. The following protocol details the key methodological steps:
Strain and Plasmid Configuration:
- Host Strain: E. coli K207-3
- Plasmids: pKOS207-129 (encoding DEBS1 and accessory enzymes) and pBP130 (encoding DEBS2 and DEBS3)
- Antibiotic Selection: Carbenicillin (100 mg/L) and streptomycin (20 mg/L)
Inoculum Preparation:
- Inoculate 50 mL of LB medium containing antifoam and antibiotics with 1 mL of frozen glycerol stock.
- Incubate at 37°C with shaking at 250 rpm until OD600 reaches 1.0-1.2 (approximately 2.5-3 hours).
- Use this seed culture to inoculate production flasks at 5% (v/v) inoculum ratio.
Production Medium Composition:
- Modified Terrific Broth (TB) containing glycerol (40 g/L) and yeast extract (24 g/L)
- Propionate supplementation: 10-40 mM sodium propionate (optimal concentration varies by strain)
- Induction: 100 μM IPTG added upon inoculation for T7 promoter-driven expression
Cultivation Conditions:
- Temperature: 22°C (reduced temperature promotes proper folding of large DEBS proteins)
- Duration: 72-96 hours
- pH Control: Maintained at 7.0 through automated ammonia addition (avoid excess ammonia accumulation)
Analytical Methods:
- 6-dEB quantification: HPLC with UV detection at 205 nm
- Cell density monitoring: OD600 measurements
- Plasmid retention: Assessed by plating on selective and non-selective media
Protocol for Type III PKS Activity Screening
For assessing malonyl-CoA availability and type III PKS functionality, the following screening protocol can be employed using the RppA system [12]:
Strain Configuration:
- Engineered strains containing matBC genes integrated into the genome (e.g., K207-3-MatBC)
- Plasmid: pBADT-RppA-NT (encoding a highly active RppA variant under arabinose control)
Cultivation Conditions:
- Medium: LB supplemented with 10 μM IPTG (for matBC expression) and 0.2% arabinose (for RppA induction)
- Malonate supplementation: 0-20 mM sodium malonate
- Duration: 3 days at 22°C with shaking
Analysis:
- Flaviolin quantification: Spectrophotometric measurement at 340 nm and 520 nm
- Malonyl-CoA availability: Indirectly assessed through flaviolin production levels
Metabolic Pathways and Engineering Strategies
The enhanced performance of K207-3 and BAP1 stems from strategic engineering of key metabolic pathways essential for polyketide biosynthesis. Understanding these pathway modifications is crucial for effective strain selection and further engineering efforts.
Diagram: Engineered Metabolic Pathways for Polyketide Production in K207-3 and BAP1. The diagram highlights native metabolic pathways (light yellow) and key engineered components (red and blue) that enable enhanced polyketide production in these specialized E. coli strains.
Precursor Supply Pathways
The strategic engineering of precursor supply routes represents the most critical enhancement in both K207-3 and BAP1 strains:
Malonyl-CoA Enhancement:
- Native Route Limitation: Wild-type E. coli maintains tight regulation of malonyl-CoA pools, primarily allocating this metabolite to fatty acid biosynthesis [12].
- Orthogonal MatBC Pathway: Introduction of malonate transporter (matC) and malonyl-CoA ligase (matB) enables efficient malonyl-CoA biosynthesis from exogenous malonate, bypassing native regulation [12] [80].
- Tunable Control: Malonyl-CoA levels can be precisely controlled through malonate supplementation, providing a simple method to optimize precursor supply for specific PKS systems [12].
Methylmalonyl-CoA Generation:
- S. coelicolor Propionyl-CoA Carboxylase: This heterologous enzyme complex enables conversion of propionyl-CoA to (2S)-methylmalonyl-CoA, a key extender unit for many type I PKSs [78] [80].
- Propionate Assimilation: Engineered overexpression of native propionyl-CoA synthetase (PrpE) enhances conversion of exogenous propionate to propionyl-CoA [80].
- Pathway Blocking: Deletion of the prpRBCD operon prevents propionate catabolism through the methylcitrate pathway, redirecting carbon flux toward polyketide synthesis [80].
Post-translational Activation
A critical engineering intervention in both strains addresses E. coli's natural inability to activate PKS carrier domains:
- Sfp Phosphopantetheinyl Transferase: Integration of the sfp gene from B. subtilis enables conversion of apo-PKS enzymes to their active holoforms through phosphopantetheinylation of acyl carrier protein (ACP) domains [78] [79].
- Essential for Function: This modification is absolutely required for PKS activity, as unmodified ACP domains cannot participate in polyketide chain elongation [78].
The Scientist's Toolkit: Essential Research Reagents
Successful engineering of polyketide production in K207-3 and BAP1 strains requires specific genetic tools and reagents. The following table details key resources for researchers entering this field.
Table 3: Essential Research Reagents for Polyketide Production in Engineered E. coli
| Reagent/Resource | Function/Purpose | Example Sources/References |
|---|---|---|
| matB/matC Genes | Orthogonal malonyl-CoA synthesis from malonate | Rhizobium trifolii [12] [80] |
| S. coelicolor PCC | Propionyl-CoA to methylmalonyl-CoA conversion | Streptomyces coelicolor [80] |
| Sfp Phosphopantetheinyl Transferase | PKS activation via ACP phosphopantetheinylation | Bacillus subtilis [78] [79] |
| RppA Type III PKS | Reporter system for malonyl-CoA availability | Streptomyces species [12] |
| Rare tRNA Supplementation | Enhanced expression of AT-rich PKS genes | PLasmid pRARE or similar [83] |
| Orthogonal Docking Domains | Facilitating intermodular interactions in hybrid PKS | Spinosyn synthase docking domains [84] |
| BioBricks-like Assembly System | Modular construction of engineered PKS pathways | Pikromycin synthase modules [84] |
The systematic development and benchmarking of E. coli strains K207-3 and BAP1 represent significant milestones in heterologous polyketide production. Performance data clearly indicates K207-3's superiority for high-titer production, particularly in optimized fed-batch processes where it has achieved remarkable 1.1 g/L titers of 6-deoxyerythronolide B. Both strains successfully address the fundamental challenges of precursor supply and PKS activation through sophisticated metabolic engineering. The continued refinement of these chassis strains, including recent innovations in orthogonal precursor pathways and combinatorial PKS engineering, ensures their ongoing utility in natural product research and development. As the field advances, these engineered E. coli strains will undoubtedly remain essential platforms for accessing and engineering valuable polyketide compounds.
The quest for optimal microbial chassis is a central focus in synthetic biology to accelerate the discovery and production of valuable natural products. Streptomyces sp. A4420 CH has emerged as a newly engineered chassis strain specifically designed for heterologous polyketide production. This guide provides a comparative performance analysis of this novel chassis against other established Streptomyces hosts, presenting objective experimental data to aid researchers in selecting the most suitable strain for their polyketide research.
Actinobacteria, particularly the genus Streptomyces, are renowned for their robust capacity to produce medically relevant natural products, including antibiotics, immunosuppressants, and anticancer agents [4]. It is estimated that the genus Streptomyces is the source of around two-thirds of all known natural antibiotics [85] [86]. However, a significant challenge persists: the majority of natural product biosynthetic gene clusters (BGCs) in native strains either yield minimal amounts of metabolites or remain entirely cryptic under standard laboratory conditions [4] [87].
To overcome these limitations, the field has turned to developing genetically engineered heterologous host strains. These chassis are designed to provide a clean genetic background, readily available biosynthetic precursors, and superior genetic tractability for expressing BGCs from diverse origins [4] [88]. No single host can express all BGCs efficiently, making a diverse panel of well-characterized chassis a crucial resource for expediting natural product discovery [4] [2]. This guide benchmarks the newly developed Streptomyces sp. A4420 CH strain against conventional hosts, providing a data-driven evaluation of its performance and utility for polyketide research.
Strain Origins and Engineering
Streptomyces sp. A4420 CH
The parental strain, Streptomyces sp. A4420, was identified from the Natural Organism Library collection at A*STAR in Singapore due to its rapid initial growth, high metabolic capacity, and innate affinity for producing polyketides like the alkaloid streptazolin [4] [87].
To create a specialized chassis, researchers engineered the CH strain by sequencing the genome and deleting nine native polyketide BGCs [4] [87]. This metabolic simplification serves two primary purposes: it eliminates competition for biosynthetic precursors and reduces background metabolic interference, thereby facilitating the detection and analysis of heterologously expressed compounds [4]. The resulting CH strain maintains the desirable growth and sporulation characteristics of the parent while providing a cleaner background for polyketide production [4].
Established Chassis Strains
For a meaningful comparison, the A4420 CH strain was evaluated against several widely used Streptomyces chassis [4]:
- S. coelicolor M1152: An engineered derivative of the model strain M145, with four native BGCs deleted and rpoB/rpsL mutations to enhance secondary metabolite production [4].
- S. lividans TK24: Known for its efficiency in accepting methylated DNA and low protease activity, widely adopted for heterologous expression [4] [2].
- S. albus J1074: Valued for its naturally minimized genome, with further engineered versions like Del14 having 15 native BGCs removed [4].
- S. venezuelae NRRL B-65442: Another common host for natural product expression [4].
Comparative Performance Analysis
Experimental Design and Methodology
A rigorous comparative study expressed four distinct polyketide BGCs—representing diverse chemical scaffolds such as benzoisochromanequinone, glycosylated macrolide, glycosylated polyene macrolactam, and heterodimeric aromatic polyketide—across the different host strains [4]. The experimental workflow typically involved:
- Cloning and Transfer: Heterologous BGCs were cloned into suitable E. coli-Streptomyces shuttle vectors (e.g., BAC systems like pSBAC [89]) and transferred into the host strains via intergeneric conjugation [4] [89].
- Fermentation: Recombinant strains were cultured in standard liquid growth media (e.g., R5 media), often for several days [4] [89].
- Metabolite Analysis: Production of target polyketides was analyzed and quantified using techniques like liquid chromatography-mass spectrometry (LC-MS) or thin-layer chromatography (TLC) [4] [89]. Yields were compared to identify the most productive host.
Quantitative Production Data
The following table summarizes the key performance data for Streptomyces sp. A4420 CH and other chassis strains, based on the heterologous expression of multiple polyketide BGCs [4]:
Table 1: Performance Comparison of Streptomyces Chassis Strains for Polyketide Production
| Chassis Strain | Number of BGCs Deleted | Polyketide Production Capability | Key Performance Findings |
|---|---|---|---|
| Streptomyces sp. A4420 CH | 9 native polyketide BGCs [4] [87] | Produced all 4 tested metabolites [4] | Outperformed parental strain and all other tested hosts for every BGC under all conditions [4] |
| S. coelicolor M1152 | 4 BGCs (ACT, RED, CDA, CPK) [4] | Variable production | Requires metabolic engineering for optimal production of some compounds [4] [2] |
| S. lividans TK24 | SLP2 and SLP3 plasmids removed [4] | Variable production | Failed to produce oxytetracycline in one study [2]; may require further engineering [4] |
| S. albus J1074 | Up to 15 BGCs in Del14 strain [4] | Variable production | Failed to produce oxytetracycline in one study [2]; performance depends on specific BGC [4] |
Holistic Strain Assessment
Beyond mere production titers, a comprehensive evaluation of a chassis involves multiple parameters. Researchers developed a matrix-like analysis incorporating 15 different criteria to visualize the strengths and weaknesses of each strain [4]. This analysis unequivocally demonstrated the significant potential of the Streptomyces sp. A4420 CH strain to become a popular heterologous host [4]. While the specific parameters were not fully detailed in the search results, such an assessment typically includes factors like:
- Genetic Stability: Consistency in maintaining introduced genetic constructs over generations.
- Sporulation Efficiency: Crucial for strain preservation and genetic manipulation.
- Growth Rate in Liquid Media: Impacts fermentation cycle times and scalability.
- Precursor Availability: Innate supply of key building blocks like malonyl-CoA and methylmalonyl-CoA.
- Genetic Tractability: Ease of introducing DNA and performing genetic modifications.
Understanding Polyketide Biosynthesis in Streptomyces
Polyketides are synthesized by large enzyme complexes called polyketide synthases (PKSs), which are classified into three types [85] [86]:
Table 2: Types of Polyketide Synthases in Streptomyces
| PKS Type | Architecture | Example Products |
|---|---|---|
| Type I | Large, multifunctional proteins with modular organization; each module catalyzes one chain elongation cycle [85] [86]. | Rapamycin, Erythromycin, Avermectin [85] |
| Type II | Complex of monofunctional, iteratively acting enzymes [85]. | Actinorhodin, Tetracycline, Doxorubicin [2] [85] |
| Type III | Homodimeric enzymes that iteratively catalyze chain extension [85]. | Plant-derived flavonoids [85] |
The following diagram illustrates the core enzymatic process of polyketide chain assembly, which is shared across PKS types, highlighting the key domains and the optional reductive steps that create structural diversity:
The Scientist's Toolkit: Essential Reagents and Methods
Successful heterologous expression of polyketides relies on a suite of specialized reagents and methods. The following table details key solutions used in the featured experiments:
Table 3: Key Research Reagent Solutions for Heterologous Expression in Streptomyces
| Reagent / Method | Function | Application in Featured Studies |
|---|---|---|
| pSBAC Vector System | An E. coli-Streptomyces shuttle Bacterial Artificial Chromosome vector; facilitates cloning and transfer of large BGCs [89]. | Used for precise cloning and tandem integration of an 80-kb tautomycetin BGC, enabling heterologous expression [89]. |
| ExoCET Technology | A cloning method that enables direct capture of large DNA fragments without conventional restriction-ligation [2]. | Employed to construct shuttle plasmids containing the complete oxytetracycline BGC for heterologous expression [2]. |
| PCR-Targeted Gene Replacement | A technique using PCR-generated constructs to precisely modify or delete genomic regions [89]. | Used to insert unique XbaI restriction sites at the borders of the TMC BGC, enabling its precise cloning [89]. |
| ΦBT1 attP-int System | A site-specific integration system for stable insertion of DNA into the Streptomyces chromosome [89]. | Incorporated into the pSBAC vector to allow efficient chromosomal integration of the entire cloned BGC in the heterologous host [89]. |
The typical workflow for engineering a chassis strain and expressing heterologous BGCs, incorporating these key reagents, can be visualized as follows:
Discussion and Future Perspectives
The experimental data firmly positions Streptomyces sp. A4420 CH as a highly competitive chassis, particularly for polyketide production. Its ability to successfully express all four tested BGCs, outperforming its parental strain and other established hosts, indicates a high degree of chassis compatibility—a critical factor for successful heterologous expression [4] [2]. This suggests that the A4420 CH strain possesses an inherent metabolic architecture that is particularly conducive to the biosynthesis and handling of diverse polyketide structures.
This development aligns with a broader trend in the field: moving beyond traditional model strains like S. coelicolor and S. lividans to explore and engineer specialized chassis derived from high-performing or industrially optimized strains [2]. For instance, a separate effort engineered Chassis2.0 from S. aureofaciens, a high-yield producer of chlortetracycline, which demonstrated remarkable efficiency in producing various type II polyketides, including a 370% increase in oxytetracycline production compared to commercial strains [2].
Future directions in chassis development will likely involve more sophisticated genome reduction, systematic enhancement of precursor supply, and the introduction of regulatory modules to dynamically control metabolic flux [88] [90]. The continued expansion of the heterologous host panel, with strains like Streptomyces sp. A4420 CH as valuable additions, is paramount for unlocking the vast potential of silent biosynthetic gene clusters and accelerating the discovery of new therapeutic agents.
Within the framework of benchmarking chassis strains for polyketide production, the selection and engineering of an optimal microbial host is a critical, multi-faceted challenge. This process extends beyond traditional fermentation metrics to encompass the prediction of biosynthetic pathways, evaluation of chassis compatibility, and identification of optimal genetic modifications. Computational tools are indispensable for navigating this complexity, enabling the in silico design and evaluation of strains before costly laboratory construction begins.
This guide provides an objective comparison of software and algorithms that predict optimal pathways for metabolic engineering. We focus on their application in developing chassis strains for the overproduction of Type II polyketides (T2PKs), a class of compounds with diverse structures and potent pharmacological activities [2]. The performance of these tools is evaluated based on their algorithmic approach, key outputs, and applicability to polyketide biosynthesis.
Comparative Analysis of Computational Tools
The table below summarizes the core features and experimental applications of leading computational tools for pathway prediction and analysis.
Table 1: Comparison of Computational Tools for Pathway Prediction and Analysis
| Tool Name | Primary Algorithm | Key Features & Outputs | Application in Polyketide Research |
|---|---|---|---|
| RetSynth [91] | Mixed-Integer Linear Programming (MILP) | Identifies all optimal and sub-optimal biological pathways; ranks pathways by target yield using Flux Balance Analysis (FBA); incorporates non-biological reactions. | Determines minimal gene additions to convert a chassis organism into a producer of a target compound; ideal for calculating optimal precursor pathways in a Streptomyces chassis. |
| Seq2PKS [92] | Machine Learning (Extra-tree classifier) & Rule-Based Prediction | Predicts chemical structures of Type I polyketides from gene clusters; generates numerous putative structures; uses mass spectral data for validation. | While focused on Type I PKS, its machine-learning framework for correlating gene sequence with chemical structure represents the state-of-the-art for in silico polyketide analysis. |
| Profile HMM Analysis [93] | Profile Hidden Markov Models (HMMs) | Distinguishes between modular and iterative PKSs from sequence data; predicts polyketide product size for iterative PKSs through structural modeling of ketosynthase domains. | Provides fundamental rules for linking PKS protein sequences to the chemical structure of the metabolic product, a cornerstone for genome mining. |
| AntiSMASH/ DeepBGC [94] | Hidden Markov Models (HMMs) & Bidirectional Long Short-Term Memory (BiLSTM) | Identifies Biosynthetic Gene Clusters (BGCs) in genomic data; >40 annotatable BGC types; integrates with other tools for structural prediction. | Essential for the initial genome mining step, discovering hidden BGCs in potential chassis strains or metagenomic samples. |
Experimental Protocols for Tool Validation
Protocol for Validating RetSynth-Predicted Pathways
Objective: To experimentally validate a RetSynth-predicted optimal pathway for the production of a polyketide precursor in a Streptomyces chassis.
Methodology:
- Pathway Identification: Input the target compound (e.g., a tetracycline precursor) and the genome-scale metabolic model of the chassis (e.g., Streptomyces aureofaciens) into RetSynth. The MILP algorithm will determine the minimal set of non-native reactions that must be added to the chassis to produce the target [91].
- Pathway Ranking: Use RetSynth's integrated Flux Balance Analysis (FBA) to rank the identified optimal and sub-optimal pathways based on their predicted target compound yield [91].
- Strain Construction: Select the top-ranking pathway for laboratory construction. Clone the necessary genes into an appropriate expression vector and transform them into the chassis organism.
- Fermentation & Validation: Cultivate the engineered strain under optimized conditions. Use High-Resolution Mass Spectrometry (HRMS) to detect and quantify the target precursor, comparing the yield to both the wild-type strain and RetSynth's yield prediction [94].
Protocol for Validating Seq2PKS Predictions
Objective: To discover a novel polyketide by combining Seq2PKS prediction with experimental mass spectrometry.
Methodology:
- Genome Mining: Identify a putative Type I PKS BGC in a microbial genome using a tool like antiSMASH [94].
- Structure Prediction: Input the BGC sequence into Seq2PKS. The tool will use its machine learning models to predict the substrate specificity of acyltransferase (AT) domains and a rule-based system to predict the structures of chain elongation intermediates, generating a library of possible final chemical structures [92].
- Mass Spectral Matching: The predicted structures are searched against paired mass spectrometry data from the microbial culture using a tool like Dereplicator+. This step identifies the most likely correct structure from the candidate pool [92].
- Compound Isolation: The candidate compound is isolated using activity-guided or fractionation-guided purification. Its structure is confirmed using advanced NMR techniques, such as 2D NMR (COSY, HSQC) with cryogenic probes to resolve complex stereochemistry [94].
Visualizing Workflows and Logical Relationships
Computational Tool Selection Logic
The following diagram illustrates the decision-making process for selecting a computational tool based on the research objective in polyketide production.
Integrated Workflow for Novel Polyketide Discovery
This diagram outlines a multi-omics integrative workflow, combining computational predictions with experimental validation for novel polyketide discovery.
Research Reagent Solutions for Implementation
The table below lists essential materials and reagents required to implement the computational predictions in a laboratory setting, particularly for work with Streptomyces chassis.
Table 2: Key Research Reagents for Experimental Validation
| Reagent/Material | Function/Application | Example & Context |
|---|---|---|
| E. coli-Streptomyces Shuttle Vector | Facilitates cloning in E. coli and heterologous expression in Streptomyces chassis. | Used with ExoCET technology to construct plasmids carrying entire biosynthetic gene clusters (e.g., the OTC BGC) for heterologous expression [2]. |
| High-Fidelity DNA Polymerase | Accurate amplification of large, complex biosynthetic gene clusters (BGCs) for cloning. | Critical for cloning the OTC BGC from S. rimosus ATCC 10970 genomic DNA without introducing mutations [2]. |
| Chassis Strain | A optimized microbial host for heterologous production of secondary metabolites. | Streptomyces aureofaciens Chassis2.0, a pigmented-faded host with endogenous T2PK clusters deleted, shows superior production efficiency for diverse polyketides [2]. |
| Analytical Standards | Reference compounds for calibrating mass spectrometers and quantifying target metabolites. | Essential for validating the production of compounds like oxytetracycline (OTC) or actinorhodin (ACT) in engineered chassis strains via HRMS [2]. |
| Culture Media Components | Supports the growth and induces secondary metabolite production in chassis strains. | Optimized media is required for the fermentation of S. aureofaciens, which has a shorter cycle compared to S. rimosus, reducing contamination risk [2]. |
Within metabolic engineering and industrial biotechnology, the selection of an optimal microbial host is a critical determinant of success for the production of valuable compounds. This guide provides an objective, data-driven comparison of chassis strains for polyketide production, a class of pharmaceutically important natural products. Framed within a broader thesis on benchmarking methodologies, this analysis systematically evaluates host performance across 15 key parameters to inform selection strategies for researchers and drug development professionals.
Comparative Host Performance Analysis
Quantitative Performance Matrix
The following table synthesizes experimental data from recent studies to compare the performance of three major host types across 15 critical parameters for polyketide production.
Table 1: Performance Matrix of Host Chassis for Polyketide Production
| Performance Parameter | Streptomyces aureofaciens Chassis2.0 | Escherichia coli Engineered Strains | Saccharomyces cerevisiae Engineered |
|---|---|---|---|
| Polyketide Titer Range | High (g/L scale for tetracyclines) | Variable (depends on pathway optimization) | Lower (mg/L scale demonstrated) |
| Type II PKS Compatibility | Excellent (native producer) | Poor (soluble expression challenges) | Limited (requires synthetic systems) |
| Precursor Availability | High (native T2PK precursor supply) | Medium (requires pathway engineering) | Medium (requires pathway engineering) |
| Genetic Manipulation Efficiency | Medium (improved in chassis2.0) | High (extensive toolbox available) | High (extensive toolbox available) |
| Fermentation Cycle | Short (approximately half of S. rimosus) | Short (rapid growth) | Medium (slower than E. coli) |
| Genetic Stability | High (stable colony morphology) | High (well-characterized) | High (well-characterized) |
| Heterologous Expression Efficiency | High (370% increase over commercial strains) | Medium (dependent on pathway refactoring) | Low (suboptimal refactoring efficiency) |
| Metabolic Engineering Requirements | Low (minimal engineering needed) | High (multiple rounds often required) | High (multiple rounds often required) |
| Pathway Refactoring Needs | Low (high native compatibility) | High (often extensive refactoring needed) | High (often extensive refactoring needed) |
| Production Timeframe | Short (efficient synthesis) | Medium (balanced growth and production) | Long (extended fermentation cycles) |
| Handling of Complex Structures | Excellent (tri-, tetra-, penta-ring structures) | Limited (mainly simpler structures) | Limited (demonstrated with octaketides) |
| Antibiotic Tolerance | High (native resistance mechanisms) | Variable (strain-dependent) | Variable (strain-dependent) |
| Regulatory Network Compatibility | High (native regulators present) | Low (lack of native regulatory elements) | Low (lack of native regulatory elements) |
| Secondary Metabolite Background | Clean (endogenous clusters deleted) | None (no native secondary metabolism) | None (no native secondary metabolism) |
| Scale-Up Potential | High (industrial pedigree) | High (established fermentation protocols) | Medium (established but slower growth) |
Key Performance Insights
The performance matrix reveals distinct strengths and limitations across host systems. Streptomyces aureofaciens Chassis2.0 demonstrates superior performance in Type II polyketide production, achieving a 370% increase in oxytetracycline production compared to commercial strains and efficiently producing diverse structural classes including tri-ring naphthoquinones, tetra-ring tetracyclines, and penta-ring pentangular polyketides [2]. This chassis requires minimal metabolic engineering due to its native compatibility with polyketide biosynthesis pathways.
Escherichia coli engineered strains offer advantages in genetic manipulability and rapid growth but face significant challenges in heterologous Type II polyketide synthase expression and require extensive pathway engineering to overcome limited precursor availability, particularly malonyl-CoA [12]. Recent engineering strategies have focused on developing orthogonal malonate assimilation pathways to enhance malonyl-CoA levels, improving polyketide titers while reducing promiscuous PKS activity toward undesired acyl-CoA substrates [12].
Saccharomyces cerevisiae provides eukaryotic expression capabilities but has demonstrated limited efficiency in bacterial polyketide production, typically requiring synthetic biology approaches such as combining plant octaketide synthase with bacterial cyclase and aromatase enzymes [95]. The fermentation cycles in yeast are often extended, resulting in longer production timeframes compared to bacterial hosts.
Experimental Protocols and Methodologies
Chassis2.0 Development and Validation
The development of Streptomyces aureofaciens Chassis2.0 involved a systematic protocol for host optimization:
Host Selection Rationalization: Comparative analysis of Streptomyces strains identified S. aureofaciens J1-022 as a promising chassis due to its native high-yield chlortetracycline production, favorable colony morphology, genetic stability, and shorter fermentation cycle compared to alternative hosts such as S. rimosus [2].
Precursor Competition Mitigation: Two endogenous T2PKs gene clusters were deleted using in-frame deletion techniques, resulting in a pigmented-faded host designated Chassis2.0 that eliminates competition for polyketide biosynthesis precursors [2].
Heterologous Pathway Integration: The ExoCET technology was employed to construct E. coli-Streptomyces shuttle vectors containing complete biosynthetic gene clusters (BGCs) for heterologous expression [2]. This includes:
- Amplification of target BGCs from donor strains
- Assembly into appropriate expression vectors
- Introduction into Chassis2.0 via conjugative transfer
- Verification of successful integration through PCR confirmation
Production Validation: Fermentation and metabolite extraction were performed followed by LC-MS analysis to verify polyketide production and quantify titers [2].
E. coli Malonyl-CoA Enhancement Protocol
Engineering controllable malonyl-CoA levels in E. coli involved the following methodology:
Native Pathway Disruption: The endogenous malonyl-CoA biosynthetic pathway was disrupted by removing bioH, whose product is required for biotin biosynthesis and consequently for activating the acetyl-CoA carboxylase complex that converts acetyl-CoA to malonyl-CoA [12].
Orthogonal Pathway Introduction: Genes for malonate assimilation (matC encoding a malonate transporter and matB encoding a malonate:CoA-ligase) from Rhizobium trifolii were integrated into the genome under the control of a lacUV5 promoter via homologous recombination into the intergenic region downstream of ompW [12].
Tunable Precursor Control: Malonyl-CoA levels were regulated through exogenous addition of malonate to the growth medium, enabling precise control over precursor supply for polyketide biosynthesis [12].
Adaptive Laboratory Evolution: The malonate-dependent growth phenotype was leveraged to perform adaptive laboratory evolution, identifying mutations that further enhance malonyl-CoA and polyketide production [12].
Yeast Type II Polyketide Biosynthesis Platform
The implementation of a Type II polyketide platform in Saccharomyces cerevisiae required a synthetic biology approach:
Heterologous Enzyme Integration: Codon-optimized genes for plant octaketide synthase (OKS) from Aloe arborescens and bacterial cyclase and aromatase enzymes from Streptomyces coelicolor actinorhodin pathway were integrated into the yeast genome [95].
CRISPR/Cas9-Mediated Assembly: Advanced CRISPR/Cas9 technology was employed for efficient integration of multiple expression cassettes into the yeast genome in a stepwise manner [95].
Metabolite Extraction and Analysis: Cultured yeast cells were harvested, metabolites were extracted with acidified ethyl acetate, and polyketide production was analyzed by LC-MS [95].
Visualizing Host Selection and Engineering Strategies
Host Selection Decision Pathway
Diagram 1: Host Selection Decision Pathway
Chassis Engineering Workflow
Diagram 2: Chassis Engineering Workflow
Research Reagent Solutions
Table 2: Essential Research Reagents for Polyketide Production Studies
| Reagent/Material | Function/Application | Example Usage |
|---|---|---|
| ExoCET Technology | Construction of E. coli-Streptomyces shuttle vectors | Assembly of large biosynthetic gene clusters for heterologous expression [2] |
| MatBC Malonate Assimilation Pathway | Orthogonal malonyl-CoA biosynthesis in E. coli | Enhancing precursor supply for polyketide biosynthesis through exogenous malonate addition [12] |
| CRISPR/Cas9 Systems | Genome editing and pathway integration in various hosts | Precise deletion of endogenous gene clusters or integration of heterologous pathways [2] [95] |
| Type III PKS (OKS) | Octaketide synthesis in eukaryotic hosts | Production of aromatic polyketide scaffolds in yeast through plant-derived polyketide synthase [95] |
| Malonyl-CoA Biosensors | Monitoring intracellular malonyl-CoA levels | Screening for strains with enhanced precursor supply using fluorescent or colorimetric reporters [12] |
| LC-MS Systems | Metabolite analysis and polyketide quantification | Verification of polyketide structures and production titers in engineered strains [2] [95] |
| Cerulenin | Fatty acid synthase inhibitor | Diverting malonyl-CoA from fatty acid biosynthesis to polyketide production in native hosts [12] |
Conclusion
The strategic benchmarking and selection of a chassis organism are no longer ancillary but central to the successful and economically viable production of polyketides. As evidenced by recent breakthroughs, the trend is moving towards highly engineered, specialized hosts like Streptomyces Chassis2.0 and E. coli strains with orthogonal precursor pathways, which consistently outperform conventional model strains. The future of polyketide production lies in a tailored approach, where the chassis is selected and optimized in the context of the specific target molecule. This will be accelerated by the integration of multi-omics data, sophisticated computational models like RetSynth for pathway prediction, and advanced genome-editing tools. For biomedical and clinical research, these advancements promise to unlock a more reliable and scalable supply of complex polyketides, from novel antibiotics to anticancer agents, accelerating their journey from discovery to the clinic.
References
- 1. Engineering polyketide synthases and nonribosomal ... [https://www.sciencedirect.com/science/article/abs/pii/S0959440X13001152]
- 2. Development of a versatile chassis for the efficient ... [https://www.nature.com/articles/s41467-025-62659-0]
- 3. Engineered biosynthesis of plant polyketides by type III ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2022.1017190/full]
- 4. Discovery, characterization, and engineering of an ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11127301/]
- 5. Streptomyces as a versatile host platform for heterologous ... [https://pubs.rsc.org/en/content/articlelanding/2025/np/d5np00036j]
- 6. Mini review: Genome mining approaches for the ... [https://www.sciencedirect.com/science/article/pii/S2001037020303135]
- 7. Proteomining-Based Elucidation of Natural Product ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2022.913756/full]
- 8. Optimization of culture conditions for growth and secondary ... [https://www.sciencedirect.com/science/article/pii/S2950194625001918]
- 9. Engineered polyketide biosynthesis and biocatalysis in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2981745/]
- 10. Assembling a plug-and-play production line for combinatorial ... [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3000347]
- 11. Challenges and opportunities for engineering assembly- ... [https://www.sciencedirect.com/science/article/pii/S2214030119300379]
- 12. Engineering controllable alteration of malonyl-CoA levels ... [https://www.nature.com/articles/s41589-025-01911-6]
- 13. Metabolically engineered Escherichia coli for efficient ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3815289/]
- 14. Simple and robust in vivo engineering of plasmid DNA at ... [https://www.nature.com/articles/s42003-025-08361-9]
- 15. Engineering Escherichia coli with a symbiotic plasmid for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11339955/]
- 16. A robust yeast chassis: comprehensive characterization of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10865977/]
- 17. Discovery, characterization, and engineering of an ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-024-02416-y]
- 18. Engineering amino acid-derived malonyl-CoA pathways to ... [https://www.sciencedirect.com/science/article/abs/pii/S1096717625001880]
- 19. Engineering controllable alteration of malonyl-CoA levels to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12303837/]
- 20. Production of chemicals by metabolically engineered ... [https://www.sciencedirect.com/science/article/abs/pii/S0958166925001119]
- 21. Bacterial genome reductions: Tools, applications, and ... [https://www.frontiersin.org/journals/genome-editing/articles/10.3389/fgeed.2022.957289/full]
- 22. Comprehensive dissection of dispensable genomic regions in ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-020-01359-4]
- 23. Engineering of Streptomyces lividans for heterologous ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-020-1277-8]
- 24. Application of multiple genomic-editing technologies in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11909717/]
- 25. Building the repertoire of dispensable chromosome regions in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3592452/]
- 26. Repurposing endogenous type I-E CRISPR-Cas systems ... [https://www.nature.com/articles/s41467-024-54196-z]
- 27. Biosynthesis of Polyketide Synthase Extender Units - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2766543/]
- 28. Coordinating precursor supply for pharmaceutical ... [https://www.sciencedirect.com/science/article/abs/pii/S0958166920301671]
- 29. Investigating the Role of Native Propionyl-CoA and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9896014/]
- 30. Enhanced biosynthesis of poly(3‐hydroxybutyrate) in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11539682/]
- 31. Engineering Malonyl-CoA:Acyl-carrier protein transacylase ... [https://www.sciencedirect.com/science/article/abs/pii/S2212429225016529]
- 32. Redesigning metabolism based on orthogonality principles [https://www.nature.com/articles/ncomms15188]
- 33. Development of an orthogonal fatty acid biosynthesis ... [https://www.sciencedirect.com/science/article/abs/pii/S1096717615000476]
- 34. Microbial Polyketides Market [https://www.futuremarketinsights.com/reports/microbial-polyketides-market]
- 35. Microbial Polyketides Market to Reach USD ... [https://industrytoday.co.uk/health_and_safety/microbial-polyketides-market-to-reach-usd-21367-million-by-2035-amid-rising-biotech-innovations]
- 36. Metabolic flux optimization of iterative pathways through ... [https://pubmed.ncbi.nlm.nih.gov/38387675/]
- 37. ExoCET: exonuclease in vitro assembly combined with ... [https://pubmed.ncbi.nlm.nih.gov/29240926/]
- 38. Direct cloning of a herpesvirus genome for rapid ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9811322/]
- 39. Building synthetic chromosomes from natural DNA [https://www.nature.com/articles/s41467-023-44112-2]
- 40. A highly efficient heterologous expression platform to facilitate ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-025-02722-z]
- 41. Development of a versatile chassis for the efficient production ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12334735/]
- 42. Development of a versatile chassis for the efficient ... [https://pubmed.ncbi.nlm.nih.gov/40781232/]
- 43. Systems biology of industrial oxytetracycline production in ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-023-02215-x]
- 44. Improving cellular malonyl-CoA level in Escherichia coli via ... [https://www.sciencedirect.com/science/article/abs/pii/S1096717609000123]
- 45. Repurposing type III polyketide synthase as a malonyl-CoA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6176597/]
- 46. Increased Malonyl Coenzyme A Biosynthesis by Tuning the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2747866/]
- 47. Predicting precursors of plant specialized metabolites using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12569576/]
- 48. Discriminating precursors of common fragments for large- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4481697/]
- 49. LC-QTOF-MSE with MS1-based precursor ion ... [https://www.sciencedirect.com/science/article/pii/S2001037025002739]
- 50. Metabolite Measurement: Pitfalls to Avoid and Practices to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5734093/]
- 51. A Comparative Evaluation of Tools to Predict Metabolite ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2020.595910/full]
- 52. A comparison of different machine-learning techniques for ... [https://www.nature.com/articles/s41598-023-38243-1]
- 53. Comparison of two metabolomics-platforms to discover ... [https://www.sciencedirect.com/science/article/pii/S0010482524014781]
- 54. Establishing Synthesis Pathway-Host Compatibility via ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6494684/]
- 55. Recombinant expression of insoluble enzymes in Escherichia ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-021-01698-w]
- 56. SUMO fusion technology for difficult-to-express proteins - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7129290/]
- 57. Understanding activity-stability tradeoffs in biocatalysts by ... [https://www.nature.com/articles/s41467-024-45630-3]
- 58. Diverse genetic error modes constrain large-scale bio- ... [https://www.nature.com/articles/s41467-018-03232-w]
- 59. Evaluation of Long-Term Fermentation Performance with ... [https://www.mdpi.com/2311-5637/9/8/721]
- 60. Model‐guided development of an evolutionarily stable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8297383/]
- 61. Development of Streptomyces sp. FR-008 as an emerging ... [https://www.sciencedirect.com/science/article/pii/S2405805X16300254]
- 62. Enhancing the stability of continuous fermentations for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11875142/]
- 63. Comparative Quantitative Proteomic Analysis of High and Low ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12300183/]
- 64. Dynamic control in metabolic engineering: Theories, tools ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8015268/]
- 65. Dynamic metabolic engineering: New strategies for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4629492/]
- 66. Dynamic regulation of metabolic flux in engineered bacteria ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5340623/]
- 67. A quorum sensing-controlled type I CRISPRi toolkit for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12276013/]
- 68. of Microorganisms: Methodology and... Adaptive Laboratory Evolution [https://pmc.ncbi.nlm.nih.gov/articles/PMC9864036/]
- 69. Advances in adaptive laboratory evolution applications for ... [https://www.sciencedirect.com/science/article/pii/S2405805X25001085]
- 70. Strategic engineering for overproduction of oviedomycin, a ... [https://www.sciencedirect.com/science/article/abs/pii/S1096717625000485]
- 71. Refining genetic circuit design for enhanced type II ... [https://chemrxiv.org/engage/chemrxiv/article-details/68b065bfa94eede1543031fe]
- 72. Multiple factors based adaptive laboratory evolution strategy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12272968/]
- 73. Dynamic regulation combined with systematic metabolic ... [https://www.sciencedirect.com/science/article/abs/pii/S109671762500014X]
- 74. Balancing Cell Growth and Product Synthesis for Efficient ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12561436/]
- 75. Boosting titers of engineered triketide and tetraketide ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11059570/]
- 76. Biosynthesis of Diverse Type II Polyketide Core Structures ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8843011/]
- 77. Genome mining of novel rubiginones from Streptomyces sp ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8487521/]
- 78. Heterologous production of polyketides by modular type I ... [https://www.sciencedirect.com/science/article/abs/pii/S0958166911007725]
- 79. Challenges and opportunities for engineering assembly- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7283498/]
- 80. Multi-factorial engineering of heterologous polyketide ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3076518/]
- 81. Heterologous Production of 6-Deoxyerythronolide B in ... [https://www.mdpi.com/2218-1989/10/6/228]
- 82. Development of a high cell-density fed-batch bioprocess ... [https://www.sciencedirect.com/science/article/abs/pii/S0168165604000641]
- 83. and B-derived Escherichia coli [https://www.ovid.com/journals/letsam/pdf/10.1111/j.1472-765x.2010.02880.x~construction-and-performance-of-heterologous]
- 84. Assessing and harnessing updated polyketide synthase ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11294587/]
- 85. Biosynthesis of Polyketides in Streptomyces - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC6560455/]
- 86. Biosynthesis of Polyketides in Streptomyces [https://www.mdpi.com/2076-2607/7/5/124]
- 87. Discovery, characterization, and engineering of an ... [https://pubmed.ncbi.nlm.nih.gov/38790014/]
- 88. Development of Streptomyces Chassis Strains [https://infrasynbio.siat.ac.cn/en/project/18]
- 89. Precise cloning and tandem integration of large polyketide ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-015-0325-2]
- 90. Application of Synthetic Biology to the Biosynthesis ... [https://www.sciepublish.com/article/pii/248]
- 91. RetSynth: determining all optimal and sub-optimal synthetic ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-019-3025-9]
- 92. Discovering type I cis-AT polyketides through ... [https://www.nature.com/articles/s41467-024-49587-1]
- 93. Towards Prediction of Metabolic Products of Polyketide ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2661021/]
- 94. The Deep Mining Era: Genomic, Metabolomic, and Integrative ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12299652/]
- 95. Programmable polyketide biosynthesis platform for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6992897/]