Comparative Analysis of Objective Functions for Flux Prediction: From FBA to Machine Learning
This article provides a comprehensive comparative analysis of objective functions used in metabolic flux prediction, a critical task for researchers, scientists, and drug development professionals.
Comparative Analysis of Objective Functions for Flux Prediction: From FBA to Machine Learning
Abstract
This article provides a comprehensive comparative analysis of objective functions used in metabolic flux prediction, a critical task for researchers, scientists, and drug development professionals. We explore the foundational principles of constraint-based modeling, including Flux Balance Analysis (FBA), and the pivotal role that the choice of objective function plays in determining accurate flux distributions. The scope extends to traditional methods like parsimonious FBA and the emerging paradigm of machine learning-based approaches, such as artificial neural networks, which offer rapid and accurate flux computations. We systematically address troubleshooting and optimization strategies for selecting and refining objective functions and conclude with robust validation and model selection frameworks to guide reliable flux analysis in biomedical and clinical research.
The Critical Role of Objective Functions in Constraint-Based Metabolic Modeling
Flux Balance Analysis (FBA) is a powerful mathematical approach for analyzing the flow of metabolites through metabolic networks. This method uses optimization to predict how a biological system, from single cells to complex communities, distributes metabolic fluxes to achieve a specific biological objective, such as maximizing growth or the production of a target metabolite [1] [2]. By relying on the stoichiometry of the network and constraints, FBA can make quantitative predictions without requiring detailed kinetic parameters, making it particularly valuable for studying genome-scale models [2].
This guide provides a comparative analysis of the core principles of FBA, with a special focus on the critical role and selection of the objective function.
Core Mathematical Principles of FBA
FBA is built upon a constraint-based modeling framework. The core idea is that an organism's metabolism must operate within physical and chemical constraints, which define a set of possible metabolic behaviors.
The Stoichiometric Matrix and Mass Balance
The metabolic network is mathematically represented by the stoichiometric matrix (S). In this matrix, each row represents a unique metabolite and each column represents a biochemical reaction. The entries in each column are the stoichiometric coefficients of the metabolites involved in that reaction (negative for consumed metabolites, positive for produced metabolites) [2].
A fundamental constraint in FBA is the steady-state assumption, which posits that the concentration of internal metabolites does not change over time. This is represented by the mass balance equation: Sv = 0 where v is the vector of all reaction fluxes in the network [1] [2]. This equation ensures that for each metabolite, the total rate of production equals the total rate of consumption.
Linear Programming and Constraints
FBA formulates metabolism as a Linear Programming (LP) problem. The steady-state equation, along with additional capacity constraints on reaction fluxes (vmin ≤ v ≤ vmax), defines the "solution space" of all possible metabolic flux distributions that the network can achieve [1] [2].
The LP problem is solved to find a single flux distribution that optimizes a defined biological goal. The general formulation is:
- Objective: Maximize/Minimize Z = cTv
- Subject to: Sv = 0 and vmin ≤ v ≤ vmax
Here, c is a vector of weights that defines the objective function, specifying which reaction(s) are to be optimized [2].
Comparative Analysis of Objective Functions
The choice of objective function is paramount, as it steers the optimization toward a particular flux distribution within the solution space. Different biological assumptions and research questions call for different objective functions. The table below summarizes commonly used objective functions and their applications.
Table: Comparison of Key Objective Functions in Flux Balance Analysis
| Objective Function | Mathematical Form (cTv) | Biological Rationale | Typical Application Context | Performance Notes |
|---|---|---|---|---|
| Maximize Biomass Production | Maximize flux through the biomass reaction | Simulates natural selection for maximal growth rate | Standard for predicting microbial growth in nutrient-rich conditions | Often produces realistic growth rates; may not predict all internal fluxes accurately [3] |
| Maximize ATP Production | Maximize total flux of ATP-generating reactions | Assumes cells evolve to maximize energy yield | Studying energy metabolism; conditions where energy is limiting | Can improve predictions in energy-limited environments or for lifespan analysis in yeast models [3] |
| Minimize Total Flux (Parsimony) | Minimize the sum of absolute values of all fluxes | Assumes cells have evolved to be metabolically efficient (use minimal protein/enzyme cost) | Finding the most efficient pathway usage; often used as a secondary objective | Can refine predictions by eliminating unrealistic flux loops; improves lifespan predictions in yeast models [3] |
| Minimize Nutrient Uptake | Minimize flux of a substrate uptake reaction (e.g., glucose) | Assumes efficiency in substrate utilization | Modeling nutrient-scarce environments | Directly optimizes for substrate use efficiency rather than a growth or energy output |
| Multi-Objective Optimization | e.g., Maximize growth, then minimize total flux (lexicographic method) | Combines multiple selective pressures | Generating more realistic, context-specific flux distributions | Can provide a more balanced and biologically realistic solution than single objectives [3] |
Experimental Protocols for FBA
The following is a generalized protocol for setting up and solving an FBA problem, which can be implemented using computational tools like the COBRA Toolbox in MATLAB or similar packages in Python [1] [2].
The following diagram illustrates the key steps in a typical FBA simulation.
Detailed Methodology
- Define the Metabolic Network Reconstruction: The foundation of any FBA study is a high-quality genome-scale metabolic reconstruction. This model is represented as a stoichiometric matrix S [2].
- Apply Physiochemical Constraints:
- Define the Biological Objective: Select an appropriate objective function based on the biological question. For example, to simulate maximal growth, the vector c is set to zero for all reactions except the biomass reaction, which is set to 1 [2].
- Solve the Linear Programming Problem: Use an LP solver to find the flux distribution v that satisfies all constraints and optimizes the objective function Z [1] [2].
- Output and Validation: The primary output is the flux vector v. The predictions, such as growth rate or metabolite secretion, should be compared with experimental data (e.g., from chemostat cultures) for validation [4] [2].
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table lists key resources required for conducting FBA studies.
Table: Essential Research Reagents and Computational Tools for FBA
| Item Name | Function/Description | Example/Note |
|---|---|---|
| Genome-Scale Metabolic Model | A computational representation of all known metabolic reactions in an organism. | Models for E. coli, S. cerevisiae, and H. sapiens are publicly available [2]. |
| Stoichiometric Matrix (S) | The core mathematical structure of the model, defining metabolite-reaction relationships. | Typically stored in a data file (e.g., SBML format) and loaded into the analysis tool [2]. |
| Linear Programming Solver | Software that performs the numerical optimization to find the optimal flux distribution. | Solvers are integrated into toolboxes like the COBRA Toolbox (for MATLAB) or Cobrapy (for Python) [1] [2]. |
| Constraint-Based Reconstruction and Analysis (COBRA) Toolbox | A suite of functions for performing FBA and other constraint-based methods. | A standard toolkit in the field; requires a MATLAB environment [2]. |
| Experimental Flux Data | Data used for validating model predictions, such as growth rates or uptake/secretion rates. | Crucial for assessing the predictive power of different objective functions [4]. |
| Python Programming Environment | An open-source platform for implementing custom FBA protocols and analyses. | Libraries like NumPy and SciPy are used for matrix operations and linear programming [1]. |
In the field of systems biology and metabolic engineering, constraint-based reconstruction and analysis (COBRA) methods have become indispensable for predicting cellular behavior. At the heart of these computational approaches lies the metabolic objective function, a mathematical representation that defines the biological goals a cell is optimizing under specific conditions. Flux Balance Analysis (FBA), the most widely used constraint-based method, relies on these objective functions to predict steady-state metabolic flux distributions through genome-scale metabolic models (GEMs) [5] [6]. The selection of an appropriate objective function is paramount, as it directly influences the accuracy of phenotypic predictions, from microbial strain improvement to drug discovery [7] [8].
While rapidly proliferating cells like microbes or cancer cells are often assumed to prioritize biomass maximization, this review demonstrates that cellular objectives are far more nuanced. Different cell types, including quiescent human cells, stem cells, and cancer cells, exhibit distinct metabolic priorities that support their specialized functions [7]. This comparative analysis examines three fundamental categories of metabolic objectives—biomass production, energy generation, and product synthesis—evaluating their formulations, applications, and performance across various biological contexts.
Comparative Analysis of Objective Function Types
Biomass Objective Function
The biomass objective function (BOF) represents the biosynthetic requirements for cellular reproduction, mathematically describing the rate at which all biomass precursors are synthesized in the correct proportions to support growth [5]. Formulating a BOF requires detailed knowledge of cellular composition, including macromolecular weights of proteins, RNA, DNA, lipids, and carbohydrates, along with associated energetic costs for polymerization [5].
Formulation Levels:
- Basic Level: Defines macromolecular content (weight fractions of protein, RNA, lipid, etc.) and their metabolic building blocks.
- Intermediate Level: Incorporates biosynthetic energy requirements (e.g., 2 ATP + 2 GTP per amino acid incorporated into protein).
- Advanced Level: Includes vitamins, cofactors, and minimal "core" cellular components essential for viability [5].
The BOF is particularly effective for predicting growth rates and essential genes in rapidly proliferating cells. However, its limitations become apparent when modeling specialized mammalian cell types or non-growth associated metabolic states [7].
Energy-Based Objectives
Energy-centric objectives prioritize ATP maximization or redox balance over biomass production, reflecting situations where cellular survival rather than proliferation is paramount. Multiple studies have demonstrated that minimizing redox potential or maximizing ATP yield per flux unit can better predict metabolic phenotypes under certain conditions [5].
Hausser et al. noted that environmental constraints create selection pressures that force phenotypic switching. For instance, late-stage cancers under hypoxic conditions tend to optimize survival, contrasting with early-stage cancers that are proliferation-optimized due to ample oxygen availability [7]. In continuous cultures with nutrient scarcity, linear maximization of ATP yield achieved higher predictive accuracy than growth maximization [5].
Product Synthesis Objectives
Biotechnological applications often employ product synthesis objectives to maximize the yield of specific metabolites. This approach is valuable in industrial microbiology for optimizing production of compounds like isopropanol-butanol-ethanol (IBE) in Clostridium species [8]. Unlike biomass objectives that represent a "selfish" cellular goal, product synthesis objectives typically represent engineering interventions where cellular metabolism is redirected toward a non-native goal.
The TIObjFind framework addresses the challenge of predicting such metabolic shifts by identifying pathway-specific weighting factors that indicate how cells prioritize reactions under different environmental conditions [8].
Table 1: Comparative Performance of Objective Functions Across Biological Systems
| Objective Function | Best Application Context | Predictive Strengths | Documented Limitations |
|---|---|---|---|
| Biomass Maximization | Rapidly proliferating cells (microbes, cancer cells) | Growth rate prediction, gene essentiality in optimal conditions | Poor performance for quiescent cells, neglects metabolic trade-offs |
| ATP Maximization | Energy-limited conditions, hypoxic environments | Survival phenotype prediction, stationary phase metabolism | May overpredict ATP-generating futile cycles |
| Redox Minimization | Aerobic respiration, oxidative stress conditions | E. coli central carbon metabolism under aerobic batch growth [5] | Limited to specific metabolic states |
| Product Synthesis | Industrial bioprocessing, metabolic engineering | High-yield strain design, pathway flux optimization | Requires genetic/regulatory interventions for implementation |
Methodologies for Objective Function Inference
Computational Frameworks
Advanced computational frameworks have been developed to infer context-specific objective functions from experimental data:
ObjFind Framework: This approach introduces Coefficients of Importance (CoIs) that quantify each reaction's contribution to an objective function. By maximizing a weighted sum of fluxes while minimizing deviations from experimental data, ObjFind interprets flux distributions in terms of optimized metabolic objectives [8].
TIObjFind Framework: Building on ObjFind, this topology-informed method integrates Metabolic Pathway Analysis (MPA) with FBA. It constructs flux-dependent weighted reaction graphs to analyze metabolic behavior across different system states, enhancing interpretability of complex networks [8].
REMI Method: The Relative Expression and Metabolomic Integration approach incorporates multi-omics data into thermodynamically curated GEMs. REMI translates differential gene expression and metabolite abundance data into differential flux constraints, significantly reducing the solution space of feasible fluxes [6].
Experimental Validation Protocols
Validating predicted objective functions requires integration of computational and experimental approaches:
13C Metabolic Flux Analysis (13C-MFA): This established experimental technique uses 13C-labeled substrates to track metabolite fluxes through central carbon metabolism, providing ground-truth data for validating computational predictions [6].
Isotopomer Analysis: Advanced mass spectrometry approaches measure isotopic labeling patterns in intracellular metabolites, enabling experimental determination of flux distributions for comparison with model predictions [5].
Multi-omics Integration: REMI and similar methods leverage transcriptomic and metabolomic data to constrain flux predictions. Performance is quantified by calculating Pearson correlation coefficients between predicted and experimentally measured fluxes, with REMI achieving r = 0.79 in E. coli models [6].
Table 2: Experimental Methods for Objective Function Validation
| Methodology | Data Output | Resolution | Integration with Modeling |
|---|---|---|---|
| 13C-MFA | Intracellular flux maps for central metabolism | Pathway-level | Gold standard for validation of predicted fluxes |
| Gene Expression Profiling | Transcript abundance for metabolic genes | Genome-wide | Constrains reaction capacity in REMI, iMAT |
| Quantitative Metabolomics | Absolute metabolite concentrations | System-wide | Enables thermodynamic constraints (TFA) |
| Flux Variability Analysis | Range of possible fluxes for each reaction | Network-wide | Identifies invariant reactions and trade-offs |
Research Workflow and Pathway Visualization
Multi-Omics Integration Workflow
The diagram below illustrates the workflow for integrating multi-omics data to infer cellular objective functions, as implemented in methods like REMI and TIObjFind.
Cellular Trade-offs and Pareto Optimality
Biological systems face fundamental trade-offs in optimizing multiple objectives simultaneously. The concept of Pareto optimality describes how cells allocate limited resources between competing goals such as growth and survival.
Table 3: Key Research Reagents and Computational Tools for Objective Function Studies
| Resource | Type | Function in Research | Example Applications |
|---|---|---|---|
| BioCyc Database | Bioinformatics Platform | Pathway/Genome Databases (PGDBs) with curated metabolic networks | Metabolic reconstruction, pathway analysis [9] |
| EcoCyc | Tier 1 PGDB | Manually curated E. coli database with 44,000+ literature citations | Gold standard for bacterial metabolic studies [9] |
| MetaCyc | Metabolic Pathway DB | Curated metabolic pathways from all domains of life (76,000+ publications) | Reference database for pathway prediction [9] |
| Pathway Tools Software | Metabolic Reconstruction | Creates organism-specific PGDBs from genome data | Generation of new metabolic models [9] |
| MetaFlux | FBA Module | Creates quantitative metabolic models from PGDBs using FBA | Constraint-based modeling and flux prediction [9] |
| 13C-Labeled Substrates | Isotopic Tracers | Enables experimental flux measurement via 13C-MFA | Validation of computational flux predictions [6] |
| Gibbs Free Energy Data | Thermodynamic Constraints | Incorporates reaction thermodynamics into FBA | Reduction of solution space in TFA [6] |
This comparative analysis demonstrates that no single objective function universally predicts metabolic behavior across all biological contexts. The performance of biomass, energy, and product synthesis objectives depends critically on cellular specialization, environmental conditions, and biological priorities. While biomass maximization effectively models proliferating microbes, energy-centric objectives better predict survival states, and product synthesis objectives drive biotechnological applications.
Advanced methods that integrate multi-omics data and identify context-specific Coefficients of Importance (CoIs) represent the future of objective function determination. Frameworks like TIObjFind and REMI significantly enhance flux prediction accuracy by incorporating regulatory constraints and thermodynamic principles [6] [8]. As systems biology continues to advance, the development of condition-specific, dynamic objective functions will be crucial for applications ranging from drug discovery to personalized medicine and sustainable bioproduction.
Why the Objective Function is Crucial for Predicting Phenotypes
In the field of computational biology, accurately predicting phenotypes from genotypes and environmental factors is a fundamental challenge with significant implications for medicine, biotechnology, and basic research. The choice of objective function—the mathematical expression that a computational model aims to optimize—is a critical determinant of the accuracy and biological relevance of these predictions. This guide compares the performance of different modeling paradigms, from traditional constraint-based methods to modern machine learning approaches, highlighting how their underlying objective functions influence predictive power.
The Fundamental Role of an Objective Function
In computational models, the objective function formally defines the presumed cellular goal. In metabolic models, for instance, this often involves maximizing biomass production or ATP yield. The core hypothesis is that cellular behavior can be predicted by assuming the organism optimizes this function. An accurate objective function leads to predictions that match experimental data; an inaccurate one can render a model biologically implausible.
The challenge is that a single, static objective function may not capture the dynamic and adaptive nature of living systems. Cells shift their metabolic priorities in response to environmental changes, and a function that works well in one condition may fail in another. This limitation has driven the development of more sophisticated frameworks for identifying and testing objective functions. [8] [10]
Comparative Analysis of Modeling Frameworks
The table below summarizes the core methodologies, key features, and primary challenges associated with different approaches to phenotype prediction.
| Modeling Approach | Core Methodology | Key Feature | Primary Challenge |
|---|---|---|---|
| Traditional FBA [8] | Linear Programming | Maximizes a single, pre-defined reaction (e.g., biomass). | Struggles to capture flux variations under different conditions. |
| TIObjFind Framework [8] [10] | Optimization + Topology Analysis | Infers objective functions from data using Coefficients of Importance (CoIs). | Requires experimental flux data for training. |
| Flux Cone Learning (FCL) [11] | Machine Learning (Supervised) | Learns the relationship between flux cone geometry and phenotypes. | Requires substantial computational resources for sampling. |
| Genomic Prediction (ML) [12] [13] | Machine Learning (e.g., SVR, GBM) | Models complex, non-linear genotype-phenotype relationships. | Performance can be affected by population structure in the data. |
Performance Benchmarking: Quantitative Comparisons
Prediction Accuracy for Metabolic Gene Essentiality
A critical test for metabolic models is accurately predicting which genes are essential for survival. The following table compares the performance of Flux Balance Analysis (FBA) and the machine learning method Flux Cone Learning (FCL) in predicting gene essentiality in E. coli. [11]
| Prediction Method | Organism | Reported Accuracy | Key Objective/Feature |
|---|---|---|---|
| Flux Balance Analysis (FBA) | E. coli | 93.5% | Biomass maximization |
| Flux Cone Learning (FCL) | E. coli | 95.0% | Geometry of the metabolic "flux cone" |
| Flux Balance Analysis (FBA) | Higher-order organisms | Lower performance | Relies on an unknown optimality objective |
Prediction Accuracy for Complex Livestock Traits
Beyond microbes, the choice of statistical objective (or model) is crucial for predicting complex polygenic traits. The table below shows the performance of various methods in predicting feed efficiency in Nellore cattle. [13]
| Prediction Method | Category | Relative Prediction Accuracy vs. ST-GBLUP |
|---|---|---|
| Multi-Trait GBLUP (MTGBLUP) | Parametric | +13.7% |
| Support Vector Regression (SVR) | Machine Learning | +14.6% |
| Multi-Layer Neural Network (MLNN) | Machine Learning | +8.9% |
| Bayesian Regression Methods | Parametric | Benchmark (lower accuracy) |
Detailed Experimental Protocols
To ensure reproducibility and provide a clear understanding of how these methods are implemented, we outline the key experimental workflows.
Protocol for the TIObjFind Framework
The TIObjFind framework integrates metabolic pathway analysis with FBA to identify context-specific objective functions. [8] [10]
a. Single-Stage Optimization:
- Step 1: The framework solves an optimization problem that minimizes the squared difference between predicted metabolic fluxes ((v)) and experimental flux data ((v^{exp})), while simultaneously maximizing a hypothesized objective function ((c^{obj} \cdot v)).
- Step 2: This step identifies a candidate set of "Coefficients of Importance" (CoIs) that define the contribution of each reaction to the cellular objective.
b. Mass Flow Graph (MFG) Generation:
- The optimized flux distribution is mapped onto a directed, weighted graph called a Mass Flow Graph. In this graph, nodes represent metabolic reactions, and edges represent the flow of metabolites between them.
c. Metabolic Pathway Analysis (MPA) via Minimum Cut:
- A graph theory algorithm (e.g., Boykov-Kolmogorov) is applied to the MFG to find the "minimum cut" between a source node (e.g., glucose uptake) and a target node (e.g., product secretion).
- This minimum cut identifies the set of reactions that are most critical for the desired metabolic output, refining the Coefficients of Importance and providing a topology-informed objective function.
Protocol for Flux Cone Learning (FCL)
Flux Cone Learning uses a data-driven approach to predict deletion phenotypes without a pre-defined objective function. [11]
a. Define the Metabolic Space:
- A Genome-Scale Model (GEM) defines the system of linear equations ((S v = 0)) and flux bounds that constitute the "flux cone" of possible metabolic states.
b. Monte Carlo Sampling:
- For each gene deletion, the corresponding reactions are constrained to zero. A Monte Carlo sampler then generates a large number (e.g., 100-5000) of random, feasible flux distributions within the resulting deformed flux cone.
- Each set of samples characterizes the geometry of the metabolic space for that specific deletion.
c. Supervised Model Training:
- The flux samples from all deletions are compiled into a feature matrix. Each sample is labeled with the corresponding experimental fitness score from a deletion screen.
- A supervised machine learning model (e.g., a Random Forest classifier) is trained on this dataset to learn the correlation between the shape of the flux cone and the phenotypic outcome.
d. Prediction and Aggregation:
- To predict the phenotype of a new deletion, the model is applied to multiple flux samples from its flux cone. The final prediction is determined by aggregating these sample-level predictions (e.g., via majority voting).
The Scientist's Toolkit: Essential Research Reagents & Solutions
Successful implementation of the methods described requires a combination of computational tools and biological data resources.
| Tool/Reagent | Function/Purpose | Relevant Method |
|---|---|---|
| Genome-Scale Model (GEM) | A mathematical representation of an organism's metabolism; the core scaffold for constraint-based methods. | FBA, TIObjFind, FCL [8] [11] |
| Experimental Flux Data ((v^{exp})) | Quantified metabolic reaction rates from experiments; used to train and validate inferred objective functions. | TIObjFind [10] |
| Gene Deletion Fitness Screen | High-throughput experimental data measuring the growth effect of gene knockouts; provides labels for supervised learning. | FCL [11] |
| Monte Carlo Sampler | Software that randomly samples the high-dimensional space of possible flux distributions in a metabolic network. | FCL [11] |
| MATLAB / Python (with pySankey) | Programming environments for implementing optimization frameworks, graph analysis, and result visualization. | TIObjFind [10] |
| Random Forest Classifier | A versatile machine learning algorithm for classification and regression tasks, known for good performance and interpretability. | FCL, Genomic Prediction [11] [13] |
The evidence demonstrates that the choice and formulation of the objective function are pivotal for accurate phenotypic prediction. While traditional FBA with a fixed objective like biomass maximization provides a strong baseline, its performance is limited when cellular priorities shift. Emerging frameworks like TIObjFind address this by inferring context-specific objective functions directly from experimental data, thereby enhancing model fidelity. Furthermore, machine learning methods like Flux Cone Learning and Support Vector Regression show that bypassing a single pre-defined objective in favor of learning the relationship between system states and outcomes can yield superior, state-of-the-art accuracy. The selection of the right objective function, therefore, remains a cornerstone of successful phenotypic prediction in computational biology.
Evolutionary Arguments and Biological Rationales for Common Objectives
Selecting appropriate objective functions remains a fundamental challenge in constraint-based metabolic modeling, particularly Flux Balance Analysis (FBA). The core premise of FBA relies on mathematical optimization to predict metabolic fluxes, requiring assumptions about cellular goals shaped by evolutionary pressures [3]. This comparative guide examines the evolutionary arguments and experimental validations supporting common objective functions, providing researchers with a structured framework for selecting biologically relevant objectives across different applications.
The evolutionary rationale for objective function selection stems from the concept that natural selection favors metabolic strategies that enhance survival and reproduction. However, this optimization process operates within complex constraints and trade-offs. As noted in critiques of evolutionary biology, "Evolutionarily, metabolism is most likely optimized for overall robustness across many conditions, rather than a single condition-specific objective" [14]. This perspective challenges simplistic assumptions about cellular optimization and underscores the need for condition-specific objective function selection.
Comparative Analysis of Common Objective Functions
Theoretical Evolutionary Rationales
Table 1: Evolutionary Arguments for Common Objective Functions in Metabolic Modeling
| Objective Function | Evolutionary Rationale | Supported Organisms/Conditions | Key Limitations |
|---|---|---|---|
| Maximal Biomass Production | Optimizes reproductive capacity by maximizing growth rate; assumes selection favors rapid proliferation | E. coli, S. cerevisiae in optimal growth conditions [3] [14] | Poor predictor under stress, nutrient limitation, or stationary phase |
| Maximal ATP Production | Maximizes energy currency for cellular maintenance and biosynthesis; reflects fundamental energy optimization | Budding yeast in early life phases [3] | May overlook biosynthetic requirements and redox balance |
| Parsimonious Enzyme Usage | Reflects protein synthesis cost optimization; conserves resources for other cellular processes | Improves lifespan predictions in yeast [3] | Requires additional constraints for accurate flux distribution |
| Multi-Objective Optimization | Mirrors evolutionary trade-offs between competing cellular goals | Condition-dependent responses in multiple organisms [3] [8] | Increased computational complexity and parameterization |
| Yield Optimization | Maximizes resource use efficiency in nutrient-limited environments | Microbes in constant nutrient environments [3] | May not predict metabolic behavior in fluctuating environments |
Experimental Validation Across Organisms
Table 2: Experimental Support for Objective Functions Across Biological Systems
| Organism/System | Optimal Objective Function | Experimental Validation Method | Key Findings |
|---|---|---|---|
| S. cerevisiae (Aging Model) | Parsimonious maximal growth with energy cost minimization | Replicative lifespan measurements and division timing [3] | Combined objectives improved lifespan predictions by increasing respiratory activity and antioxidative capacity |
| E. coli | Condition-dependent: Maximal energy or biomass production | C-based flux data fitting across conditions [3] | Most accurate objectives varied with environmental conditions |
| C. acetobutylicum (Fermentation) | Pathway-specific weighted objectives | Fluxomic data comparison using TIObjFind framework [8] | Stage-specific metabolic priorities required different objective weightings |
| A. thaliana (Cold Acclimation) | Flux sampling without predefined objective | Metabolite measurements, CO₂ uptake, carbon allocation tracking [14] | Eliminated observer bias; revealed fumarate and GABA importance in cold response |
| Multi-species IBE system | Hybrid objective with importance coefficients | Experimental product secretion rates [8] | Weighted combination of fluxes better captured community metabolic interactions |
Methodological Approaches for Objective Function Identification
Computational Frameworks and Algorithms
Several advanced computational frameworks have been developed to identify appropriate objective functions, moving beyond simple assumptions:
The TIObjFind (Topology-Informed Objective Find) framework integrates Metabolic Pathway Analysis (MPA) with FBA to systematically infer metabolic objectives from experimental data [8]. This method determines Coefficients of Importance (CoIs) that quantify each reaction's contribution to cellular objectives, using network topology and pathway structure to analyze metabolic behavior across different system states.
The REMI (Relative Expression and Metabolomic Integrations) method represents another approach, integrating relative gene expression, metabolite abundance, and thermodynamic constraints into genome-scale models [6]. This multi-omic integration significantly reduces the solution space of feasible fluxes and improves prediction accuracy.
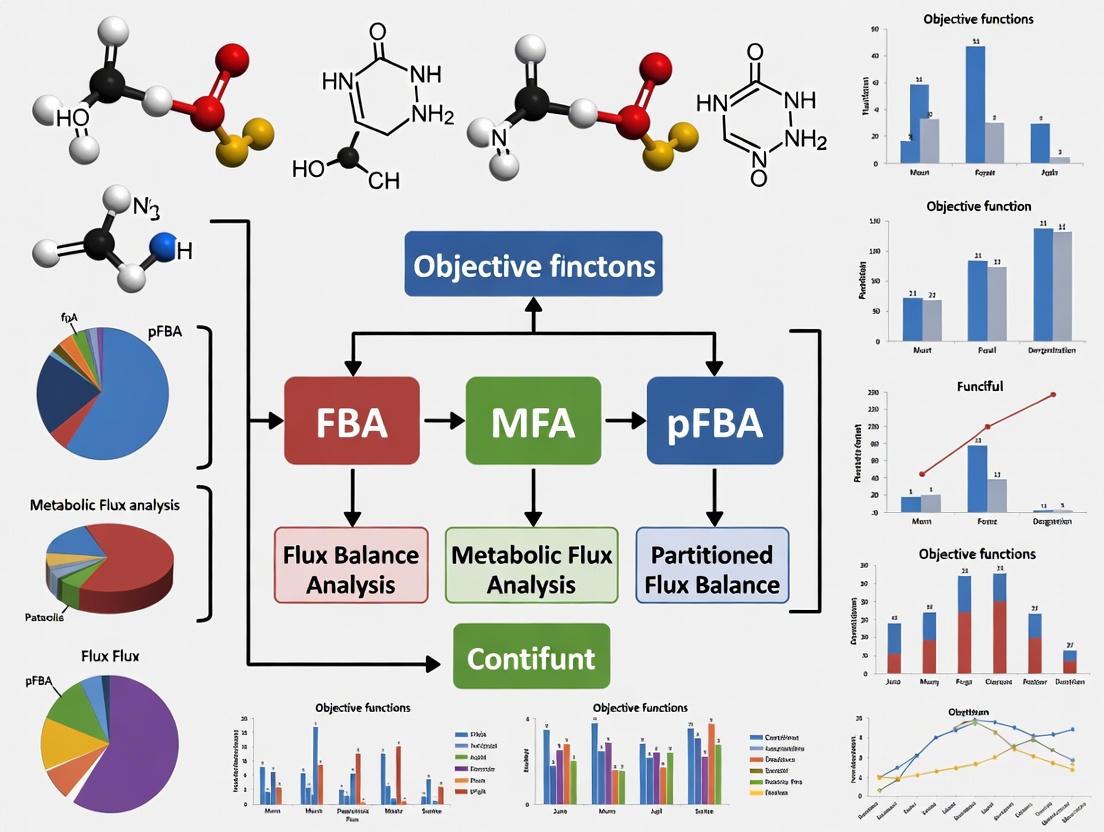
Figure 1: Comparative Workflows for Traditional FBA versus Flux Sampling Approaches
Flux Sampling as an Objective-Free Alternative
Flux sampling has emerged as a powerful alternative to objective-dependent methods, particularly for studying metabolism under changing environmental conditions [14]. This approach generates probability distributions of steady-state reaction fluxes without assuming a specific cellular objective, thereby eliminating observer bias.
The Coordinate Hit-and-Run with Rounding (CHRR) algorithm has demonstrated superior efficiency for flux sampling, being 2.5-8 times faster than alternative algorithms depending on model complexity [14]. This method enables comprehensive exploration of metabolic solution spaces, providing insights into network robustness and flexibility without predefined objectives.
Experimental Protocols for Objective Function Validation
Multi-Scale Model Validation for Aging Studies
For replicative aging studies in yeast, researchers have employed a multi-scale mathematical model integrating cellular metabolism, nutrient sensing, and damage accumulation [3]. The protocol involves:
- Model Construction: Develop enzyme-constrained FBA models of central carbon metabolism including ROS creation
- Regulatory Network Integration: Implement Boolean representations of key signaling pathways (Snf1, PKA, TOR, Yap1, Sln1)
- Dynamic Simulation: Connect optimal fluxes to ODE models of damage accumulation and growth
- Validation Metrics: Compare predicted number of cell divisions and generation time against experimental measurements
- Objective Function Testing: Systematically test individual and combined objective functions against lifespan data
This approach confirmed that maximal growth is essential for realistic lifespans, while parsimonious solutions or additional energy cost optimization further improved predictions [3].
Flux Sampling Protocol for Environmental Responses
For studying metabolic responses to environmental changes such as temperature acclimation in plants, the following flux sampling protocol is recommended [14]:
- Model Selection: Choose appropriate genome-scale metabolic model (e.g., A. thaliana models)
- Experimental Constraints: Incorporate measured CO₂ uptake and organic carbon accumulation data
- Sampling Implementation: Apply CHRR algorithm with appropriate thinning constants
- Convergence Diagnostics: Verify using Raftery & Lewis and IPSRF diagnostics
- Solution Space Analysis: Compare flux distributions between conditions
- Biological Interpretation: Identify key metabolic changes supporting acclimation
This protocol revealed how regulated interplay between diurnal starch and organic acid accumulation defines plant acclimation to cold, confirming fumarate accumulation and predicting GABA's role in metabolic signaling [14].
Table 3: Key Research Reagents and Computational Tools for Objective Function Studies
| Resource Category | Specific Tools/Methods | Primary Function | Application Context |
|---|---|---|---|
| Constraint-Based Modeling Software | COBRA Toolbox, FlexFlux | Implement FBA, FVA, and regulatory constraints | Metabolic network simulation and analysis [8] |
| Flux Sampling Algorithms | CHRR, ACHR, OPTGP | Explore solution spaces without objective functions | Analysis of metabolic robustness and plasticity [14] |
| Multi-Omics Integration Methods | REMI, GIM3E, iReMet-Flux | Incorporate transcriptomic, metabolomic data | Context-specific model construction [6] |
| Objective Function Identification | TIObjFind, ObjFind | Infer cellular objectives from experimental data | Data-driven objective function discovery [8] |
| Validation Techniques | 13C-MFA, Isotopomer Analysis | Experimental flux measurement | Objective function validation [6] |
| Convergence Diagnostics | Raftery & Lewis, IPSRF | Assess flux sampling convergence | Quality control for sampling studies [14] |
The evolutionary arguments supporting objective function selection continue to evolve with advancing computational methods and experimental techniques. While traditional objectives like maximal biomass production remain valid for optimal growth conditions, more sophisticated approaches including multi-objective optimization, condition-specific weighting, and objective-free flux sampling provide more biological realism for complex environments.
The fundamental insight from evolutionary biology—that natural selection optimizes for robustness across multiple conditions rather than maximal performance in any single condition—should guide objective function selection [14]. By aligning computational models with these evolutionary principles, researchers can enhance predictive accuracy and biological relevance in metabolic modeling for basic research and applied biotechnology.
Challenges in Predicting Individual Fluxes and Enzyme Activities
Predicting individual metabolic fluxes and enzyme activities remains a significant hurdle in systems biology, despite comprehensive knowledge of metabolic network structures. The core challenge stems from the complex interplay of multiple regulatory layers that control metabolic flux, which is the rate at which metabolites are converted in a biochemical network. While the stoichiometry of metabolic networks is well-established for many organisms, the dynamic behavior of metabolism cannot yet be adequately described, predicted, or engineered [15].
This prediction challenge is primarily rooted in several key factors: the influence of kinetic interactions and allosteric control mechanisms that are difficult to comprehensively characterize in vivo; the disconnect between in vitro enzyme properties and their actual behavior within the cellular environment; and the fundamental difficulty in determining whether changes in metabolic flux are driven by alterations in enzyme levels or by other regulatory mechanisms [16] [15]. Unknowns in metabolic flux behavior particularly arise from these kinetic interactions, making it infeasible to exhaustively test every possible enzyme-metabolite interaction in vitro [15].
Comparative Analysis of Flux Prediction Methodologies
Various computational approaches have been developed to address the challenge of flux prediction, each with distinct methodological foundations and limitations. The table below provides a structured comparison of the primary methodologies discussed in the literature.
Table 1: Comparison of Metabolic Flux Prediction Methodologies
| Method | Core Principle | Level of Expression Data Integration | Key Challenges |
|---|---|---|---|
| Flux Balance Analysis (FBA) [4] [17] [3] | Constraint-based optimization of an objective function (e.g., biomass) under steady-state assumptions. | Not inherently integrated; can be used as a constraint. | Choice of objective function significantly impacts results; does not directly incorporate enzyme kinetics or regulation [17] [3]. |
| Enzyme-Constrained FBA [17] [3] | Extends FBA by incorporating constraints based on measured or estimated enzyme usage and capacity. | Proteomic data can constrain enzyme usage (e_i) [3]. | Requires comprehensive enzyme abundance and kinetic (k_cat) data, which is often incomplete [3]. |
| Flux Potential Analysis (FPA) [16] | Integrates relative enzyme levels from the reaction of interest and its network neighbors, weighted by proximity. | Individual reaction & network neighborhood. | Correlates weakly with flux changes; suboptimal predictive power [16]. |
| Enhanced FPA (eFPA) [16] | Improved FPA that integrates expression data at the pathway level rather than for single reactions or the entire network. | Pathway-level. | Outperforms FPA and alternatives; optimal balance between reaction-specific and network-wide analysis [16]. |
The choice of the objective function in FBA-based methods is particularly crucial, as it determines how fluxes are distributed across the network. Studies have systematically tested various objectives, including maximal growth (biomass production), minimal substrate uptake, and maximal ATP production, confirming that this choice is critical for generating realistic predictions of physiological states, such as the replicative lifespan in yeast [17] [3]. No single consensus objective function exists, and the best choice may be condition-dependent [3].
Experimental Data and Performance Benchmarking
The development of enhanced Flux Potential Analysis (eFPA) was guided by benchmarking against experimental data. A key dataset fulfilling the requirements for a statistically meaningful analysis came from Saccharomyces cerevisiae (yeast), providing flux estimates for 232 metabolic reactions and associated enzyme levels across 25 different nutrient limitation conditions [16].
A central finding from this systematic evaluation was that flux changes correlate more strongly with pathway-level changes in enzyme levels than with changes in the expression of individual enzymes or network-wide expression profiles [16]. This discovery informed the eFPA algorithm, which integrates enzyme expression data at this optimal pathway level.
Table 2: Key Experimental Findings from Flux-Enzyme Correlation Studies
| Study System | Key Measured Variables | Central Finding | Impact on Prediction |
|---|---|---|---|
| S. cerevisiae (Yeast) [16] | - Fluxomic data (232 reactions)- Proteomic data (156 enzymes)- 25 growth conditions | Flux changes are best predicted from changes in enzyme levels of pathways, not individual reactions or the whole network. | Led to the development of eFPA, which uses pathway-level integration for superior predictions [16]. |
| S. cerevisiae (Yeast) [17] [3] | - Replicative lifespan (cell divisions)- Generation time- Metabolic flux distributions | The choice of FBA objective function (e.g., maximal growth) is crucial for predicting realistic replicative lifespans. | Connects flux prediction objectives to long-term cellular outcomes like ageing; suggests combining objectives (e.g., parsimonious maximal growth) [17] [3]. |
| Human Tissues [16] | - Proteomic and Transcriptomic data- Predicted tissue metabolic function | eFPA consistently predicts tissue metabolic function using either proteomic or transcriptomic data. | Demonstrates method's robustness and applicability to human data, even handling data sparsity and noisiness in single-cell RNA-seq data [16]. |
The performance of eFPA demonstrates its advantage over other methods. It consistently generates robust predictions of tissue metabolic function in human data using either proteomic or transcriptomic datasets and efficiently handles the sparsity and noisiness inherent in single-cell gene expression data [16].
Detailed Experimental Protocols for Method Validation
Protocol for Yeast Flux-Enzyme Correlation Analysis
This protocol is derived from the study that developed and validated eFPA [16].
- Data Acquisition: Obtain a curated dataset with simultaneous measurements of metabolic fluxes and enzyme levels from the same samples. The benchmark yeast dataset included flux estimates for 232 reactions and 156 enzyme levels across 25 chemostat conditions with different nutrient limitations (e.g., glucose, leucine, nitrogen, phosphate) and growth rates [16].
- Data Preprocessing: Adjust raw flux values for growth rate variations by dividing each flux by the corresponding specific growth rate. This yields relative flux values that are comparable across conditions. Enzyme abundance is typically already normalized as a proportion of total protein [16].
- Correlation Analysis:
- Calculate correlation coefficients between flux and the level of its cognate enzyme for individual reactions.
- Calculate correlation coefficients between flux and a composite expression score for the pathway in which the reaction resides.
- Statistically compare the correlation strengths to determine which level (individual vs. pathway) is more predictive of flux changes [16].
- Algorithm Optimization: Use the findings to optimize the parameters of a predictive algorithm (e.g., FPA). The key is to define the optimal "distance factor" or network neighborhood size (pathway level) over which enzyme expression data should be integrated for the most accurate flux prediction [16].
Protocol for Testing Objective Functions in a Multi-Scale Model
This protocol is based on the work that connected FBA objective functions to yeast replicative ageing [17] [3].
- Model Setup: Employ a multi-scale mathematical model (e.g., yMSA for yeast) that integrates:
- An enzyme-constrained FBA model of central carbon metabolism.
- A regulatory network (e.g., using Boolean logic for nutrient sensing and stress pathways).
- A dynamic model of damage accumulation and cell growth [3].
- Define Optimization Strategy: Implement a two-stage lexicographic optimization:
- First Optimization: Solve the FBA problem for a primary objective function (e.g., maximize biomass reaction).
- Second Optimization: Constrain the solution to the optimal value from the first step (allowing a small flexibility factor, ε) and then optimize a second objective (e.g., minimize total flux, representing enzyme usage parsimony) [3].
- Simulate Ageing: Run the integrated model with different objective function combinations (e.g., maximal growth, maximal ATP, minimal NADH, and their parsimonious versions) to simulate the entire replicative lifespan of a yeast cell [17] [3].
- Output Analysis: For each simulation, record key observables:
- Replicative Lifespan (RLS): The total number of cell divisions.
- Generation Time: The time between divisions.
- Metabolic Flux Distributions: Particularly in different metabolic phases (e.g., early vs. late life) [3].
- Validation: Compare the simulated RLS and generation times against established experimental data for wild-type yeast cells to determine which objective function produces the most realistic physiological predictions [17].
Diagram 1: Workflow for developing and validating flux prediction methods.
Table 3: Essential Research Reagents and Computational Tools for Flux Prediction Research
| Reagent / Resource | Function / Description | Relevance in Flux Studies |
|---|---|---|
| Chemostat Cultures [16] | A bioreactor that maintains microbial cells in steady-state growth at a fixed dilution rate. | Essential for acquiring consistent and reproducible omics data (fluxomic, proteomic) across multiple controlled growth conditions. |
| Stoichiometric Genome-Scale Model (GEM) [17] [3] | A computational reconstruction of an organism's metabolism, detailing reaction stoichiometry and network connectivity. | Serves as the core structural framework for constraint-based methods like FBA and eFPA. |
| Enzyme-Abundance Datasets [16] | Quantitative measurements of protein levels, typically via mass spectrometry. | Used as constraints in ecFBA or as input data for correlation analysis and predictive algorithms like eFPA. |
| Fluxomic Data [16] | Experimental measurements of intracellular metabolic reaction rates. | Serves as the "ground truth" gold standard for validating and benchmarking the accuracy of flux prediction methods. |
| Curated Yeast Benchmark Dataset [16] | A publicly available dataset containing paired flux and enzyme abundance measurements across 25 conditions. | A critical resource for the initial development, parameterization, and validation of new predictive algorithms. |
| Multi-Scale Modelling Framework (e.g., yMSA) [3] | An integrated computational model combining metabolism, regulation, and physiology. | Allows for testing the physiological consequences of different flux distributions and objective functions on outcomes like ageing. |
Diagram 2: Interrelationship between core methodological challenges and broader research directions.
From Traditional FBA to Cutting-Edge Machine Learning Flux Predictors
In the field of systems biology, Flux Balance Analysis (FBA) serves as a fundamental constraint-based modeling approach for analyzing metabolic networks at the genome scale. FBA calculates flow of metabolites through a metabolic network, enabling prediction of organism's growth, metabolic production, and physiological properties. The core principle of FBA involves solving for a flux distribution that satisfies mass-balance and steady-state constraints while optimizing a specified cellular objective. The selection of an objective function is therefore crucial as it represents the biological goal driving the metabolic behavior and ultimately determines the predicted flux distribution.
While numerous objective functions have been proposed, three have emerged as particularly influential: maximal growth (biomass production), ATP production, and parsimonious solutions (minimization of total flux or enzyme usage). These functions are motivated by different evolutionary hypotheses about cellular optimization principles. This guide provides a comparative analysis of these common objective functions, examining their underlying assumptions, performance characteristics, and applicability across different biological contexts and organism types.
Comparative Analysis of Common Objective Functions
The table below summarizes the key characteristics, applications, and limitations of the three primary objective functions discussed in this guide.
Table 1: Comparison of Common Objective Functions in Flux Balance Analysis
| Objective Function | Underlying Principle | Typical Applications | Performance Highlights | Key Limitations |
|---|---|---|---|---|
| Maximal Growth (Biomass) | Maximizes biomass production, reflecting evolutionary pressure for rapid reproduction | - Microbial growth prediction- Nutrient-rich conditions- Standard FBA benchmarks | - Essential for realistic yeast replicative lifespans [3]- Accurate for E. coli and yeast in standard conditions [3] | - Often unrealistic under substrate excess [18]- Overestimates growth in mammalian cells [19] |
| ATP Production | Maximizes or minimizes ATP yield, representing energy efficiency goals | - Energy metabolism studies- Conditions with energy constraints- Multi-objective optimization | - Improves lifespan predictions in yeast when combined with growth [3]- Condition-dependent accuracy [3] | - Rarely optimal as sole objective [3] |
| Parsimonious Solution | Minimizes total flux or enzyme usage, representing resource efficiency | - Enzyme-limited conditions- Substrate excess scenarios- Multi-stage optimizations | - Increases respiratory activity in yeast [3]- Enhances antioxidative activity in early life [3]- Better fits C. butyricum glycerol culture [18] | - Requires precise flexibility constraints to maintain feasibility [3] |
Experimental Validation and Performance Data
Quantitative Performance Across Organisms
Experimental validations across multiple organisms demonstrate how the performance of objective functions varies significantly with biological context and environmental conditions.
Table 2: Experimental Performance Metrics of Objective Functions Across Organisms
| Organism | Condition | Objective Function | Performance Metric | Result | Reference |
|---|---|---|---|---|---|
| S. cerevisiae (Yeast) | Replicative ageing | Maximal growth | Replicative lifespan | Essential for realistic lifespans | [3] |
| S. cerevisiae (Yeast) | Replicative ageing | Parsimonious + maximal growth | Number of cell divisions | ~23 divisions (reference cell) | [3] |
| S. cerevisiae (Yeast) | Replicative ageing | Parsimonious + maximal growth | Average generation time | ~1.5 hours | [3] |
| C. butyricum | Glycerol culture | Maximal growth | Biomass yield error | 300% overestimation | [18] |
| C. butyricum | Glycerol culture | Maximal growth | PDO yield error | 100% error | [18] |
| C. butyricum | Glycerol limitation | Biomass per enzyme usage | Growth prediction | Accurate phenotype state | [18] |
| C. butyricum | Glycerol excess | Growth + minimized enzyme/ATP usage | Growth prediction | Accurate phenotype state | [18] |
| CHO cells | Standard culture | Maximal growth | Growth prediction | Significant overestimation | [19] |
Detailed Experimental Protocols
Protocol: Multi-Scale Modeling of Yeast Replicative Ageing
The enzyme-constrained FBA approach integrated within a multi-scale model of yeast replicative ageing provides a robust framework for evaluating objective functions [3].
Methodology:
- Metabolic Modeling: Implement an enzyme-constrained FBA model of central carbon metabolism with constraints on total enzyme pool
- Regulatory Network Integration: Connect metabolic outputs to a vector-based Boolean model of nutrient sensing pathways (Snf1, PKA, TOR, Yap1, Sln1)
- Dynamic Simulation: Feed optimal fluxes into an ODE model of damage accumulation and cell growth, solved iteratively over time
- Division Tracking: Monitor biomass production until FBA becomes infeasible (cell death), recording division count and generation time
Key Constraints and Parameters:
- Total enzyme pool limited by σfPtot (average saturation σ × fraction of enzymes covered f × total protein content Ptot)
- Non-metabolic damage formation rate (f0) = 0.0001
- Damage repair rate (r0) = 0.0005
- Regulation factor = 0.04
Lexicographic Optimization Strategy: The approach utilizes successive optimizations with controlled flexibility [3]:
- Optimize primary objective (e.g., maximal growth)
- Constrain primary objective to optimal value with flexibility factor ε₁ ≤ 1
- Optimize secondary objective (e.g., flux minimization) within this constrained solution space
- Allow flexibility ε₂ ≤ 1 for subsequent regulation steps to maintain feasibility
Protocol: Genome-Scale Model Reconstruction and Validation for Clostridium butyricum
The iCbu641 model reconstruction and validation demonstrates condition-dependent performance of objective functions [18].
Model Reconstruction:
- Draft Construction: Build initial model from RAST annotation (641 genes, 365 enzymes, 671 reactions, 606 metabolites)
- Gap Analysis: Identify 303 blocked metabolites using GapFind
- Curation: Add 59 reactions based on experimental fermentation evidence and curated GSM models
- Biomass Reaction Formulation: Adapt from C. beijerinckii with inclusion of proton formation for charge balance
Final Model Specifications:
- 641 genes, 891 reactions, 701 metabolites
- Elemental composition per C atom: CH₁.₆₂₄O₀.₄₅₆N₀.₂₁₆P₀.₀₃₃S₀.₀₀₄₇
- Includes PDO dehydrogenase (EC.1.1.1.202) and glycerol dehydratase (EC.4.2.1.30)
Validation Approach:
- Compare flux distribution predictions with experimental proteomic data (84% agreement achieved)
- Evaluate phenotype states under different culture conditions
- Test robustness through enzyme deletions and biomass composition variations
Visualization of Methodologies and Metabolic Relationships
Flux Balance Analysis with Multi-Stage Optimization
FBA Multi-Stage Optimization Workflow: This diagram illustrates the lexicographic optimization approach where a primary objective is optimized first, followed by a secondary objective within a constrained solution space with defined flexibility factors [3].
Multi-Scale Integration of Metabolism and Ageing
Multi-Scale Model Integration: This workflow shows how metabolic models are integrated with regulatory networks and dynamic damage accumulation to simulate cellular ageing processes, enabling evaluation of how objective functions impact lifespan [3].
Research Reagent Solutions and Computational Tools
Table 3: Essential Research Reagents and Computational Tools for Objective Function Validation
| Item | Type | Function/Application | Example Implementation |
|---|---|---|---|
| Enzyme-Constrained FBA | Computational Framework | Incorporates enzyme kinetics and capacity limitations into FBA | Total enzyme pool constraint: σfPtot [3] |
| Lexicographic Optimization | Computational Method | Solves multi-objective optimization with priority ranking | Two-stage approach with flexibility factors ε₁, ε₂ [3] |
| Genome-Scale Model iCbu641 | Metabolic Model | Clostridium butyricum-specific network for PDO production | 641 genes, 891 reactions, 701 metabolites [18] |
| Boolean Regulatory Network | Computational Model | Simulates nutrient sensing and stress response pathways | Snf1, PKA, TOR, Yap1, Sln1 pathways [3] |
| Dynamic ODE Model | Computational Model | Simulates damage accumulation and cellular growth over time | Parameters: f0=0.0001, r0=0.0005 [3] |
| TIObjFind Framework | Computational Tool | Identifies context-specific objective functions from data | Uses Coefficients of Importance (CoIs) [10] |
| Uptake-rate Objective Functions (UOFs) | Computational Approach | Minimizes non-essential nutrient uptake for mammalian cells | Resolves essential amino acid limitations in CHO cells [19] |
| ThermOptCOBRA | Computational Tool | Ensures thermodynamic feasibility in flux predictions | Eliminates thermodynamically infeasible cycles [20] |
The comparative analysis presented in this guide demonstrates that the performance of objective functions in FBA is highly context-dependent, varying with organism type, environmental conditions, and biological process being studied. Maximal growth serves as a reliable objective for microbial systems under standard conditions but frequently fails in mammalian cells or under substrate excess. ATP production objectives rarely stand alone but can significantly enhance predictions when combined with other objectives. Parsimonious solutions consistently improve predictions across diverse contexts by incorporating constraints on cellular resources.
For researchers designing FBA studies, the following evidence-based recommendations emerge:
- For microbial growth prediction in standard conditions, maximal growth with parsimonious enzyme usage provides the most accurate results
- For mammalian cells or conditions with multiple essential nutrients, uptake-rate objective functions (UOFs) overcome limitations of traditional biomass maximization
- For substrate excess conditions or enzyme-limited scenarios, parsimonious solutions that minimize total flux or enzyme usage better capture cellular physiology
- For dynamic processes like ageing, multi-objective approaches combining growth with energy production or flux minimization yield the most biologically realistic simulations
The ongoing development of data-driven frameworks like TIObjFind that automatically infer objective functions from experimental data represents a promising direction for the field, potentially moving beyond predefined objective functions to context-specific optimization principles [10].
Multi-Objective Optimization and Hybrid Approaches
The accurate prediction of cellular behavior, particularly metabolic fluxes, is a cornerstone of modern systems biology and drug development. This process is inherently a multi-objective optimization problem (MOOP), where researchers must balance conflicting goals such as maximizing biomass production, minimizing energy expenditure, and optimizing product yield simultaneously [8]. Traditional single-objective approaches often fail to capture the complex trade-offs that cells make in response to environmental changes, leading to inaccurate predictions. The emergence of hybrid approaches that combine mechanistic models with machine learning represents a paradigm shift in computational biology, offering enhanced predictive power while maintaining biological plausibility [21].
This comparative analysis examines the landscape of multi-objective optimization methodologies for flux prediction, with particular emphasis on their application in drug discovery and metabolic engineering. We objectively evaluate the performance of three prominent frameworks—neural-mechanistic hybrids, topology-informed optimization, and evolutionary algorithms—providing researchers with a comprehensive guide to selecting appropriate methodologies for specific research scenarios. The performance of these approaches is assessed through standardized benchmarking tasks and quantitative metrics, enabling direct comparison of their respective strengths and limitations in addressing the complex challenges of biological system optimization.
Comparative Analysis of Multi-Objective Optimization Frameworks
Performance Benchmarking of Optimization Approaches
Table 1: Quantitative Performance Comparison of Multi-Objective Optimization Frameworks
| Optimization Framework | Prediction Accuracy (%) | Computational Efficiency | Key Performance Metrics | Typical Applications |
|---|---|---|---|---|
| Neural-Mechanistic Hybrid (AMN) | N/A | Training time: 25.7s [22] | CPU usage: 10.55% [22]; Outperforms FBA in quantitative phenotype predictions [21] | Microbial growth prediction; Gene knockout phenotype prediction [21] |
| DBI-LSTM-2AM-PSO | 95.53 [22] | Fitness value: 0.47 [22] | F1 score: 91.41%; MSE: 0.049 [22] | Renewable energy prediction; Distributed power generation systems [22] |
| Evolutionary Algorithm (MoGA-TA) | Success rate significantly improved over NSGA-II [23] | Maintains population diversity; Prevents premature convergence [23] | Dominating hypervolume; Geometric mean; Internal similarity [23] | Drug molecule optimization; Multi-property molecular design [23] |
| Topology-Informed (TIObjFind) | Good match with experimental data [8] | Identifies pathway-specific weighting factors [8] | Reduces prediction errors; Captures stage-specific metabolic objectives [8] | Metabolic network analysis; Cellular response prediction under changing conditions [8] |
Experimental Protocols and Methodologies
Neural-Mechanistic Hybrid Approach (AMN)
The Artificial Metabolic Network (AMN) framework embeds Flux Balance Analysis (FBA) within artificial neural networks to overcome the gradient backpropagation limitation of traditional simplex solvers [21]. The experimental protocol involves:
- Network Architecture: A trainable neural layer followed by a mechanistic layer (Wt-solver, LP-solver, or QP-solver) [21]
- Training Process: The neural layer computes initial flux values (V0) from medium uptake flux bounds (Vin) or medium compositions (Cmed)
- Constraint Integration: Custom loss functions surrogate FBA constraints while enabling gradient computation
- Validation: Comparison of predicted fluxes (Vout) with reference fluxes from FBA simulations or experimental data
This approach demonstrates systematic outperformance over constraint-based models, requiring training set sizes orders of magnitude smaller than classical machine learning methods [21]. The hybrid architecture successfully captures metabolic enzyme regulation and predicts gene knockout effects on phenotype.
DBI-LSTM-2AM-PSO for Renewable Energy Systems
The DBI-LSTM-2AM-PSO model combines deep learning with improved particle swarm optimization for distributed power generation systems [22]:
- Prediction Component: Dense Bidirectional Long Short-Term Memory with Attention Mechanism (DBI-LSTM-AM) performs time-series forecasting of energy demand
- Optimization Component: Adaptive Linear Decreasing Inertia Weight Particle Swarm Optimization with Mutation Strategy (ALD-MPSO) simultaneously optimizes economic efficiency, environmental benefits, and system reliability
- Integration: The fused model achieves 95.53% prediction accuracy with 25.7s training time and low computational resource consumption (10.55% CPU usage) [22]
Experimental validation demonstrates superior performance over benchmark algorithms across multiple metrics including mean squared error (0.049) and F1 score (91.41%) [22].
MoGA-TA for Molecular Optimization
The Multi-objective Genetic Algorithm with Tanimoto similarity and Adaptive acceptance probability (MoGA-TA) addresses drug molecule optimization through:
- Similarity Calculation: Tanimoto similarity-based crowding distance captures molecular structural differences [23]
- Population Update: Dynamic acceptance probability strategy balances exploration and exploitation during evolution [23]
- Optimization Process: Decoupled crossover and mutation strategy in chemical space continues until predefined stopping conditions
- Evaluation Metrics: Success rate, dominating hypervolume, geometric mean, and internal similarity assess algorithm performance [23]
Benchmark evaluation across six molecular optimization tasks demonstrates significant improvements in efficiency and success rate compared to NSGA-II and GB-EPI [23].
Visualization of Methodological Relationships and Workflows
Figure 1: Methodology Taxonomy for Multi-Objective Optimization. This diagram illustrates the hierarchical relationship between broad optimization categories and their specific implementations discussed in this review, highlighting the diverse methodological approaches available for flux prediction and biological system optimization.
Figure 2: TIObjFind Framework Workflow. This workflow diagram outlines the three key steps in the Topology-Informed Objective Find methodology, demonstrating how experimental data and network topology are integrated to identify critical pathways and compute Coefficients of Importance for metabolic objective functions [8] [10].
Table 2: Key Research Reagent Solutions for Multi-Objective Optimization Studies
| Resource Category | Specific Tools | Function and Application | Implementation Details |
|---|---|---|---|
| Constraint-Based Modeling | Flux Balance Analysis (FBA) [8] [21] | Predicts metabolic flux distributions at steady state | Requires stoichiometric matrix, flux bounds, objective function |
| Metabolic Pathway Analysis | TIObjFind [8] | Identifies objective functions aligning with experimental data | MATLAB implementation with maxflow package [10] |
| Deep Learning Architectures | DBI-LSTM-AM [22] | Time-series forecasting of energy demand | Combines Bi-LSTM, Dense layers, and Attention Mechanism |
| Hybrid Neural-Mechanistic | Artificial Metabolic Networks (AMN) [21] | Enhances constraint-based model predictions | Embeds FBA within neural networks; enables gradient backpropagation |
| Multi-Objective Evolutionary Algorithms | NSGA-II [23], MoGA-TA [23] | Optimizes multiple molecular properties simultaneously | Uses non-dominated sorting and crowding distance |
| Molecular Similarity Metrics | Tanimoto Coefficient [23] | Quantifies structural similarity between molecules | Based on fingerprint comparisons; range 0-1 |
| Optimization Algorithms | ALD-MPSO [22] | Adaptive particle swarm optimization for multiple objectives | Mutation strategy prevents premature convergence |
| Data Sources | KEGG [8], EcoCyc [8], ChEMBL [23] | Provides metabolic pathways and molecular data | Foundation for stoichiometric models and benchmarking |
This comparative analysis demonstrates that the selection of appropriate multi-objective optimization strategies must be guided by specific research contexts and constraints. Neural-mechanistic hybrid models offer superior performance for quantitative phenotype predictions when sufficient training data is available, effectively bridging the gap between mechanistic understanding and predictive power [21]. Topology-informed approaches like TIObjFind provide critical insights into pathway contributions and adaptive cellular responses, making them particularly valuable for metabolic engineering applications where elucidation of biological mechanisms is prioritized [8] [10]. Evolutionary algorithms excel in molecular optimization tasks where multiple physicochemical properties must be balanced simultaneously, with enhanced techniques like MoGA-TA addressing the critical challenge of maintaining diversity in chemical space exploration [23] [24].
The continuing evolution of multi-objective optimization methodologies points toward increased integration of mechanistic constraints with machine learning approaches, offering researchers an expanding toolkit for addressing the complex challenges in flux prediction and drug development. As these hybrid frameworks mature, they promise to significantly accelerate the discovery and optimization cycle while providing deeper insights into the fundamental principles governing biological systems.
Flux analysis is a critical computational technique for quantifying the flow of molecules, energy, or information through biological, chemical, and engineering systems. In metabolic engineering, it describes the rates at which nutrients are converted into biomass and products through biochemical reactions. In engineering systems, it can represent heat or particle flow. Accurately predicting these fluxes is fundamental to optimizing bioprocesses, understanding cellular physiology, and designing efficient industrial systems. Traditional methods, particularly in metabolic engineering, have relied heavily on constraint-based modeling approaches like Flux Balance Analysis (FBA), which predict steady-state flux distributions by assuming the cell optimizes an objective, such as biomass maximization [8] [25] [21].
However, these mechanistic models face challenges, including an inherent inability to fully capture the complex regulatory mechanisms of cells and a frequent reliance on difficult-to-measure input parameters, such as nutrient uptake rates [21]. The emergence of machine learning (ML) offers powerful new tools to overcome these limitations. ML models can learn complex, non-linear relationships directly from experimental data, leading to more accurate and generalizable predictions. This guide provides a comparative analysis of two prominent ML frameworks in flux analysis: the versatile Artificial Neural Network (ANN) and the specialized ML-Flux, detailing their performance, experimental protocols, and applications to help researchers select the appropriate tool for their specific objectives.
Performance Comparison of ANN and ML-Flux Frameworks
The performance of ANN and ML-Flux varies significantly depending on the application domain, data availability, and specific prediction task. The following tables summarize their key characteristics and quantitative performance metrics based on recent studies.
Table 1: Overall Framework Characteristics and Application Scope
| Feature | ANN Framework | ML-Flux Framework |
|---|---|---|
| Primary Application Domain | Diverse: Membrane desalination, nuclear reactor safety, metabolic modeling [26] [27] [28] | Specialized: Central Carbon Metabolism in biological systems [29] |
| Core Methodology | Neural networks learning input-output relationships from data; often used in hybrid mechanistic-ML models [26] [21] | Pre-trained neural networks mapping isotope labeling patterns directly to metabolic fluxes [29] |
| Key Input Features | System-specific parameters (e.g., temperatures, flow rates, control rod positions, medium composition) [26] [28] [21] | Mass Isotopomer Distribution (MID) from 13C-tracer experiments [29] |
| Typical Output | System-specific fluxes (e.g., permeate flux, critical heat flux), growth rates, or operational parameters [26] [30] [21] | Net and exchange fluxes in metabolic networks [29] |
| Major Advantage | Flexibility; can be integrated with mechanistic models for improved generalization with small datasets [21] | High speed and accuracy for 13C-Metabolic Flux Analysis (13C-MFA); can impute missing labeling data [29] |
Table 2: Quantitative Performance Metrics from Experimental Studies
| Framework & Model | Application / Task | Performance Metrics | Reference |
|---|---|---|---|
| ANN (ANFIS-C4 Hybrid) | Flux prediction in water desalination (DCMD) | Training: 100% accuracy, RMSE=0.0522Testing: 99.73% accuracy, RMSE=0.7121 | [26] |
| ANN (Classification Model) | Critical Heat Flux (CHF) prediction in a CANDU reactor | RMSE: ~2.5%; Reduced overfitting compared to regression ANN | [27] |
| ANN (Hybrid Lookup Table) | CHF prediction in vertical tubes | rRMSE: 9.3%, outperforming standalone ML models and lookup tables | [30] |
| Hybrid Neural-Mechanistic (AMN) | Growth rate prediction for E. coli and P. putida | Outperformed standard FBA; required orders of magnitude less training data than pure ML | [21] |
| ML-Flux | Flux prediction in Central Carbon Metabolism | >90% of predictions more accurate than conventional MFA software; computation is consistently faster | [29] |
Framework Architecture and Workflow
The ANN Framework: Versatility through Hybrid Modeling
Artificial Neural Networks are a class of ML algorithms that learn complex relationships through interconnected layers of nodes. In flux analysis, ANNs are often not used in isolation but as part of a hybrid mechanistic-ML architecture. This combines the data-driven learning power of ML with the established scientific principles of mechanistic models, improving predictive power even with small training datasets [21].
A prominent example is the Artificial Metabolic Network (AMN), which embeds a mechanistic FBA model within a neural network. The neural network layer learns to predict optimal uptake flux constraints from environmental conditions, which are then fed into the mechanistic layer to compute the steady-state metabolic phenotype [21]. This hybrid approach overcomes a key FBA limitation—the inaccurate estimation of uptake fluxes from extracellular concentrations.
Diagram: Workflow of a Hybrid Neural-Mechanistic Model for Metabolic Flux Prediction
The ML-Flux Framework: Specialized Speed for 13C-MFA
ML-Flux is a specialized framework designed to accelerate and improve the accuracy of 13C-Metabolic Flux Analysis (13C-MFA), a gold-standard method for determining intracellular metabolic fluxes. Unlike traditional 13C-MFA, which uses iterative, computationally expensive optimization to fit fluxes to experimental isotope labeling data, ML-Flux uses pre-trained neural networks to directly map Mass Isotopomer Distributions (MIDs) to metabolic fluxes [29].
The framework employs two key neural network models: a Partial Convolutional Neural Network (PCNN) that imputes missing isotope labeling patterns in experimental data, and an Artificial Neural Network (ANN) that takes the complete set of MIDs as input and outputs the predicted metabolic fluxes. This creates a streamlined, highly efficient pipeline that bypasses the need for repeated model simulations and optimizations.
Diagram: ML-Flux Workflow for Metabolic Flux Quantitation
Experimental Protocols and Methodologies
Protocol for Developing an ANN for Flux Prediction
The development of a robust ANN model for flux prediction involves a standardized sequence of steps, from data collection to model deployment.
- Data Collection and Curation: Assemble a comprehensive dataset encompassing a wide range of input conditions and corresponding output fluxes. For a desalination membrane flux prediction model, inputs may include feed temperature, coolant flow rate, and salinity [26]. For a metabolic model, inputs could be environmental conditions like medium composition [21]. The dataset must be cleaned and normalized.
- Dataset Partitioning: Randomly split the dataset into three subsets:
- Training Set (~70%): Used to adjust the weights of the neural network.
- Validation Set (~15%): Used to tune hyperparameters (e.g., learning rate, number of layers) and prevent overfitting during training.
- Test Set (~15%): Used only once for the final evaluation of the model's generalization performance.
- Model Architecture Selection and Training: Choose an appropriate network architecture (e.g., feedforward, convolutional). Train the model using an optimization algorithm (e.g., Adam, SGD) to minimize a loss function, such as Mean Squared Error (MSE) between predictions and actual flux values. For hybrid models, the loss function also incorporates mechanistic constraints [21].
- Model Validation and Benchmarking: Rigorously evaluate the trained model on the test set using metrics like Root Mean Square Error (RMSE) and prediction accuracy. Compare its performance against existing empirical correlations, mechanistic models, or other ML algorithms to establish its superiority [26] [30].
- Deployment and Real-Time Monitoring: Integrate the validated model into the target application, such as a real-time monitoring system for a nuclear reactor [28] [30] or a bioprocess control system.
Protocol for Applying ML-Flux to 13C-MFA
ML-Flux simplifies the traditional 13C-MFA workflow by replacing the iterative optimization steps with a single forward pass through a pre-trained neural network.
- Tracer Experiment and Data Acquisition:
- Cell Cultivation: Grow cells in a defined medium where a key carbon source (e.g., glucose) is replaced with a 13C-labeled tracer (e.g., [1,2-13C2]-glucose).
- Metabolite Extraction and Measurement: Harvest cells during mid-exponential growth and extract intracellular metabolites. Measure the Mass Isotopomer Distributions (MIDs) of proteinogenic amino acids or central carbon metabolites using Mass Spectrometry (MS) [29].
- Data Preprocessing and Imputation:
- Input the measured MIDs into the pre-trained Partial CNN (PCNN) component of ML-Flux. This network imputes any missing MID values for metabolites that were not reliably measured, creating a complete input vector for the next stage [29].
- Flux Prediction:
- Feed the completed set of MIDs into the pre-trained ANN model. The network outputs the predicted net and exchange fluxes for the entire metabolic network model it was trained on (e.g., Central Carbon Metabolism) [29].
- Validation and Analysis:
- While ML-Flux is inherently fast, it is good practice to validate key predictions by comparing the simulated MIDs generated from the predicted fluxes against the original experimental data to ensure consistency. The predicted flux map can then be analyzed to draw biological conclusions.
Essential Research Reagents and Computational Tools
Successful implementation of these ML frameworks requires a combination of wet-lab reagents and computational resources.
Table 3: Key Research Reagents and Tools for Flux Analysis
| Category | Item / Tool | Specific Example / Function | Relevance |
|---|---|---|---|
| Wet-Lab Reagents | 13C-Labeled Tracers | [1,2-13C2]-glucose, 13C-glutamine | Creates unique isotope labeling patterns for ML-Flux input [29] |
| Analytical Instrumentation | Mass Spectrometry (MS) | Measures Mass Isotopomer Distributions (MIDs) from tracer experiments [29] | |
| Biological Media Components | Defined chemical media for microbial cultivation | Provides controlled environmental conditions for training hybrid ANN models [21] | |
| Computational Tools & Models | Genome-Scale Model (GEM) | E. coli iML1515, P. putida iJN1463 | Provides the structural metabolic network for FBA and hybrid AMN models [21] |
| ML-Flux Web Resource | metabolicflux.org | Provides pre-trained models for direct flux prediction from MIDs [29] | |
| Programming Frameworks | TensorFlow, PyTorch, Cobrapy | Libraries for building ANNs and performing constraint-based modeling [27] [21] | |
| Thermodynamic Data | Gibbs free energy of metabolites (ΔG) | Used as constraints in models like REMI and TFA to improve flux prediction accuracy [6] |
The integration of machine learning into flux analysis represents a significant leap forward. The choice between a versatile ANN and the specialized ML-Flux framework depends entirely on the research goal. For problems requiring integration with mechanistic models, prediction of diverse outputs, or operation with limited data, the hybrid ANN approach is a powerful and flexible solution. In contrast, for high-throughput, highly accurate quantification of fluxes in central metabolism using 13C-tracer data, ML-Flux offers unmatched speed and precision.
Future developments will likely see a deeper fusion of mechanistic and ML models, improved generalization of models like ML-Flux to larger metabolic networks and diverse organisms, and the increasing use of transfer learning to adapt pre-trained models to new, specific conditions with minimal additional data. These advancements will further solidify the role of ML as an indispensable tool in the systems biologist's and engineer's toolkit.
Data-Driven Objective Function Identification with TIObjFind
Flux Balance Analysis (FBA) serves as a cornerstone of computational systems biology, enabling researchers to predict metabolic fluxes in various organisms. However, the predictive accuracy of FBA heavily depends on the selection of an appropriate biological objective function, which represents the cellular goal driving metabolic activity [4]. Traditional FBA implementations often utilize static objectives such as biomass maximization, which may not accurately capture cellular behavior under dynamic environmental conditions or in complex multi-species systems [10] [8].
To address this fundamental limitation, novel computational frameworks have emerged that leverage experimental data to infer context-specific objective functions. This comparative analysis examines TIObjFind (Topology-Informed Objective Find), a recently developed optimization framework that integrates Metabolic Pathway Analysis (MPA) with FBA to identify metabolic objectives [10] [8]. We evaluate its performance against traditional FBA and other contemporary approaches, providing researchers with a comprehensive assessment of methodological capabilities, experimental requirements, and practical applications in metabolic engineering and drug discovery.
Traditional Flux Balance Analysis (FBA)
FBA operates on the principle of stoichiometric mass balance, constraining the solution space within a metabolic network and identifying flux distributions that optimize a predefined cellular objective [4]. The method assumes steady-state metabolic operation and utilizes linear programming to compute optimal flux distributions. Common biological objectives include biomass production, ATP generation, or synthesis of specific metabolites. While FBA has demonstrated considerable utility in predicting metabolic phenotypes, particularly in microbial systems, its reliance on a single, pre-specified objective function represents a significant limitation when modeling complex biological behaviors or adaptive cellular responses [10].
The ObjFind Framework
The ObjFind framework represents an initial approach to objective function identification, introducing Coefficients of Importance (CoIs) that quantify each metabolic flux's contribution to a composite objective function [10] [8]. This method formulates a multi-objective optimization problem that maximizes a weighted sum of fluxes while minimizing the sum of squared deviations from experimental flux data. Each coefficient cj reflects a reaction's relative importance, with higher values indicating closer alignment between experimental fluxes and their maximum theoretical potential. While ObjFind demonstrated improved alignment with experimental data compared to traditional FBA, it exhibits tendencies toward overfitting and requires comprehensive isotopomer analysis for experimental flux determination [8].
TIObjFind: Topology-Informed Objective Find
TIObjFind extends the ObjFind concept by integrating Metabolic Pathway Analysis (MPA) with FBA, creating a topology-informed framework that enhances biological interpretability while reducing overfitting risks [10]. The methodology employs three key steps: (1) reformulating objective function selection as an optimization problem minimizing differences between predicted and experimental fluxes; (2) mapping FBA solutions to a Mass Flow Graph (MFG) for pathway-based interpretation; and (3) applying a minimum-cut algorithm to extract critical pathways and compute pathway-specific Coefficients of Importance [10]. This approach selectively evaluates fluxes in key pathways rather than the entire network, significantly improving interpretability of complex metabolic networks and capturing metabolic flexibility during environmental adaptations [8].
Flux Cone Learning: A Machine Learning Alternative
Flux Cone Learning (FCL) represents a fundamentally different approach that employs Monte Carlo sampling and supervised learning to predict deletion phenotypes based on the geometry of the metabolic space [11]. This method utilizes the observation that gene deletions perturb the shape of the flux cone—the high-dimensional polytope defined by stoichiometric constraints—and correlates these geometric changes with experimental fitness scores using machine learning classifiers. Unlike FBA-based approaches, FCL operates without optimality assumptions, potentially offering advantages for complex organisms where cellular objectives are poorly defined [11].
Table 1: Comparative Overview of Methodological Approaches
| Feature | Traditional FBA | ObjFind | TIObjFind | Flux Cone Learning |
|---|---|---|---|---|
| Core Principle | Linear programming with predefined objective | Weighted sum of fluxes with CoIs | MPA-integrated FBA with pathway-specific CoIs | Monte Carlo sampling with machine learning |
| Objective Function | Single, user-defined | Data-inferred combination | Topology-informed, pathway-weighted | Not required |
| Experimental Data Requirements | Limited (growth rates, uptake/secretion) | Extensive (isotopomer flux data) | External flux measurements | Fitness data from deletion screens |
| Network Topology Utilization | Implicit via stoichiometry | Limited | Explicit via Mass Flow Graph | Implicit via flux cone geometry |
| Key Output | Optimal flux distribution | Flux distribution + CoIs | Flux distribution + pathway CoIs | Phenotype predictions |
| Computational Demand | Low | Moderate | High | Very High |
Experimental Protocols and Methodologies
TIObjFind Implementation Workflow
The TIObjFind framework implements a structured computational workflow to infer metabolic objectives:
Step 1: Single-Level Optimization Reformulation Researchers reformulate the traditional FBA problem using duality theory, transforming it into a single-level optimization that minimizes the difference between predicted and experimental fluxes while maximizing an inferred metabolic goal. This formulation incorporates thermodynamic, mass balance, and uptake constraints, with dual variables (ui and g) reflecting the sensitivity of the optimal objective value to constraint modifications [31].
Step 2: Mass Flow Graph Construction The computed flux distributions from FBA solutions are mapped onto a Mass Flow Graph where primal reactions become metabolites in the dual network, and primal metabolites serve as constraints. Self-loops represent autocatalytic reactions, visually capturing internal metabolic fluxes and their interconnections [31].
Step 3: Pathway Importance Normalization The framework applies a minimum-cut algorithm (typically Boykov-Kolmogorov for computational efficiency) to the Mass Flow Graph to identify critical pathways. The resulting edge weights are normalized to determine pathway-specific Coefficients of Importance, leading to a refined objective reaction flux distribution [10] [31].
Figure 1: TIObjFind Computational Workflow. The framework integrates experimental data with stoichiometric models to derive pathway-specific objective functions through graph-based analysis.
Flux Cone Learning Methodology
The Flux Cone Learning approach implements a distinct four-component workflow for phenotype prediction:
Model Preparation: Start with a genome-scale metabolic model (GEM) defined by stoichiometric constraints S·v = 0 and flux bounds Vimin ≤ vi ≤ Vimax. Gene deletions are implemented through gene-protein-reaction maps that zero out appropriate flux bounds [11].
Monte Carlo Sampling: Generate multiple random flux samples (typically 100-5000 per deletion) from the metabolic space of each gene deletion variant using appropriate sampling algorithms. This creates a high-dimensional feature set representing the shape of each deletion's flux cone [11].
Supervised Learning: Train machine learning models (random forests perform optimally) using flux samples as features and experimental fitness measurements as labels. All samples from the same deletion cone receive identical labels, creating an expanded training dataset [11].
Prediction Aggregation: Apply majority voting or averaging to aggregate sample-wise predictions into deletion-wise phenotype forecasts, generating final essentiality predictions or production capabilities [11].
Performance Comparison and Experimental Validation
Predictive Accuracy Assessment
Gene Essentiality Prediction: In comprehensive evaluations using E. coli metabolic models, Flux Cone Learning achieved approximately 95% accuracy in predicting metabolic gene essentiality across multiple carbon sources, outperforming traditional FBA which reached 93.5% accuracy [11]. FCL demonstrated particular improvements in classifying essential genes (6% enhancement) and non-essential genes (1% enhancement) compared to FBA. The method maintained strong performance even with sparse sampling, matching FBA accuracy with as few as 10 samples per deletion cone [11].
Flux Prediction Alignment: TIObjFind demonstrated superior alignment with experimental flux data in case studies involving Clostridium acetobutylicum fermentation and multi-species isopropanol-butanol-ethanol (IBE) production systems [8]. The framework successfully captured stage-specific metabolic objectives and adaptive cellular responses, reducing prediction errors while improving consistency with experimental observations. The topology-informed approach particularly excelled in identifying metabolic shifts during phase transitions in fermentation processes [10] [8].
Table 2: Quantitative Performance Comparison Across Methodologies
| Performance Metric | Traditional FBA | ObjFind | TIObjFind | Flux Cone Learning |
|---|---|---|---|---|
| Gene Essentiality Accuracy (E. coli) | 93.5% | Not Reported | Not Reported | 95% |
| Essential Gene Classification | Baseline | Not Reported | Not Reported | +6% |
| Non-Essential Gene Classification | Baseline | Not Reported | Not Reported | +1% |
| Flux Prediction Error Reduction | Baseline | Moderate | Significant | Not Applicable |
| Stage-Specific Adaptation Capture | Limited | Moderate | Strong | Not Reported |
| Minimum Data Requirements | Low | High | Moderate | Very High |
| Computational Time | Fastest | Fast | Moderate | Slowest |
Application Case Studies
Clostridium acetobutylicum Fermentation: TIObjFind was applied to glucose fermentation by C. acetobutylicum to determine pathway-specific weighting factors [8]. By applying different weighting strategies to Coefficients of Importance, researchers demonstrated substantial impacts on flux prediction accuracy, significantly reducing errors while improving alignment with experimental data. The framework successfully identified shifting metabolic priorities throughout different fermentation stages, demonstrating capabilities in capturing dynamic metabolic adaptations [10] [8].
Multi-Species IBE System: In a more complex application, TIObjFind analyzed a multi-species system comprising C. acetobutylicum and C. ljungdahlii for isopropanol-butanol-ethanol production [8]. The method employed Coefficients of Importance as hypothesis coefficients within objective functions to assess cellular performance, demonstrating strong agreement with observed experimental data while effectively capturing stage-specific metabolic objectives that would be missed by traditional FBA approaches [8].
Cross-Organism Essentiality Prediction: Flux Cone Learning was validated across organisms of varying complexity, including E. coli, Saccharomyces cerevisiae, and Chinese Hamster Ovary cells [11]. The method consistently outperformed FBA in metabolic gene essentiality prediction, with particular advantages in higher-order organisms where cellular objectives are poorly defined or nonexistent. This demonstrates the method's versatility beyond microbial systems [11].
Research Reagent Solutions
Table 3: Essential Research Resources for Implementation
| Resource Category | Specific Tools/Platforms | Function/Purpose |
|---|---|---|
| Computational Environments | MATLAB with maxflow package [10] | Primary implementation platform for TIObjFind with graph analysis capabilities |
| Programming Languages | Python with pySankey package [10] | Visualization and supplementary analysis |
| Metabolic Databases | KEGG, EcoCyc [8] | Foundational sources for pathway, genomic, and reaction information |
| Metabolic Modeling Tools | COBRA Toolbox, FlexFlux [8] | Constraint-based reconstruction and analysis |
| Machine Learning Frameworks | Scikit-learn, TensorFlow (for FCL) [11] | Implementation of random forests and neural networks for phenotype prediction |
| Data Sources | Community Innovation Survey (CIS) [32] | Firm-level innovation data for predictive modeling |
| Sampling Algorithms | Monte Carlo Samplers (for FCL) [11] | Generation of flux distributions for machine learning features |
This comparative analysis demonstrates that data-driven approaches for objective function identification significantly enhance metabolic prediction capabilities compared to traditional FBA. TIObjFind provides substantial advantages in contexts where understanding pathway-level contributions and adaptive metabolic responses is critical, particularly in bioprocessing applications with dynamic environmental conditions. Its integration of metabolic pathway analysis with flux balance analysis creates a biologically interpretable framework that aligns computational predictions with experimental observations while maintaining mechanistic insights.
Flux Cone Learning represents a paradigm shift from optimization-based to geometry-based prediction, excelling in gene essentiality forecasting without requiring explicit objective function specification. This approach appears particularly valuable for complex organisms where cellular objectives remain poorly characterized or in applications focused specifically on deletion phenotype prediction.
The selection between these methodologies should be guided by research objectives, data availability, and computational resources. TIObjFind offers superior capabilities for mapping metabolic adaptations and identifying driving objectives in dynamic systems, while Flux Cone Learning provides best-in-class essentiality prediction without optimality assumptions. Both approaches represent significant advances over traditional FBA, enabling more accurate, context-specific prediction of metabolic behaviors across diverse biological systems and applications.
Flux Balance Analysis (FBA) is a powerful constraint-based method for studying genome-scale metabolic networks. A fundamental aspect of FBA is the requirement of an objective function, which the model optimizes to predict metabolic fluxes. The choice of this objective function is crucial, as it determines the predicted flux distribution and, consequently, any downstream biological interpretations derived from the model [3] [4]. While evolutionary arguments often guide the selection of objectives such as maximizing biomass (growth) or energy (ATP) production, the direct connection between these choices and long-term cellular processes like aging has remained less explored [3].
This case study investigates how different objective functions in FBA impact the prediction of replicative lifespan (RLS) in budding yeast (Saccharomyces cerevisiae). The yeast RLS, defined as the number of mitotic divisions a mother cell undergoes before death, serves as a valuable model for eukaryotic aging [33] [34]. We leverage a multi-scale mathematical model that integrates enzyme-constrained FBA with modules for regulatory networks and damage accumulation to simulate how metabolic objectives influence aging [3]. By systematically comparing objective functions, this analysis provides a framework for selecting appropriate modeling strategies to study aging and other complex cellular processes.
Methodological Framework: Integrating Metabolism with Lifespan Analysis
Multi-Scale Model of Yeast Metabolism and Aging (yMSA)
The core of this analysis utilizes a multi-scale mathematical model (yMSA) that links metabolic activity to replicative aging [3]. The model comprises three integrated components:
- Metabolic Module: An enzyme-constrained FBA model of the central carbon metabolism. This module calculates optimal metabolic fluxes (
v) constrained by enzyme usage (e), which is limited by the total cellular enzyme pool [3]. - Regulatory Network: A Boolean model representing key signaling pathways (Snf1, PKA, TOR, Yap1, Sln1) that respond to metabolic states and constrain enzyme usage further [3].
- Dynamic Damage Accumulation Model: An ordinary differential equation (ODE) model that tracks the accumulation of protein damage over time. Damage arises from reactive oxygen species (ROS) generated by metabolism and is asymmetrically distributed during cell division. The model simulates cell growth and division, with lifespan ending when damage levels preclude metabolic feasibility [3].
Experimental Protocol for Model Simulation
The simulation protocol for assessing replicative lifespan is as follows [3]:
- Model Initialization: The metabolic FBA model is initialized with a specific objective function and constraints reflecting nutrient conditions.
- Flux Calculation: The model solves the linear programming problem to find an optimal flux distribution. A two-stage lexicographic optimization is often used, where a primary objective (e.g., maximal growth) is first optimized, and then a secondary objective (e.g., flux minimization) is pursued within a flexible boundary of the primary solution [3].
- Regulatory Feedback: The optimal fluxes inform the input layer of the Boolean regulatory network. The regulatory output, in turn, imposes stricter constraints on enzyme usage in the metabolic model [3].
- Dynamic Simulation: The constrained metabolic fluxes are used to update the ODE model for one time step, calculating damage accumulation, biomass growth, and maintenance costs [3].
- Cell Division and Lifespan Tracking: Steps 2-4 are repeated. When sufficient biomass is synthesized, cell division occurs. The replicative lifespan is recorded as the number of successful divisions before the model becomes infeasible due to excessive damage [3].
Objective Functions Tested
The study systematically tested several common objective functions and their combinations [3]:
- Maximal growth: Maximizing the flux through the biomass reaction.
- Maximal/Minimal ATP production: Maximizing or minimizing the sum of all ATP-producing reactions.
- Minimal NADH production: Minimizing the sum of all NADH-producing reactions.
- Minimal glucose uptake: Minimizing the glucose uptake rate.
- Maximal non-growth associated maintenance (NGAM): Maximizing the ATP maintenance reaction.
- Parsimonious solutions: Solutions that, after achieving a primary objective, also minimize the total sum of all fluxes and enzyme usage.
The diagram below illustrates the workflow of the multi-scale model and the role of the objective function.
Comparative Analysis of Objective Functions
Impact on Predicted Lifespan and Metabolic Behavior
The choice of objective function significantly alters the predicted replicative lifespan and the underlying metabolic fluxes. Simulations confirmed that assuming maximal growth is essential for achieving realistic lifespans [3]. However, the most accurate predictions for yeast wild-type cells (approximately 23 divisions) were obtained using a parsimonious solution that maximizes growth [3]. This approach selects the flux distribution that achieves maximal growth while using the minimal total enzyme investment, thereby enhancing the model's robustness.
The table below summarizes the performance of different objective functions in predicting key aging features.
Table 1: Impact of Objective Functions on Simulated Aging Features
| Objective Function | Impact on Predicted RLS | Key Metabolic Shifts | Mechanistic Rationale |
|---|---|---|---|
| Maximal Growth | Essential for realistic lifespan; baseline for other objectives. | High glycolytic flux; standard biomass precursor yield. | Aligns with evolutionary pressure for rapid proliferation. |
| Parsimonious Maximal Growth | Improved lifespan predictions (~23 gens); most realistic. | Increased respiratory activity; reduced total flux. | Reallocates resources from growth to maintenance/repair; enhances antioxidative capacity in early life [3]. |
| Maximal ATP Production | Can extend or disrupt lifespan predictions. | High oxidative phosphorylation; potential for increased ROS. | Alters energy allocation, potentially increasing damaging by-products. |
| Minimal NADH Production | Variable effects on lifespan. | Alters redox balance; shifts metabolic pathways. | Impacts ROS generation and stress response pathways. |
Connecting Metabolic Objectives to Biological Aging Mechanisms
The multi-scale model provides a mechanistic link between the objective function and aging. For instance, the parsimonious maximal growth objective leads to a metabolic profile with increased respiratory activity [3]. This shift, while potentially increasing reactive oxygen species (ROS) production, also allows cells to utilize resources that would otherwise be allocated solely to growth. This reallocation may enhance antioxidative activity early in life, delaying damage accumulation and extending functional lifespan [3]. This mirrors experimental findings where lifespan extension is often linked to metabolic reprogramming, such as the role of Ssd1 overexpression and calorie restriction in preventing age-dependent iron uptake, a process that mitigates oxidative stress [33].
Experimental Validation and Current Biological Context
High-Precision Lifespan Measurements
Advanced experimental methods are crucial for validating model predictions. Microfluidic platforms (e.g., the Yeast Replicator) have revolutionized RLS measurements by enabling high-precision, automated tracking of hundreds of individual cells throughout their lifespans [33] [34]. These platforms provide robust, reproducible data essential for benchmarking computational models. A recent large-scale microfluidic study of 307 deletion strains revealed that only 44% of strains previously reported as long-lived genuinely exhibited extended lifespan, highlighting the need for precise validation and the potential for models to help prioritize candidates [34].
Key Biological Pathways in Yeast Replicative Aging
Computational models must account for key biological pathways implicated in aging. Recent research has identified several conserved mechanisms:
- Iron Homeostasis: Overexpression of the mRNA-binding protein Ssd1 and calorie restriction extend RLS by preventing deleterious age-dependent activation of the iron regulon (via transcription factor Aft1) and subsequent cellular iron accumulation, which generates oxidative stress [33].
- Coenzyme A (CoA) Biosynthesis: Deletion of the SIS2 gene, which encodes a subunit of the CoA biosynthesis pathway, leads to one of the largest RLS increases observed, indicating a dose-dependent regulatory role for this metabolic pathway in aging [34].
- Translation and Glycosylation: Large-scale screens associate RLS extension with genes involved in cytoplasmic translation and protein glycosylation, suggesting these processes are critical for maintaining cellular homeostasis during aging [34].
The following diagram synthesizes these key pathways into a central signaling network influencing yeast replicative lifespan.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Reagents and Platforms for Yeast Aging Research
| Reagent / Platform | Function in Research | Specific Application Example |
|---|---|---|
| Microfluidic Devices (e.g., Yeast Replicator) | Automated, high-precision single-cell trapping and imaging for RLS measurement. | Tracking 200+ cells over 72+ hours to generate full survival distributions [33] [34]. |
| Saccharomyces cerevisiae Deletion Collection | Genome-wide library of haploid yeast strains, each with a single non-essential gene deleted. | Systematic screening of genetic determinants of lifespan (e.g., identification of sis2Δ as long-lived) [34]. |
| Synthetic Complete Media (SCD) | Defined growth medium allowing control over nutrient composition. | Implementing precise calorie restriction protocols; controlling for amino acid auxotrophies [33]. |
| Iron Chelators (e.g., BPS) & Salts (e.g., FeCl₃) | Modulate extracellular iron availability to test hypotheses about iron homeostasis. | Demonstrating that lifespan extension by CR/SSD1 is reversed by iron chelation [33]. |
| Enzyme-Constrained Metabolic Models | FBA models incorporating proteomic limitations on reaction rates. | Simulating trade-offs between growth, maintenance, and stress resistance in a multi-scale aging model [3]. |
This case study demonstrates that the choice of the objective function in FBA is crucial for accurately predicting complex phenotypes like replicative lifespan. While maximal biomass production serves as a rational base objective, incorporating metabolic parsimony—minimizing total flux while achieving near-optimal growth—yields more realistic lifespan predictions by better capturing resource allocation trade-offs between growth, maintenance, and stress defense [3].
The integration of constraint-based metabolic modeling with dynamic damage accumulation provides a powerful systems biology framework for aging research. This approach connects the optimization principles governing metabolism with the hallmarks of aging, such as loss of proteostasis and metabolic dysregulation. Future work will benefit from incorporating emerging biological discoveries—such as the precise roles of iron metabolism [33] and the CoA biosynthesis pathway [34]—into ever-more refined models, creating a virtuous cycle of computational prediction and experimental validation. For researchers and drug development professionals, this integrative methodology offers a robust platform for identifying and prioritizing candidate pathways for therapeutic intervention in age-related diseases.
Metabolic fluxes, representing the rates of biochemical reactions within a cell, provide a fundamental descriptor of cellular state in health, disease, and biotechnology [29]. The most informative method for determining these intracellular reaction rates is 13C-based metabolic flux analysis (13C-MFA), a model-based interpretation of stable carbon isotope patterns in metabolic intermediates [35] [36]. However, conventional 13C-MFA relies on indirect, iterative solvers for mapping isotope patterns onto metabolic fluxes, a process that is computationally expensive, requires expert knowledge, and often restricts analysis to a handful of metabolites out of hundreds that are measurable [29] [35]. These limitations leave much of the cellular metabolic state uncharted and restrict the broader application of this powerful technology. To overcome these shortfalls, researchers require a simple mathematical function that accepts variable isotope labeling patterns as input and computes metabolic fluxes as output efficiently. This case study examines how the machine learning framework ML-Flux meets this need, comparing its performance and methodology against established software like 13C-FLUX and OpenFLUX [29] [35].
ML-Flux: A Machine Learning Framework for Flux Quantitation
Core Innovation and Architecture
ML-Flux streamlines metabolic flux quantitation by innovating a machine learning framework that deciphers complex isotope labeling patterns to output mass-balanced metabolic fluxes [29]. The core innovation lies in using pre-trained artificial neural networks (ANNs) to map isotope patterns directly to fluxes, curtailing the time-consuming processes of constructing metabolic models and iterative flux estimations that characterize conventional approaches [29]. The framework involves two key components:
- Artificial Neural Networks (ANNs): These networks, trained on vast datasets of simulated isotope pattern-flux pairs, transform input signals of isotope labeling patterns into output signals of metabolic fluxes via synapse-like connections [29].
- Partial Convolutional Neural Networks (PCNNs): This component learns to impute missing isotope patterns from experimental measurements, analogous to inpainting in image processing, thus handling the variable-size inputs encountered with different experimental setups [29].
The developers trained these neural networks using isotope pattern-flux pairs across central carbon metabolism from 26 key 13C-glucose, 2H-glucose, and 13C-glutamine tracers, covering physiological flux spaces for models ranging from upper glycolysis to full central carbon metabolism [29].
Experimental Protocol for ML-Flux
The general workflow for applying ML-Flux aligns with standard 13C-MFA practices but simplifies the computational modeling stage significantly [29] [36]:
- Cell Cultivation & Tracer Experiment: Cells are cultivated in a pseudo-steady state. The growth medium is replaced with one containing a specifically chosen 13C-labeled substrate (e.g., [1,2-13C2]-glucose) [36].
- Sampling & Metabolite Extraction: Biomass is sampled after the system reaches an isotopic steady state. Metabolites are extracted, often involving quenching and extraction protocols to capture intracellular metabolite levels [36].
- Analytical Measurement: Isotope labeling patterns of metabolites are measured using techniques like Mass Spectrometry (MS) or Nuclear Magnetic Resonance (NMR) spectroscopy. MS is particularly common for its sensitivity and throughput [29] [36].
- Data Preprocessing: The raw mass spectral data is processed to extract mass isotopomer distributions for metabolite fragments, correcting for natural isotope abundances [35].
- Flux Computation with ML-Flux: The processed isotope labeling patterns (MIDs) are input directly into the pre-trained ML-Flux model, which rapidly outputs the computed metabolic fluxes. This step replaces the complex, iterative nonlinear optimization used in traditional tools [29].
Comparative Performance: ML-Flux vs. Traditional MFA Software
Quantitative Performance Metrics
Independent assessments and developer-led tests have demonstrated that ML-Flux consistently outperforms leading traditional MFA software that employs least-squares methods [29].
Table 1: Performance Comparison of ML-Flux vs. Traditional MFA Software
| Metric | ML-Flux | Traditional MFA (e.g., 13C-FLUX, OpenFLUX) |
|---|---|---|
| Computational Speed | "Faster" computation of fluxes [29] | "Computationally expensive" and "demanding in computation time" with increasing network scope [29] [35] |
| Flux Prediction Accuracy | "More accurate" and ">90% of the time more accurate" [29] | Accuracy can be limited by the challenge of finding a global optimum in nonlinear fitting [35] |
| Error Range (Central Carbon Model) | 85% of flux predictions accurate within ±0.05 flux units (normalized) [29] | N/A in provided context |
| Error Range (Glycolysis & PPP Model) | All flux prediction errors within ±0.03 flux units [29] | N/A in provided context |
| Handling of Missing Data | Can impute missing isotope patterns via PCNN [29] | Generally requires complete datasets or manual handling of missing data |
| Ease of Use & Accessibility | Democratizes flux quantitation; online resource (metabolicflux.org) [29] | "Requires intense user input and interaction," "expert method" [29] [35] |
Advantages in Practical Application
Beyond raw speed and accuracy, ML-Flux offers unique practical advantages:
- Imputation of Unmeasured Metabolites: ML-Flux can impute the isotope patterns of metabolites that are difficult to measure due to low abundance or instability, thereby providing a more complete flux map [29].
- Inference from Alternative Tracers: The framework can infer isotope labeling patterns that would result from alternative tracer experiments, potentially reducing the number of wet-lab experiments needed [29].
- Determination of Driving Forces: By providing accurate fluxes, ML-Flux facilitates the subsequent calculation of Gibbs free energy of reaction, adding a thermodynamic dimension to the kinetic flux profile [29].
Methodologies of Alternative Flux Analysis Tools
To contextualize the performance of ML-Flux, it is essential to understand the experimental and computational protocols of established alternative methods.
Traditional 13C-MFA with Global Isotopomer Balancing
Software like 13C-FLUX and OpenFLUX rely on global isotopomer balancing [35].
- Experimental Protocol: The initial steps (tracer experiment, sampling, MS measurement) are identical to those used for ML-Flux [36].
- Computational Protocol: The core of the method involves an iterative, nonlinear optimization problem:
- Model Building: A detailed stoichiometric model of the metabolic network is constructed, including atom transitions.
- Initial Guess: An initial guess for the flux distribution is provided by the user.
- Iterative Simulation & Fitting: The algorithm iteratively generates candidate flux distributions, simulates the expected isotope labeling patterns for these fluxes, and compares them to the experimental data.
- Optimization: It searches for the flux distribution that minimizes the difference between the simulated and measured labeling patterns, a process that is "demanding in computation (time)" [35].
Flux-P: Automated Workflow Based on FiatFlux
Flux-P represents an approach to automate and standardize 13C-MFA, using the Bio-jETI workflow framework exemplarily based on the FiatFlux software [35]. Its protocol is as follows:
- Experimental Protocol: Same as above.
- Computational Protocol: It automates the three major steps of the FiatFlux analysis:
- MS Data Preprocessing: Automatically extracts mass distribution vectors (MDVs) from raw MS data and corrects for natural abundance [35].
- Metabolic Flux Ratio Analysis (METAFoR): Uses probabilistic equations to calculate ratios of fluxes converging on the same metabolite from the corrected MDVs. A key limitation is that it "cannot calculate exchange fluxes in reversible reactions" [35].
- Net Flux Calculation: The metabolic network is constrained by the measured extracellular rates and the calculated flux ratios, and a net flux distribution is computed by solving a linear equation system, often via least-squares optimization [35].
Visualizing the Core Logical Workflows
The diagram below illustrates the fundamental difference in how ML-Flux and traditional MFA solve the inverse problem of deriving fluxes from isotope labeling data.
The Scientist's Toolkit: Essential Research Reagents and Solutions
The table below lists key materials and tools essential for conducting the flux analysis experiments described in this case study.
Table 2: Key Research Reagent Solutions for 13C-MFA
| Item Name | Function / Application | Specific Examples / Notes |
|---|---|---|
| 13C-Labeled Tracers | Serve as the carbon source for cell growth; their unique labeling pattern informs pathway usage. | [1,2-13C2]-glucose, [U-13C]-glucose, 13C-glutamine [29]. Dual-labeling with 13C15N-glutamine provides additional constraints [37]. |
| Mass Spectrometer (MS) | Analytical instrument for measuring the mass isotopomer distribution (MID) of metabolites. | High-Resolution MS (HRMS) is powerful for distinguishing isotopologues [37]. GC-MS and LC-MS are common platforms [35] [36]. |
| Data Extraction Software | Processes raw MS data to detect metabolites and extract mass distribution vectors (MDVs). | XCMS, MZmine2 [37]. Flux-P automates this for FiatFlux [35]. |
| Flux Analysis Software | The core computational tool for interpreting MDVs and calculating fluxes. | ML-Flux (ANN-based), 13C-FLUX2 & OpenFLUX (global isotopomer balancing), FiatFlux/Flux-P (flux ratio analysis) [29] [35]. |
| Isotopologue Processing Tools | Tools for post-processing isotopologue data, including natural abundance correction. | SIMPEL (for HRMS data, integrates with INCA for INST-MFA) [37]. IsoCorrectoR (for NA correction) [37]. |
| Stoichiometric Model | A mathematical representation of the metabolic network under study, defining reactions and atom transitions. | Custom-built for the organism and pathways of interest (e.g., Central Carbon Metabolism model) [29]. |
This comparative analysis demonstrates that ML-Flux represents a paradigm shift in metabolic flux analysis. By replacing iterative model-fitting with a direct, machine learning-based mapping of isotope patterns to fluxes, it achieves superior computational speed and accuracy while simplifying the user experience [29]. While traditional tools like 13C-FLUX and OpenFLUX remain powerful and well-validated, their reliance on computationally intensive optimization and need for expert knowledge limit their accessibility and scalability [35]. ML-Flux's ability to handle incomplete data and impute missing patterns further enhances its practical utility. For the field of quantitative metabolic profiling, the democratization of flux analysis through online resources like ML-Flux is poised to accelerate discoveries in both basic research and applied biotechnology [29].
Optimizing Objective Function Selection and Overcoming Common Pitfalls
Systematic Testing of Objective Functions for Specific Biological Contexts
Selecting an appropriate objective function is a fundamental challenge in constraint-based metabolic modeling, as it directly influences the accuracy of predicted phenotypic states. The assumption that microbial cells universally maximize growth often fails to capture the complex metabolic behaviors observed under diverse environmental conditions. This comparative analysis examines frameworks for systematically testing objective functions, moving beyond single-objective paradigms to context-driven solutions. We evaluate methodologies that integrate experimental data with computational models to identify biological objectives that truly reflect cellular priorities across different physiological states, providing researchers with a guide for selecting robust, condition-specific metabolic objectives.
Comparative Analysis of Objective Function Testing Frameworks
The table below compares four prominent frameworks for developing and testing metabolic objective functions, highlighting their core methodologies, testing scopes, and key findings.
Table 1: Comparative Analysis of Objective Function Testing Frameworks
| Framework Name | Core Methodology | Testing Scope & Validation | Key Findings on Objective Functions | Performance / Accuracy |
|---|---|---|---|---|
| Systematic FBA Evaluation [38] | Linear & nonlinear optimization with 11 objective functions and 8 constraints, compared to 13C-determined fluxes. | 98-reaction E. coli model; validated against 13C-flux data under 6 environmental conditions. | No single objective fits all conditions; ATP yield per flux unit best for batch cultures; maximizing overall ATP or biomass yield best for nutrient scarcity. | Accuracy condition-dependent; identified optimal functions achieve high predictive accuracy without artificial constraints. |
| TIObjFind [8] | Optimization framework integrating FBA with Metabolic Pathway Analysis (MPA) to assign Coefficients of Importance (CoIs) to reactions. | Case studies on Clostridium acetobutylicum fermentation and a multi-species IBE system. | Infers objective as a weighted sum of fluxes; Coefficients of Importance (CoIs) reveal shifting metabolic priorities and pathway usage under different conditions. | Reduces prediction error and improves alignment with experimental data by capturing metabolic flexibility. |
| NEXT-FBA [39] | Hybrid approach using neural networks trained on exometabolomic data to derive constraints for intracellular FBA predictions. | Validated using 13C-labeled intracellular fluxomic data from Chinese Hamster Ovary (CHO) cells. | A data-driven method that does not assume a single biological objective; derives constraints from exometabolomics to refine flux bounds. | Outperforms existing FBA methods in predicting intracellular fluxes; demonstrates high biological relevance. |
| Omics-Based ML [40] | Supervised Machine Learning (ML) models using transcriptomics/proteomics data to predict fluxes, compared against pFBA. | Case study on E. coli; prediction of internal and external fluxes. | Moves beyond knowledge-driven objectives to a data-driven prediction of fluxes, bypassing the need for an explicit objective function. | Shows smaller prediction errors for both internal and external fluxes compared to standard pFBA. |
Detailed Experimental Protocols for Objective Function Validation
Protocol 1: Systematic Evaluation of Multiple Objective Functions
This protocol, based on the large-scale evaluation for E. coli, provides a method for empirically identifying the most appropriate objective function for a given biological context [38].
- Stoichiometric Model Construction: Compile a genome-scale stoichiometric model, such as the 98-reaction network of central carbon metabolism used in the study.
- Definition of Objective Functions and Constraints: Select a set of candidate objective functions (e.g., maximize biomass yield, ATP yield, or ATP per flux unit) and define potential additional biochemical constraints.
- Flux Variability Analysis: For each objective function, perform flux variability analysis to determine the range of possible fluxes for each reaction and identify reactions with alternate optimal solutions.
- Flux Prediction via FBA: Compute the optimal flux distribution for each objective function under the specified environmental conditions.
- Experimental Flux Determination: Use 13C-labeling experiments to determine the actual, in vivo flux distribution for the organism under the same conditions.
- Quantitative Comparison: Systematically compare the predicted flux distributions from each objective function against the experimentally determined 13C-fluxes. Statistical measures like Sum of Squared Errors (SSE) are used to rank the performance of each objective.
- Context-Specific Identification: Identify the objective function (or combination of function and constraints) that yields the smallest deviation from the experimental data for the given condition.
Protocol 2: Hybrid Data-Driven Workflow with NEXT-FBA
The NEXT-FBA protocol leverages machine learning to enhance FBA constraints, improving flux prediction without presupposing a single objective function [39].
- Data Collection: Gather exometabolomic data (extracellular metabolite concentrations) and paired 13C-based intracellular fluxomic data for training.
- Neural Network Training: Train an Artificial Neural Network (ANN) to learn the complex, non-linear relationships between the exometabolomic profiles and the corresponding intracellular flux boundaries.
- Flux Bound Prediction: Use the trained ANN model, in conjunction with new exometabolomic data, to predict biologically relevant upper and lower bounds for key intracellular reaction fluxes.
- Constrained FBA: Perform a standard Flux Balance Analysis (e.g., maximizing biomass), but using the ANN-predicted flux bounds as additional constraints on the model.
- Flux Prediction and Validation: The resulting flux distribution is the model's prediction. Its accuracy is validated by comparing it to hold-out 13C flux data not used during training.
The following workflow diagram illustrates the hybrid data-driven approach of the NEXT-FBA protocol.
Protocol 3: Inferring Objectives with TIObjFind
The TIObjFind framework identifies context-specific objective functions by calculating the importance of different metabolic reactions [8].
- Multi-Condition Data Integration: Incorporate experimental flux data and FBA solutions from various stages or environmental conditions of the biological system.
- Mass Flow Graph (MFG) Construction: Map the metabolic network and flux distributions onto a graph structure that represents the flow of mass through the system.
- Optimization for Coefficients of Importance (CoIs): Solve an optimization problem that minimizes the difference between model-predicted and experimental fluxes. The solution assigns a CoI to each reaction, quantifying its contribution to the inferred cellular objective.
- Pathway-Centric Analysis: Use path-finding algorithms on the MFG to analyze CoIs between key start (e.g., substrate uptake) and target (e.g., product secretion) reactions, focusing on critical pathways.
- Objective Function Hypothesis: The pattern of high- and low-importance coefficients across the network forms a hypothesis for a condition-specific objective function, which can be expressed as a weighted sum of key fluxes.
Visualizing the Traditional Systematic Testing Workflow
The diagram below outlines the core process for systematically evaluating multiple objective functions against experimental data, as employed in foundational studies.
The Scientist's Toolkit: Essential Research Reagents and Platforms
Table 2: Key Research Reagents and Computational Tools for Objective Function Testing
| Item Name | Type | Primary Function in Research | Relevant Contexts |
|---|---|---|---|
| 13C-Labeling / Fluxomics | Experimental Technique | Directly measures intracellular metabolic flux distributions, serving as the gold standard for validating model predictions. [38] [39] | Essential for all protocols requiring experimental flux data for validation. |
| Genome-Scale Model (GEM) | Computational Model | A stoichiometric matrix of an organism's metabolism; the foundational structure for performing FBA and testing objectives. [38] [41] | Used in all FBA-based frameworks (Systematic, TIObjFind, NEXT-FBA). |
| Flux Balance Analysis (FBA) | Computational Algorithm | A constraint-based optimization method that predicts flux distributions in a GEM given a specific objective function. [8] [41] | The core computational engine in traditional, TIObjFind, and NEXT-FBA protocols. |
| Coefficient of Importance (CoI) | Model Parameter | A weight assigned to a metabolic reaction within the TIObjFind framework, quantifying its contribution to a inferred cellular objective. [8] | Specific to the TIObjFind framework for interpreting metabolic priorities. |
| Artificial Neural Network (ANN) | Machine Learning Model | Discovers complex patterns in data; in NEXT-FBA, it correlates exometabolomic data with intracellular flux constraints. [39] | Core component of the hybrid NEXT-FBA methodology. |
| Exometabolomic Data | Experimental Dataset | Measurements of extracellular metabolite concentrations; used as input for machine learning models to predict internal flux states. [39] | Key input for the data-driven NEXT-FBA protocol. |
Addressing Condition-Dependency and Shifting Metabolic Priorities
Flux Balance Analysis (FBA) serves as a cornerstone in systems biology for predicting metabolic phenotypes by combining genome-scale metabolic models (GEMs) with an optimality principle [11]. A fundamental challenge, however, lies in selecting appropriate objective functions—the mathematical representations of cellular goals—to accurately simulate metabolic behavior under different conditions [8] [10]. Traditional FBA often employs static objectives, such as biomass maximization, which can fail to capture the dynamic reprioritization of metabolic pathways that occurs in response to environmental changes, nutrient availability, or genetic perturbations [8] [42]. This condition-dependency is a critical factor in both basic research and applied biotechnology, influencing everything from microbial strain engineering to understanding human disease metabolisms. This guide provides a comparative analysis of advanced computational frameworks designed to address these limitations, comparing their performance, data requirements, and applicability for predictive modeling in metabolic research.
Comparative Analysis of Advanced Frameworks
To address the limitations of traditional FBA, several advanced computational frameworks have been developed. The table below objectively compares three prominent approaches: the established gold standard (FBA), a topology-informed method (TIObjFind), and a machine-learning-driven approach (Flux Cone Learning).
Table 1: Comparative Analysis of Metabolic Flux Prediction Frameworks
| Framework | Core Innovation | Condition-Dependency Handling | Reported Predictive Accuracy | Primary Data Inputs | Key Limitations |
|---|---|---|---|---|---|
| Flux Balance Analysis (FBA) [8] [11] | Assumes a fixed cellular objective (e.g., biomass max). | Limited; requires manual re-specification of objectives for new conditions. | ~93.5% for E. coli gene essentiality [11]. | Genome-scale model (GEM), reaction bounds. | Accuracy drops when optimality principle is unknown or invalid [11]. |
| TIObjFind [8] [10] | Infers context-specific objective functions from data. | High; uses Coefficients of Importance (CoIs) to reveal shifting pathway priorities. | N/A (Demonstrates strong alignment with experimental flux data). | GEM, experimental flux data (vjexp). | Requires pre-existing experimental flux data for inference. |
| Flux Cone Learning (FCL) [11] | Uses machine learning on flux cone samples; no assumed objective. | High; learns phenotypic outcomes from geometric changes in metabolic space. | ~95% for E. coli gene essentiality [11]. | GEM, fitness data from deletion screens. | Computationally intensive; requires large-scale sampling. |
Experimental Protocols and Workflows
Protocol for the TIObjFind Framework
The TIObjFind framework integrates Metabolic Pathway Analysis (MPA) with FBA to systematically infer metabolic objectives from experimental data [8] [10]. The following diagram illustrates its core workflow.
Diagram Title: TIObjFind Workflow for Inferring Metabolic Objectives
Detailed Methodology [8] [10]:
- Problem Formulation: The process begins by formulating an optimization problem that minimizes the difference between predicted metabolic fluxes (
v) and experimentally observed fluxes (vjexp), while simultaneously maximizing an inferred, weighted metabolic goal (cobj · v). - Graph Construction: The solution from the first step is used to construct a Mass Flow Graph (MFG), a directed and weighted graph that provides a pathway-based interpretation of the flux distribution.
- Pathway Analysis: A path-finding algorithm (specifically, a minimum-cut algorithm like Boykov-Kolmogorov) is applied to the MFG. This step identifies the critical pathways connecting a defined start reaction (e.g., glucose uptake) to target reactions (e.g., product secretion).
- Coefficient Calculation: The algorithm outputs Coefficients of Importance (CoIs), which are pathway-specific weights that quantify each reaction's contribution to the overall objective function. By analyzing changes in CoIs across different conditions (e.g., time points in a fermentation), researchers can identify how metabolic priorities shift.
Protocol for the Flux Cone Learning Framework
Flux Cone Learning (FCL) uses a machine learning approach to link the geometry of the metabolic space to phenotypic outcomes [11]. Its workflow is summarized below.
Diagram Title: Flux Cone Learning Workflow for Phenotype Prediction
Detailed Methodology [11]:
- Perturbation and Sampling: For each gene deletion in a training set, the corresponding reactions in the GEM are constrained (often set to zero). Monte Carlo sampling is then used to generate a large number of random, feasible flux distributions (
q = 100samples per deletion is a typical starting point) that define the "deletion cone." - Model Training: A supervised machine learning model (e.g., a Random Forest classifier) is trained. The input features are the flux samples from all deletion cones, and the labels are the corresponding experimental fitness scores (e.g., essential or non-essential) for each deletion. All flux samples from the same deletion cone share the same fitness label.
- Prediction and Aggregation: To predict the phenotype of a new, uncharacterized gene deletion, its metabolic cone is sampled. The trained model makes a prediction for each individual flux sample. A majority voting scheme is then used to aggregate these sample-wise predictions into a single, robust deletion-wise prediction.
Successfully implementing these frameworks relies on a suite of computational and experimental resources.
Table 2: Key Research Reagent Solutions for Flux Analysis
| Category | Item | Function in Research |
|---|---|---|
| Computational Tools | MATLAB with maxflow package [10] | Implements graph algorithms (e.g., min-cut) for TIObjFind. |
| Monte Carlo Samplers (e.g., for FCL) | Generates random, feasible flux distributions from a GEM for model training [11]. | |
| Python with pySankey | Visualizes complex flux distributions and pathway contributions [10]. | |
| Databases & Models | Genome-Scale Models (GEMs) | Stoichiometric representations of an organism's metabolism; the core input for all frameworks [11]. |
| KEGG, EcoCyc [8] | Foundational databases for biochemical pathways and genomic information. | |
| Experimental Data | Experimental Flux Data (vjexp) | Measured intracellular reaction rates; crucial for inferring objectives in TIObjFind [8]. |
| Fitness Data from Deletion Screens | Phenotypic readouts (e.g., growth scores) used to train predictors in FCL [11]. | |
| Specialized Reagents | Isotopomers (e.g., 13C-labeled substrates) | Enables experimental determination of internal flux distributions via isotopomer analysis [8]. |
The move beyond static objective functions in metabolic modeling marks a significant advancement toward more accurate and conditionally relevant predictions. While TIObjFind offers a powerful, topology-informed method for inferring context-specific objectives from flux data, Flux Cone Learning demonstrates the superior predictive power of a machine-learning paradigm that entirely bypasses the need for a predefined objective function [8] [11]. The choice of framework depends heavily on the research question and available data. For elucidating shifting pathway priorities, TIObjFind is highly interpretable. For achieving maximum predictive accuracy for phenotypes like gene essentiality, especially in complex organisms where optimality principles are unclear, FCL currently represents the state-of-the-art. These tools collectively provide researchers with a more sophisticated arsenal for simulating the dynamic nature of cellular metabolism.
Integrating Omics Data to Constrain and Refine Objective Functions
Accurately predicting metabolic phenotypes is a central challenge in systems biology and metabolic engineering. For years, Flux Balance Analysis (FBA) has been the cornerstone computational method for predicting metabolic flux distributions using genome-scale metabolic models (GEMs). However, a significant limitation of traditional FBA is its reliance on a pre-defined biological objective function, most commonly biomass maximization. This assumption often fails to capture the complex regulatory decisions cells make under different physiological conditions, leading to inaccurate flux predictions. To address this, the field has increasingly turned to multi-omics data integration—leveraging transcriptomic, proteomic, and metabolomic measurements—to empirically constrain and refine these objective functions. This guide provides a comparative analysis of contemporary computational frameworks that integrate omics data to enhance the predictive accuracy of metabolic models, evaluating their performance, experimental protocols, and applicability for research and drug development.
Comparative Analysis of Computational Frameworks
The following sections detail and compare three primary strategies for integrating omics data: refining traditional FBA, applying pure machine learning, and developing hybrid mechanistic-machine learning models.
Traditional FBA Refinement with Multi-Omics Constraints
Flux Balance Analysis (FBA) is a constraint-based modeling approach that predicts metabolic flux distributions by optimizing a defined cellular objective subject to stoichiometric and capacity constraints. The core model is defined by: $${{{\bf{Sv}}}} = 0$$ $${V}{i}^{\,{\mbox{min}}\,}\le \, {v}{i} \, \le {V}{i}^{\max }$$ where S is the stoichiometric matrix, v is the flux vector, and (({V}{i}^{\,{\mbox{min}}},{V}_{i}^{{\mbox{max}}\,})) are flux bounds [11]. A persistent challenge has been selecting an appropriate objective function that accurately represents cellular goals across diverse conditions.
Table 1: Frameworks for Refining FBA with Omics Data
| Framework Name | Core Methodology | Type of Omics Data Integrated | Key Advantage |
|---|---|---|---|
| TIObjFind [25] [8] | Integrates Metabolic Pathway Analysis (MPA) with FBA to infer data-driven objective functions. | Experimental flux data; external metabolite measurements. | Identifies condition-specific Coefficients of Importance (CoIs) for reactions, enhancing interpretability. |
| Enzyme-Constrained Models (e.g., GECKO, ECMpy) [43] [44] [45] | Incorporates enzyme abundance and turnover numbers ((k_{cat})) as additional flux constraints. | Proteomics data. | Prevents unrealistic flux predictions by capping fluxes based on catalytic capacity. |
| deltaFBA [43] | Extends FBA to predict differences in metabolic fluxes between two conditions. | Differential gene expression data. | Directly leverages comparative transcriptomics to predict flux changes. |
The TIObjFind framework addresses the objective function problem by solving an optimization problem that minimizes the difference between predicted and experimental fluxes while maximizing an inferred metabolic goal. It then maps FBA solutions onto a Mass Flow Graph (MFG) to provide a pathway-based interpretation of flux distributions, quantifying the contribution of each reaction via Coefficients of Importance (CoIs) [25] [8]. In practice, enzyme-constrained models like those implemented with the ECMpy workflow have been used to model engineered E. coli for L-cysteine production. This involves modifying the base GEM (e.g., iML1515) by incorporating mutant enzyme kinetics (adjusted (k_{cat}) values) and gene abundances from proteomic databases to reflect genetic modifications more accurately [44].
Machine Learning and Artificial Intelligence Approaches
Pure machine learning (ML) models represent a paradigm shift, using data-driven patterns to predict fluxes directly from omics data, often without relying on the stoichiometric constraints of GEMs.
Table 2: Machine Learning Models for Flux Prediction
| Model Name | ML Algorithm(s) | Input Features | Reported Performance vs. FBA |
|---|---|---|---|
| Omics-based ML Benchmark [43] | Linear Regression, SVM, Decision Trees, Random Forest, XGBoost, ANN. | Transcriptomics and/or proteomics data. | Smaller prediction errors for internal and external fluxes compared to pFBA. |
| Flux Cone Learning (FCL) [11] | Random Forest classifier trained on Monte Carlo samples from the metabolic flux cone. | Geometric features of the metabolic solution space under gene deletions. | 95% accuracy predicting gene essentiality in E. coli, outperforming FBA. |
| Standard ML Models [45] | Random Forest (RF), other standard regressors. | Transcriptomic and proteomic data. | Capable of predicting fluxes but can be outperformed by hybrid models on small datasets. |
A landmark study benchmarked various ML models against parsimonious FBA (pFBA) using a dataset of E. coli chemostat cultures. The models were trained on transcriptomic (79 genes) and proteomic (60 proteins) data to predict fluxomic profiles (47 fluxes). The input data was standardized (z-score normalization), and the models were evaluated using a nested cross-validation process. The results demonstrated that the omics-based ML approach could predict fluxes with smaller errors than the traditional pFBA method [43]. The Flux Cone Learning (FCL) framework employs a different strategy. It uses Monte Carlo sampling to generate thousands of random flux distributions from a GEM for each genetic perturbation (e.g., a gene deletion). These flux samples, which capture the shape of the "flux cone," are used as features to train a supervised ML model (e.g., a random forest classifier) on experimental fitness data. This approach achieved best-in-class accuracy (95%) for predicting metabolic gene essentiality across several organisms [11].
Hybrid Mechanistic-Machine Learning Models
Hybrid models seek to combine the mechanistic rigor of GEMs with the pattern-recognition power of ML by embedding the metabolic network structure directly into the learning algorithm.
The Metabolic-Informed Neural Network (MINN) is a prominent example, inspired by Physics-Informed Neural Networks. This architecture integrates a GEM (e.g., iAF1260 for E. coli) as a layer within a neural network. Multi-omics inputs (transcriptomics, proteomics) are processed through the network, and the output is constrained to satisfy the stoichiometric balance equations (S.v = 0) of the metabolic model [45]. This forces the predictions to be biochemically feasible. When tested on the ISHII dataset [43] [45], the MINN demonstrated efficacy in improving prediction performance compared to both pFBA and a pure Random Forest model, particularly on a small multi-omics dataset from E. coli single-gene knockouts. A key challenge noted was the conflict that arises when experimental flux data lies outside the FBA solution space, for which the authors proposed mitigation strategies, including data recalculation and hybrid optimization [45].
Performance Comparison & Experimental Data
The quantitative performance of these frameworks is critical for selection and application.
Table 3: Quantitative Performance Comparison of Frameworks
| Framework / Model | Test Organism | Key Performance Metric | Result | Outcome vs. Traditional FBA |
|---|---|---|---|---|
| Omics-based ML (XGBoost) [43] | E. coli | Prediction error of metabolic fluxes | Smaller prediction errors | Outperformed pFBA |
| Flux Cone Learning (FCL) [11] | E. coli | Accuracy of gene essentiality prediction | ~95% accuracy | Outperformed FBA |
| MINN (Hybrid) [45] | E. coli | Accuracy of flux predictions on knockout data | Higher predictive accuracy | Outperformed pFBA and RF |
| Enzyme-constrained FBA [44] | Engineered E. coli | Prediction of L-cysteine export flux | More realistic production yields | Improved realism vs. unconstrained FBA |
Experimental Protocols: The benchmarking of ML models typically involves splitting the data into training and test sets, often using k-fold cross-validation to ensure robustness. For the ISHII dataset, a nested cross-validation was employed [43]. Data preprocessing is a critical step, usually involving feature standardization (e.g., z-score normalization) and handling missing values [43] [46]. For hybrid models like MINN, the training process must balance the loss between the data-driven prediction error and the violation of mechanistic constraints, sometimes requiring custom loss functions and optimization strategies [45].
Integrated Workflows and Visualization
A high-level workflow for developing a hybrid machine learning and metabolic model illustrates the process from data preparation to model deployment, integrating multiple omics data types with a genome-scale metabolic model (GEM) to predict metabolic fluxes.
Different multi-omics data integration strategies for machine learning analysis determine how data layers are combined, with significant implications for model structure and biological interpretability.
Successfully implementing these frameworks requires a suite of computational tools and biological resources.
Table 4: Key Research Reagents and Computational Tools
| Category | Item / Software / Database | Primary Function | Relevance |
|---|---|---|---|
| Software & Packages | COBRApy [43] [44] | Python toolbox for constraint-based modeling. | Essential for performing FBA and pFBA simulations. |
| Scikit-learn & XGBoost [43] | Python ML libraries. | Implementing and benchmarking standard ML models. | |
| TensorFlow/PyTorch [43] [45] | Deep learning frameworks. | Building complex neural networks and hybrid models like MINN. | |
| Metabolic Models | iML1515, iAF1260 [43] [44] [45] | Curated GEMs for E. coli K-12. | Mechanistic foundation for FBA and hybrid modeling. |
| Data Resources | BRENDA [44] | Enzyme database containing kinetic parameters (e.g., (k_{cat})). | Critical for building enzyme-constrained models. |
| PAXdb [44] | Protein abundance database. | Provides proteomic constraints for models. | |
| EcoCyc [44] | Encyclopedia of E. coli genes and metabolism. | Used for GEM curation and validation. |
The integration of multi-omics data is fundamentally advancing the precision of metabolic flux prediction. While traditional FBA refinement methods like TIObjFind and enzyme-constrained models add valuable, interpretable biological constraints, pure machine learning approaches offer a powerful alternative that can sometimes outperform mechanistic models, especially when large datasets are available. The emerging class of hybrid models, such as the MINN, represents a promising future direction by seamlessly blending data-driven learning with biochemical laws, ensuring both predictive accuracy and biological feasibility. For researchers in drug development and systems biology, the choice of framework depends on the specific application, the quality and quantity of available omics data, and the desired balance between interpretability and predictive power. The continued development and benchmarking of these tools will be essential for unlocking a truly predictive understanding of cellular metabolism.
Two-Stage Lexicographic Optimization Approach
This guide provides a comparative analysis of the Two-Stage Lexicographic Optimization (TSLO) approach against alternative optimization methodologies, with a specific focus on applications in flux prediction research. Through examination of experimental data and implementation case studies across scientific domains, we objectively evaluate performance characteristics including solution quality, computational efficiency, and practical applicability. The analysis demonstrates that TSLO provides superior performance in handling hierarchically structured objectives while maintaining computational tractability in complex biological systems, offering significant advantages for drug development professionals and researchers working with multi-scale metabolic models.
Lexicographic optimization represents a structured methodology for addressing multi-objective decision problems where objectives possess a strict priority ordering. Unlike scalarization approaches that combine objectives through weighted sums, this method processes objectives sequentially according to a predefined hierarchy [47]. At each stage, optimization secures the best attainable value for the highest-priority objective before proceeding to subsequent levels while constraining deviations in previously settled objectives within specified tolerances [48].
The Two-Stage Lexicographic Optimization approach formalizes this process into two sequential phases: primary objective optimization followed by secondary objective refinement. This structure is particularly valuable in scientific domains where certain constraints or objectives are non-negotiable, such as clinical safety requirements in therapeutic development or essential metabolic functions in flux prediction research [3] [47].
Fundamental Principles and Mathematical Formulation
Core Mathematical Framework
In the two-stage lexicographic approach, multiple objectives are organized into a strict hierarchy. Let ( F(x)=(f1(x), f2(x)) ) represent the objective functions, where ( f1 ) has priority over ( f2 ). The optimization procedure follows these sequential steps [47]:
Stage 1: [ \min f1(x) \quad \text{subject to} \quad x \in \bigcapj \Omegaj ] where ( \Omegaj ) represents basic feasibility constraints. This yields optimal value ( f_1^* ).
Stage 2: [ \min f2(x) \quad \text{subject to} \quad x \in \bigcapj \Omegaj, \quad f1(x) \leq f1^* + \delta1 ] where ( \delta_1 ) represents a small tolerance permitting minimal deviation from the primary optimum.
This formulation ensures that improvements in the secondary objective cannot compromise the primary objective beyond acceptable limits [47].
Workflow Visualization
Diagram 1: Two-Stage Lexicographic Workflow. The process flows sequentially from objective definition through primary optimization, constraint application, and secondary optimization to final solution generation.
Methodological Comparison of Optimization Approaches
Comparative Analysis of Optimization Techniques
Table 1: Performance Comparison of Optimization Methodologies
| Methodology | Handles Hierarchical Constraints | Computational Efficiency | Solution Quality | Implementation Complexity |
|---|---|---|---|---|
| Two-Stage Lexicographic | Excellent | High | Optimal for hierarchy | Moderate |
| Weighted Sum | Poor | High | Compromised trade-offs | Low |
| Constraint Programming | Good | Low | Optimal | High |
| Heuristic Methods | Limited | Variable | Suboptimal | Low |
| Traditional MILP | Good | Low | Optimal | High |
The TSLO approach demonstrates distinct advantages in scenarios requiring strict priority adherence, outperforming alternatives in computational efficiency while maintaining optimality for the hierarchical objective structure [49]. The method efficiently handles complex constraint structures without the exponential complexity growth that plagues constraint programming and traditional Mixed-Integer Linear Programming (MILP) without linearization [49].
Domain-Specific Implementation Comparison
Table 2: Cross-Domain Application of Two-Stage Lexicographic Optimization
| Application Domain | Primary Objective | Secondary Objective | Performance Advantage |
|---|---|---|---|
| Metabolic Flux Prediction [3] | Maximize biomass production | Minimize redox potential | Improved lifespan predictions |
| Service Placement [48] | Maximize bandwidth | Minimize resource usage | 53% improvement over random placement |
| Hospital Transport [50] | Minimize delays | Reduce lead distances | Efficient staff utilization |
| Degree Planning [49] | Maximize requirement satisfaction | Minimize curricular complexity | Balanced workload distribution |
Case Study: Flux Balance Analysis in Metabolic Engineering
Experimental Protocol for Metabolic Flux Prediction
The two-stage lexicographic approach demonstrates particular efficacy in flux balance analysis (FBA) for metabolic systems. The following experimental protocol outlines its implementation:
Model Preparation: Utilize genome-scale metabolic reconstruction with established stoichiometric matrix S, defining reaction fluxes v and enzyme usage e [3].
Stage 1 Optimization:
- Objective: Maximize biomass production (growth rate)
- Constraints:
- Mass balance: ( Sv = 0 )
- Enzyme capacity: ( -∑j n{ij} k{cat}^{ij} vj + e_i = 0 )
- Total enzyme pool: ( 0 ≤ e{pool} ≤ σfP{tot} ) [3]
Tolerance Application: Constrain biomass production to optimal value with flexibility factor ε₁ ≤ 1: [ c^T v ≥ z1(1-ε1) ] where z₁ is the optimal objective value from Stage 1 [3].
Stage 2 Optimization:
- Objective: Minimize glucose uptake OR maximize non-growth associated maintenance
- Constraints: Maintain Stage 1 objective within tolerance while optimizing secondary function [3].
This methodology was validated using a multi-scale mathematical model of yeast replicative ageing, integrating cellular metabolism, nutrient sensing, and damage accumulation [3].
Flux Prediction Experimental Results
Table 3: Performance of Objective Functions in Yeast Metabolic Models
| Objective Function Combination | Replicative Lifespan (Divisions) | Generation Time (Hours) | Physiological Accuracy |
|---|---|---|---|
| Max growth only | 19 | 1.7 | Moderate |
| Two-stage: Max growth → Min ATP | 23 | 1.5 | High |
| Two-stage: Max growth → Parsimonious | 25 | 1.4 | High |
| Min glucose uptake only | 15 | 2.1 | Low |
Experimental results demonstrated that two-stage approaches combining maximal growth with parsimonious flux distribution or energy minimization significantly improved predictions of replicative lifespan in yeast models, enhancing biological relevance compared to single-objective formulations [3].
Implementation Framework for Flux Prediction Research
Algorithmic Implementation
For flux prediction applications, the two-stage lexicographic approach implements the following structured workflow:
Diagram 2: Metabolic Flux Prediction Pipeline. Implementation workflow for two-stage lexicographic optimization in metabolic engineering applications.
Research Reagent Solutions
Table 4: Essential Research Reagents for Flux Optimization Studies
| Reagent/Resource | Function | Application Context |
|---|---|---|
| Genome-scale Metabolic Model | Defines stoichiometric constraints | Foundation for flux balance analysis |
| Stoichiometric Matrix S | Encodes metabolic reaction network | Mass balance constraints in FBA |
| Enzyme Capacity Constraints | Limits maximum reaction rates | Implementation of kcat values |
| Total Enzyme Pool (Ptot) | Constrains cellular protein resources | Represents proteome allocation limits |
| Objective Function Coefficients | Defines biological optimization targets | Biomass composition, ATP demand |
Performance Analysis and Comparative Evaluation
Quantitative Performance Metrics
Experimental comparisons across domains demonstrate consistent performance advantages for the TSLO approach:
In community network service placement problems, the TSLO method achieved a 53% improvement in bandwidth gain compared to random placement approaches and a 10% improvement over the best-known bandwidth-aware placement algorithm in the literature [48]. When enhanced with biased randomization techniques, these improvements increased to 58% and 20% respectively [48].
In computational efficiency comparisons, the two-stage approach enabled more effective rescheduling in robust scheduling applications by leveraging optimal substructure imposed by lexicographic optimality [51]. This structural advantage facilitated approximate rescheduling with bounded performance guarantees, characterized by a price of robustness parameterized by uncertainty degree [51].
Advantages and Limitations
Key Advantages:
- Maintains strict priority adherence for critical objectives
- Computationally efficient through sequential linearization
- Provides predictable and interpretable solution structures
- Enables tractable solutions for complex multi-scale systems
Identified Limitations:
- Requires explicit priority ranking of objectives
- Performance dependent on appropriate tolerance selection
- May yield inferior results when objectives are strongly correlated
- Limited exploration of intermediate trade-off solutions
The Two-Stage Lexicographic Optimization approach represents a mathematically rigorous and computationally efficient methodology for multi-objective optimization problems with inherent hierarchical structure. In flux prediction research and related biological applications, this approach demonstrates consistent advantages in maintaining essential system functions while achieving secondary optimization targets. The experimental evidence across domains confirms that TSLO provides superior performance compared to traditional weighted-sum and heuristic approaches, particularly in scenarios requiring strict adherence to priority constraints. For researchers and drug development professionals working with complex biological systems, this method offers a structured framework for balancing competing objectives while maintaining computational tractability.
Dealing with Large Solution Spaces and Flux Variability
Predicting metabolic fluxes—the rates at which metabolic reactions occur—is fundamental for understanding cellular behavior in fields ranging from biotechnology to drug development. A significant challenge in this domain is dealing with large solution spaces and flux variability, where multiple flux distributions can equally satisfy cellular constraints. This article provides a comparative analysis of computational frameworks designed to address this challenge, evaluating their performance in predicting reliable fluxes from complex metabolic networks.
Each method approaches the problem of solution space redundancy differently: Flux Variability Analysis (FVA) quantifies the range of possible fluxes for each reaction, 13C Metabolic Flux Analysis (13C-MFA) uses isotopic tracers to constrain the system, machine learning models learn pattern-to-flux relationships from data, and objective function discovery frameworks algorithmically determine cellular goals. The following sections compare these alternatives' methodologies, performance, and applicability, providing researchers with a guide for selecting the appropriate tool for their flux prediction challenges.
Comparative Analysis of Computational Frameworks
The table below summarizes the core characteristics, performance, and primary applications of the major computational frameworks for flux prediction.
Table 1: Comparative Overview of Flux Prediction Methods
| Method | Core Approach | Key Performance Metric | Handling of Flux Variability | Primary Application Context |
|---|---|---|---|---|
| Flux Variability Analysis (FVA) | Optimization-based; solves LP problems to find min/max flux ranges [52] | Reduced LPs required by ~40%; computation time reduced proportionally [52] | Directly quantifies feasible flux ranges | Identifying essential reactions, network flexibility analysis [52] |
| 13C-MFA with 13CFLUX(v3) | Isotope labeling simulation & parameter fitting [53] | >90% accuracy; >1000x faster than iterative methods [29] | Constrains solution space using experimental isotopic labeling data [53] | Metabolic engineering, quantitative systems biology [53] |
| Machine Learning (ML-Flux) | Neural networks mapping isotope patterns to fluxes [29] | >90% accuracy; handles variable-size input with missing data [29] | Learns flux patterns from training data; imputes missing patterns [29] | Rapid flux prediction from partial omics data [29] |
| Objective Function Discovery (TIObjFind) | Integrates MPA with FBA; infers objective coefficients [10] | Aligns predictions with experimental data; reduces overfitting [10] | Identifies context-specific objectives reducing solution space [10] | Multi-condition metabolic studies, adaptive response analysis [10] |
| Bayesian 13C-MFA | Multi-model inference with Markov Chain Monte Carlo [54] | Robust to model uncertainty; quantifies flux probability [54] | Provides probability distributions over flux values [54] | Scenarios with model selection uncertainty, bidirectional flux analysis [54] |
Detailed Methodologies and Experimental Protocols
Flux Variability Analysis with Improved Algorithms
Traditional FVA quantifies the feasible ranges of reaction fluxes by solving numerous linear programming (LP) problems—specifically, 2n+1 LPs for a network with n reactions [52]. The improved FVA algorithm reduces computational burden through a solution inspection procedure that leverages the basic feasible solution property of LPs [52]. The experimental protocol involves:
Phase 1: Solve a single LP to find the maximum objective value (Z₀) for the biological imperative (e.g., biomass maximization) [52]: Equation 1: Z₀ = max cᵀv, subject to Sv = 0, and v_low ≤ v ≤ v_high
Phase 2: Determine flux ranges while maintaining optimality within a factor μ [52]: Equation 2: max/min v_i, subject to Sv = 0, cᵀv ≥ μZ₀, and v_low ≤ v ≤ v_high
Solution Inspection: After solving each LP, check if any flux variables hit their bounds. If so, skip the dedicated optimization for that bound, reducing the total LPs needed [52].
Benchmarking on 112 metabolic network models showed this approach reduced the number of LPs required by approximately 40%, with corresponding decreases in computation time [52].
13C Metabolic Flux Analysis with 13CFLUX(v3)
13CFLUX(v3) employs a high-performance C++ engine with a Python interface for isotopically stationary and nonstationary metabolic flux analysis [53]. The experimental workflow involves:
Model Preparation: Define the metabolic network, atom transitions, and measurement configuration using the FluxML modeling language [53].
Isotope Labeling Simulation: Simulate isotopic labeling patterns using either Elementary Metabolite Units (EMUs) or cumomers as state-space representations. The system automatically selects the most dimension-reduced representation [53].
Parameter Optimization: Fit model parameters to experimental isotope labeling data using nonlinear optimization [53].
Statistical Analysis: Employ Bayesian inference or classical methods for uncertainty quantification [53].
The software's architecture enables efficient simulation of large-scale labeling systems exceeding 1000 dimensions, with substantial performance gains over previous versions [53].
Machine Learning Approach with ML-Flux
ML-Flux bypasses traditional iterative optimization by training neural networks to directly map isotope labeling patterns to metabolic fluxes [29]. The protocol involves:
Data Generation: Simulate training data by sampling fluxes from physiological ranges and computing corresponding mass isotopomer distributions (MIDs) for various tracer configurations [29].
Network Training: Train artificial neural networks (ANNs) using flux-MID pairs. Log-uniform flux sampling has been shown to produce the best-performing models [29].
Model Application: Apply trained networks to experimental MIDs, with the PCNN component handling missing data through imputation [29].
Validation: Compare predictions against held-out test data and results from conventional 13C-MFA software [29].
This approach demonstrated >90% accuracy with computational speeds orders of magnitude faster than iterative least-squares methods [29].
Objective Function Discovery with TIObjFind
TIObjFind addresses the challenge of selecting appropriate objective functions in FBA by inferring Coefficients of Importance (CoIs) for reactions [10]. The methodology involves:
Problem Formulation: Reformulate objective function selection as an optimization problem minimizing the difference between predicted and experimental fluxes while maximizing an inferred metabolic goal [10].
Mass Flow Graph Construction: Map FBA solutions onto a directed, weighted graph representing metabolic flux distributions [10].
Pathway Analysis: Apply a minimum-cut algorithm (Boykov-Kolmogorov) to identify critical pathways and compute Coefficients of Importance [10].
Flux Prediction: Use the weighted objective function for context-specific flux predictions [10].
This approach has been successfully applied to Clostridium acetobutylicum fermentation and multi-species systems, demonstrating improved alignment with experimental data [10].
Workflow Visualization of Key Methods
The following diagrams illustrate the core workflows for the primary methods discussed, highlighting their distinct approaches to handling flux variability.
Diagram 1: Flux Variability Analysis (FVA) workflow with solution inspection.
Diagram 2: Machine Learning Flux (ML-Flux) prediction workflow.
Diagram 3: TIObjFind framework for objective function discovery.
Research Reagent Solutions for Flux Analysis
The table below details essential computational tools and resources for implementing the flux analysis methods discussed in this guide.
Table 2: Key Research Reagent Solutions for Metabolic Flux Analysis
| Tool/Resource | Type | Primary Function | Compatibility/Requirements |
|---|---|---|---|
| 13CFLUX(v3) [53] | Software Platform | High-performance 13C-MFA simulation | Python 3.9-13, C++17 compiler; Docker containers available |
| COBRApy [52] | Software Toolbox | Constraint-based modeling, FBA, FVA | Python environment |
| ML-Flux [29] | Machine Learning Framework | Flux prediction from isotope patterns | Online web resource (metabolicflux.org) |
| TIObjFind [10] | MATLAB Framework | Objective function discovery | MATLAB with maxflow package |
| FluxML [53] | Modeling Language | Representing metabolic networks & experiments | Used with 13CFLUX(v3) platform |
| Eigen Library [53] | Numerical Library | Sparse matrix operations | C++ (used by 13CFLUX backend) |
| SUNDIALS CVODE [53] | ODE Solver | Isotopically nonstationary MFA | C++ (integrated in 13CFLUX) |
The comparative analysis presented in this guide demonstrates that method selection for dealing with large solution spaces and flux variability depends heavily on the specific research context. For researchers requiring comprehensive flux flexibility analysis, improved FVA algorithms provide computational efficiency gains. When isotopic tracer experiments are feasible, 13C-MFA methods—particularly the Bayesian approaches and high-performance 13CFLUX(v3) platform—offer rigorous flux estimation with uncertainty quantification. For rapid prediction from omics data or when dealing with missing measurements, ML-Flux presents a promising data-driven alternative. Finally, for multi-condition studies where cellular objectives may shift, TIObjFind offers a principled approach to objective function discovery.
Emerging methodologies, including quantum interior-point methods for flux balance analysis [55] and flux-sum coupling analysis [56], represent the continuing evolution of this field. As metabolic models grow in size and complexity—encompassing multi-species communities and dynamic temporal processes—the computational efficiency and statistical robustness of these tools will become increasingly critical for applications in metabolic engineering and drug development.
Benchmarking Performance: Validation Frameworks and Model Selection
The prediction of metabolic reaction rates, or fluxes, is fundamental to advancing our understanding of cellular processes in systems biology and metabolic engineering. Flux Balance Analysis (FBA) has emerged as one of the most important techniques for estimating these fluxes, utilizing optimization criteria to select flux distributions from a feasible space delimited by metabolic reactions and constraints [4]. Similarly, 13C-Metabolic Flux Analysis (13C-MFA) employs isotopic labeling data to estimate intracellular fluxes [57]. Both methods operate under the steady-state assumption, where reaction rates and metabolic intermediate levels remain constant. However, these approaches generate predictions rather than direct measurements, creating an essential need for robust statistical validation methods to assess their reliability and accuracy.
The statistical validation of flux predictions ensures that computational models accurately represent biological reality, which is particularly crucial when these models inform metabolic engineering strategies or biological conclusions. Despite advances in metabolic modeling techniques, validation and model selection methods have been underappreciated and underexplored in the field [57]. Goodness-of-fit tests provide a statistical framework for evaluating how well model-derived fluxes align with experimental data, serving as critical tools for model selection and refinement. Within this context, the χ2-test of goodness-of-fit has become the most widely used quantitative validation approach in 13C-MFA, though it has specific limitations and requires complementary validation strategies [57] [58].
This review examines the current landscape of statistical validation methods for flux predictions, with particular emphasis on goodness-of-fit tests and their application in comparing objective functions for flux prediction research. We provide comparative analysis of different validation approaches, detailed experimental protocols, and resources to assist researchers in selecting appropriate validation frameworks for their specific applications.
Flux Prediction Methods: A Comparative Framework
Constraint-Based Modeling Approaches
Constraint-based modeling frameworks represent the cornerstone of metabolic flux prediction, with two primary methodologies dominating the field:
Flux Balance Analysis (FBA) utilizes linear optimization to identify flux maps that maximize or minimize an objective function representing biological goals such as growth rate maximization or product formation [57]. The core FBA formulation solves for flux distributions (v) subject to stoichiometric constraints (S·v = 0) and capacity constraints (vmin ≤ v ≤ vmax). A critical determinant in FBA outcomes is the objective function, which embodies hypotheses about what cellular systems have evolutionarily optimized [57]. Comparative studies have evaluated numerous objective functions—including maximal biomass production, ATP maximization, and flux minimization—to determine which produces flux distributions most consistent with experimental data [4].
13C-Metabolic Flux Analysis (13C-MFA) works backward from measured isotopic label distributions in metabolites to infer flux maps by minimizing differences between measured and simulated mass isotopomer distributions [57]. This approach incorporates atom mapping information that describes carbon atom transitions through metabolic networks, providing additional constraints that enable more precise flux estimation than FBA alone. Recent advances in 13C-MFA include parallel labeling experiments that employ multiple tracers simultaneously to generate more precise flux maps [57].
The Centrality of Objective Functions in Flux Prediction
The choice of objective function represents a fundamental assumption in FBA that significantly influences resulting flux predictions. As Schuetz et al. demonstrated, different objective functions can produce markedly different flux distributions, with maximal energy (ATP) or biomass production often providing the most accurate descriptions of experimental data [17]. However, the optimal objective function may be condition-dependent, varying across different environmental contexts or organism types [4].
Table 1: Common Objective Functions in Flux Balance Analysis
| Objective Function | Biological Rationale | Typical Applications | Key References |
|---|---|---|---|
| Biomass Maximization | Represents cellular growth as an evolutionary priority | Microbial growth simulations, biotechnology | [57] [17] |
| ATP Maximization | Assumes energy production as primary cellular goal | Energy metabolism studies, hypoxic conditions | [17] |
| Minimization of Metabolic Adjustment (MOMA) | Assumes minimal redistribution after perturbation | Prediction of knockout mutant metabolism | [57] |
| Parsimonious Enzyme Usage | Minimizes total flux as proxy for enzyme efficiency | Conditions with enzyme synthesis constraints | [17] |
| Product Yield Maximization | Optimizes for specific metabolite production | Metabolic engineering, bioproduction | [59] |
The critical importance of objective function selection was further highlighted by Schnitzer et al., who demonstrated that the choice of objective function significantly affects predictions of replicative lifespan in yeast models, with maximal growth being essential for realistic lifespan predictions [17]. This connection between objective function choice and physiological outcomes underscores the necessity of rigorous validation against experimental data.
Goodness-of-Fit Tests for Flux Prediction Validation
Fundamental Concepts and Statistical Framework
Goodness-of-fit evaluation forms the statistical foundation for validating flux predictions. In general terms, goodness of fit describes how well observed data align with values expected under a specific statistical model [60]. These tests quantify the discrepancy between observed and expected values, enabling researchers to determine whether differences are statistically significant or likely due to random variation. For flux predictions, goodness-of-fit tests assess how well model-generated fluxes match experimentally determined fluxes or measurements of related system properties.
The general approach involves formulating two competing hypotheses:
- Null Hypothesis (H₀): The model adequately fits the data, with discrepancies due to random variation
- Alternative Hypothesis (Hₐ): The model does not adequately fit the data, with discrepancies being systematic
A test statistic quantifies the overall discrepancy, and its value is compared to a reference distribution to determine whether to reject the null hypothesis [60]. The following sections detail specific goodness-of-fit tests relevant to flux prediction validation.
The Chi-Square (χ²) Test of Goodness-of-Fit
The chi-square test represents the most widely used goodness-of-fit test in 13C-MFA, providing a quantitative method for comparing observed and expected isotopic labeling patterns [57] [58]. The test statistic is calculated as:
χ² = Σ[(Oi - Ei)² / E_i]
Where Oi represents the observed frequency (e.g., of a specific mass isotopomer), Ei represents the expected frequency predicted by the model, and the summation occurs across all measured bins [61]. The resulting value is compared to a chi-square distribution with (k - c) degrees of freedom, where k represents the number of non-empty bins and c the number of estimated parameters [61].
In 13C-MFA applications, the chi-square test specifically evaluates the fit between measured mass isotopomer distributions (MIDs) and those simulated from candidate flux maps [57]. A statistically non-significant chi-square value (typically assessed at α = 0.05) indicates that the model adequately explains the experimental data, while a significant value suggests model inadequacy.
Despite its widespread use, the chi-square test in 13C-MFA has important limitations. The test assumes that measurement errors follow a normal distribution with known variances, which may not always hold in practice [57]. Additionally, the test can be insensitive to specific forms of model misspecification, particularly when applied to large-scale models with many degrees of freedom [57] [58].
Alternative Goodness-of-Fit Measures
While the chi-square test dominates 13C-MFA validation, other goodness-of-fit measures provide valuable alternatives or complementary approaches:
R-squared (R²) measures the percentage of variance in the dependent variable explained by the model, providing an intuitive 0-100% scale for model performance [60]. In flux prediction contexts, R² can quantify how well FBA predictions explain variations in measured exchange fluxes or 13C-MFA derived fluxes.
Standard Error of the Regression (S) represents the average absolute difference between observed and predicted values in the units of the response variable [60]. For flux predictions, this could express the typical deviation in mmol/(gDW·h) between predicted and measured fluxes.
Akaike's Information Criterion (AIC) facilitates model comparison by balancing goodness of fit against model complexity, penalizing the addition of unnecessary parameters [60]. The AIC formula is:
AIC = 2k - 2ln(L)
Where k represents the number of parameters and L the likelihood of the model given the data. When comparing multiple models, lower AIC values indicate better balance between fit and complexity [60].
G-test represents a likelihood-ratio test increasingly used as an alternative to Pearson's chi-square test for categorical data [61]. The test statistic is:
G = 2Σ[Oi · ln(Oi / E_i)]
This test is particularly useful when sample sizes are small or when expected frequencies are low [61].
Table 2: Goodness-of-Fit Tests for Flux Prediction Validation
| Test/Metric | Application Context | Strengths | Limitations |
|---|---|---|---|
| Chi-square Test | 13C-MFA model validation, isotopic labeling data | Well-established, provides p-value for hypothesis testing | Sensitive to sample size, assumes known measurement errors |
| R-squared (R²) | Overall model fit for continuous flux measurements | Intuitive interpretation, scale-independent | Can be inflated by adding parameters regardless of relevance |
| Standard Error (S) | Absolute fit of flux predictions | In original units, directly interpretable | Difficult to compare across studies with different units |
| Akaike Information Criterion (AIC) | Comparison of alternative model structures | Penalizes complexity, facilitates model selection | Requires multiple models, no absolute goodness-of-fit measure |
| G-test | Alternative to chi-square for categorical data | Better performance with small samples | Less familiar to many researchers |
Experimental Protocols for Validation Studies
Protocol for Comparative Evaluation of Objective Functions
Comprehensive evaluation of objective functions requires systematic comparison against experimental data following this established protocol:
Select Reference Dataset: Obtain experimentally determined fluxes, typically from 13C-MFA studies or direct flux measurements. Ensure the dataset covers diverse metabolic pathways and conditions relevant to the intended application [4].
Define Candidate Objective Functions: Compile a set of biologically plausible objective functions for testing. Common candidates include:
- Biomass maximization
- ATP production maximization
- Minimization of total flux (parsimonious FBA)
- Product synthesis maximization
- Combinations through multi-objective optimization [17]
Implement FBA Simulations: For each objective function, perform FBA calculations using identical stoichiometric models, constraints, and computational frameworks to generate flux predictions [4].
Calculate Goodness-of-Fit Metrics: Quantify the agreement between predicted and experimental fluxes using multiple metrics:
- Chi-square statistics for categorical data
- R-squared values for variance explanation
- Root Mean Square Error (RMSE) for absolute differences
- AIC for model comparison [60]
Statistical Analysis: Perform appropriate statistical tests to determine whether differences in model performance are statistically significant. For nested models, use F-tests; for non-nested models, use AIC or related information criteria [60].
Condition-Specific Validation: Repeat the evaluation across different environmental conditions (e.g., carbon sources, nutrient limitations) to test objective function robustness [4] [17].
Protocol for 13C-MFA Model Validation Using Chi-Square Test
The standard protocol for validating 13C-MFA models with chi-square tests involves these key steps:
Experimental Design: Conduct isotopic tracing experiments using one or more 13C-labeled substrates (e.g., [1-13C]glucose, [U-13C]glucose). Parallel labeling experiments with multiple tracers provide more comprehensive information for flux estimation [57].
Mass Isotopomer Measurement: Quantify mass isotopomer distributions (MIDs) of intracellular metabolites using mass spectrometry or NMR techniques. Technical replicates are essential for estimating measurement errors [57].
Flux Estimation: Estimate metabolic fluxes by minimizing the difference between measured and simulated MIDs using appropriate optimization algorithms. The residual sum of squares (SSR) forms the basis for chi-square calculation [57].
Chi-Square Calculation: Compute the chi-square statistic as: χ² = Σ[(MIDmeasured - MIDsimulated)² / σ²] Where σ² represents the variance of measurement errors for each MID [57] [58].
Statistical Evaluation: Compare the calculated chi-square value to the critical value from a chi-square distribution with degrees of freedom equal to the number of measured MID points minus the number of estimated parameters. A p-value > 0.05 typically indicates acceptable model fit [57].
Sensitivity Analysis: Perform sensitivity analysis to identify reactions with high flux uncertainty and potentially refine the model structure [57].
Visualization of Validation Workflows and Relationships
The following diagrams illustrate key workflows and relationships in flux prediction validation, created using Graphviz DOT language with appropriate color contrast and styling.
Figure 1: Workflow for flux prediction validation. The process begins with data collection and proceeds through model setup, prediction, and statistical evaluation. Models failing goodness-of-fit tests require refinement or alternative objective functions.
Figure 2: Relationship between goodness-of-fit tests and their applications in flux prediction validation. Different tests serve distinct purposes across 13C-MFA, FBA, and model selection contexts.
Research Reagent Solutions for Flux Studies
The following table details essential research reagents and computational tools used in flux prediction and validation studies.
Table 3: Essential Research Reagents and Tools for Flux Studies
| Reagent/Tool | Function/Application | Specifications | Example Uses |
|---|---|---|---|
| 13C-labeled Substrates | Isotopic tracing for MFA | [1-13C]glucose, [U-13C]glucose, other positional isomers | Experimental input for 13C-MFA flux determination [57] |
| Mass Spectrometry | Measurement of mass isotopomer distributions | LC-MS, GC-MS systems with high mass accuracy | Quantifying isotopic labeling patterns for 13C-MFA [57] |
| Flux Analysis Software | Computational flux estimation | 13C-MFA packages (e.g., INCA, OpenFlux) | Flux estimation from isotopic labeling data [57] |
| Constraint-Based Modeling Tools | FBA simulation and analysis | COBRA Toolbox, CellNetAnalyzer, custom code | Implementing FBA with different objective functions [4] [17] |
| Genome-Scale Metabolic Models | Stoichiometric networks for FBA | Organism-specific models (e.g., iML1515, yeast 8.0) | Providing biochemical constraints for flux predictions [57] |
| Statistical Software | Goodness-of-fit testing | R, Python (scipy), MATLAB | Implementing chi-square tests, AIC calculation, other metrics [60] |
The statistical validation of flux predictions through goodness-of-fit tests represents a critical component of metabolic modeling workflows. The chi-square test remains the gold standard for 13C-MFA validation, while a diverse set of metrics including R-squared, AIC, and standard error provide complementary perspectives on model performance. The choice of objective function in FBA represents a fundamental assumption that significantly influences flux predictions, with different functions performing optimally under different biological contexts.
Robust validation requires multiple goodness-of-fit measures evaluated across diverse experimental conditions. No single test provides a complete picture of model performance, emphasizing the need for comprehensive validation strategies. As the field advances, integrating newer approaches such as comparative flux sampling analysis [59] with traditional goodness-of-fit tests promises to enhance our ability to discriminate between alternative model structures and select those with greatest biological fidelity.
The continued refinement of validation methodologies will strengthen confidence in constraint-based modeling approaches, ultimately facilitating more reliable applications in basic biological discovery and biotechnological engineering.
Comparing FBA Predictions Against 13C-MFA Estimated Fluxes
Quantifying intracellular metabolic fluxes is crucial for understanding cellular physiology in systems biology, metabolic engineering, and biomedical research. Flux Balance Analysis (FBA) and 13C-Metabolic Flux Analysis (13C-MFA) have emerged as the two primary computational frameworks for predicting and estimating these fluxes, yet they differ fundamentally in their approaches and applications [57] [62]. FBA is a constraint-based modeling approach that predicts flux distributions by assuming the cellular metabolic network optimizes a biological objective function, such as maximizing growth rate or biomass production [57] [63]. In contrast, 13C-MFA is an experimentally driven method that integrates isotopic tracer data with computational modeling to estimate fluxes by minimizing the difference between measured and simulated metabolite labeling patterns [64] [62]. This comparison guide examines the performance characteristics, validation methodologies, and appropriate applications of these complementary approaches, providing researchers with a framework for selecting and implementing these tools in metabolic flux studies.
The fundamental distinction between these methods lies in their core operating principles. FBA requires a stoichiometric model of the metabolic network and uses linear programming to identify flux distributions that optimize a specified objective function within physico-chemical constraints [57] [65]. Alternatively, 13C-MFA requires experimental data from isotopic labeling experiments where cells are fed with 13C-labeled substrates (e.g., glucose or glutamine), after which the labeling patterns of intracellular metabolites are measured using mass spectrometry or NMR techniques [62]. These labeling patterns are then used to infer intracellular fluxes through model-based regression analysis [64] [62]. While FBA is primarily a predictive tool, 13C-MFA is an estimation approach that directly leverages experimental data to determine flux values, making their comparative analysis particularly valuable for validating model predictions and refining metabolic networks.
Fundamental Methodological Differences
Core Principles and Data Requirements
The methodological foundations of FBA and 13C-MFA stem from different philosophies about how metabolic fluxes should be determined. FBA operates on the principle that metabolic networks have evolved to optimize certain functions, and it predicts fluxes based on stoichiometric constraints and an assumed biological objective [57] [63]. The solution space in FBA is defined by mass balance constraints (the stoichiometric matrix), thermodynamic constraints, and measured external fluxes, with the optimal solution selected through linear optimization [57] [65]. A significant challenge in FBA is the existence of multiple optimal solutions that satisfy the same objective function equally well, requiring additional techniques such as Flux Variability Analysis or random sampling to characterize the range of possible flux maps [57] [63].
In contrast, 13C-MFA is fundamentally an parameter estimation problem where fluxes are determined by fitting experimental isotopic labeling data to a metabolic network model [62] [66]. The method works because different flux distributions produce distinct isotopic labeling patterns in intracellular metabolites when cells are fed with 13C-labeled substrates [64] [62]. The analysis involves minimizing the residuals between measured and simulated mass isotopomer distributions through iterative adjustment of flux values [57] [62]. Unlike FBA, 13C-MFA can accurately resolve fluxes through parallel pathways and reversible reactions, and can quantify metabolic cycles and exchange fluxes, providing a more detailed view of central carbon metabolism [62] [66].
Table 1: Fundamental Characteristics of FBA and 13C-MFA
| Characteristic | Flux Balance Analysis (FBA) | 13C-Metabolic Flux Analysis (13C-MFA) |
|---|---|---|
| Primary Basis | Stoichiometric constraints and optimization principles | Experimental isotopic labeling data and model fitting |
| Data Requirements | Stoichiometric model, external flux measurements (optional) | 13C-labeling data, external fluxes, metabolic network with atom transitions |
| Mathematical Framework | Linear programming (optimization) | Nonlinear least-squares regression |
| Key Assumptions | Steady-state metabolism, optimal cellular behavior | Metabolic and isotopic steady state |
| Network Scale | Genome-scale models (hundreds to thousands of reactions) | Central carbon metabolism (dozens to hundreds of reactions) |
| Flux Resolution | Net fluxes only | Net and exchange (reversible) fluxes |
Experimental and Computational Workflows
The implementation of FBA and 13C-MFA involves distinct experimental and computational workflows. The FBA workflow begins with model reconstruction, where a stoichiometric representation of the metabolic network is assembled from genomic and biochemical data [57]. Constraints are applied based on measured uptake and secretion rates, and an objective function is selected, most commonly biomass maximization for growing cells [63] [65]. The model is then solved using linear programming, and the resulting flux predictions are validated through comparison with experimental data, such as growth rates or gene essentiality [57] [65].
The 13C-MFA workflow initiates with experimental design, where appropriate 13C-tracers are selected to maximize information gain about the metabolic pathways of interest [62] [66]. Cells are cultured with the labeled substrates until metabolic and isotopic steady state is reached, after which metabolites are extracted and their labeling patterns are measured using analytical techniques such as GC-MS or LC-MS [62]. The labeling data, along with measured external fluxes, are integrated with a metabolic network model that includes atom mapping information to simulate carbon atom rearrangements through metabolic pathways [57] [62]. Fluxes are estimated by iteratively adjusting flux values to achieve the best fit between simulated and measured labeling data, followed by statistical evaluation of the goodness-of-fit and flux confidence intervals [62] [66].
Diagram 1: Comparative workflows of FBA and 13C-MFA approaches to flux determination.
Quantitative Comparison of Flux Predictions
Case Study: E. coli under Aerobic and Anaerobic Conditions
A comprehensive comparative analysis of FBA predictions and 13C-MFA estimated fluxes was conducted for wild-type E. coli (K-12 MG1655) grown aerobically and anaerobically in glucose-limited minimal medium [63]. This study employed a consistent metabolic network model for both analyses, allowing direct comparison of the resulting flux maps. The 13C-MFA results revealed that the fraction of maintenance ATP consumption in total ATP production was approximately 14% higher under anaerobic (51.1%) compared to aerobic conditions (37.2%) [63]. FBA predictions suggested this increased ATP utilization was consumed by ATP synthase to secrete protons from fermentation. Furthermore, 13C-MFA indicated the TCA cycle operates non-cyclically in aerobically growing cells, with submaximal growth attributed to limitations in oxidative phosphorylation [63].
The study demonstrated that FBA successfully predicted product secretion rates in aerobic cultures when constrained with both glucose and oxygen uptake measurements [63]. However, the most frequently predicted values of internal fluxes obtained through sampling of the feasible solution space showed substantial differences from 13C-MFA derived fluxes [63]. This highlights a key limitation of FBA: while it may accurately predict external phenotypes (e.g., secretion rates), its internal flux predictions may deviate significantly from experimentally determined values. The synergy between both approaches revealed physiological insights that would not have been apparent from either method alone, such as the submaximal efficiency of ATP production and the incomplete operation of the TCA cycle [63].
Table 2: Comparative Flux Values for E. coli Central Metabolism (Aerobic Conditions)
| Metabolic Pathway/Reaction | FBA Predicted Flux (mmol/gDCW/h) | 13C-MFA Estimated Flux (mmol/gDCW/h) | Relative Difference (%) |
|---|---|---|---|
| Glycolysis | |||
| Glucose uptake | 8.2 | 8.2 (constrained) | 0.0 |
| Pyruvate production | 16.4 | 15.9 | 3.1 |
| Pentose Phosphate Pathway | |||
| G6PDH flux | 1.1 | 2.3 | 52.2 |
| TCA Cycle | |||
| Citrate synthase | 5.8 | 2.1 | 64.3 |
| Isocitrate dehydrogenase | 5.8 | 2.1 | 64.3 |
| α-ketoglutarate dehydrogenase | 5.8 | 1.3 | 77.6 |
| Anaplerotic Reactions | |||
| PEP carboxylase | 1.5 | 3.2 | 53.1 |
| Pyruvate carboxylase | 0.0 | 0.8 | 100.0 |
Statistical Validation and Goodness-of-Fit
A critical aspect of comparing FBA predictions and 13C-MFA estimates is the statistical validation of the results. In 13C-MFA, the goodness-of-fit is typically evaluated using the χ²-test, which compares the minimized weighted sum of squared residuals (SSRES) between measured and simulated labeling data to a theoretical χ² distribution [57] [66]. Additionally, flux confidence intervals are determined through statistical evaluation of the parameter sensitivity, often using Monte Carlo sampling or parameter continuation methods [62] [66]. These statistical measures provide quantitative assessment of the precision and reliability of the flux estimates.
For FBA, validation approaches are more varied and less standardized. Common techniques include comparing predicted versus actual growth rates on different substrates, testing the model's ability to predict gene essentiality, and comparing internal flux predictions with 13C-MFA results when available [57] [65]. The MEMOTE (MEtabolic MOdel TEsts) pipeline has been developed to provide standardized testing of metabolic models, ensuring appropriate stoichiometry and consistency with format standards [65]. However, unlike 13C-MFA, FBA does not inherently provide statistical confidence intervals for its predictions, making quantitative assessment of prediction uncertainty challenging.
Experimental Protocols for Method Comparison
Protocol for Parallel FBA and 13C-MFA Analysis
To directly compare FBA predictions with 13C-MFA estimated fluxes, researchers should follow an integrated experimental and computational protocol:
Strain and Culture Conditions: Use wild-type E. coli K-12 MG1655 (or other relevant model organism) cultured in defined minimal medium (e.g., M9) with glucose (2 g/L) as sole carbon source [63]. Perform parallel aerobic and anaerobic cultivations at 37°C with appropriate monitoring of growth parameters (optical density, cell counts).
External Flux Measurements: During mid-exponential growth phase, measure substrate uptake and product secretion rates using analytical methods such as enzymatic assays, HPLC, or NMR [63] [62]. For aerobic cultures, measure oxygen uptake rates; for anaerobic cultures, measure CO2 and H2 production if applicable. Calculate specific uptake/secretion rates (nmol/10⁶ cells/h) using the growth rate and concentration changes [62].
13C-Labeling Experiments: Cultivate cells with specifically 13C-labeled glucose tracers (e.g., [1-13C]glucose, [U-13C]glucose, or mixture designs) [63] [62]. Harvest cells during isotopic steady state (typically after 3-5 generations for microbial systems). Extract intracellular metabolites and measure mass isotopomer distributions using GC-MS or LC-MS [62] [66].
Metabolic Network Modeling: Construct a consistent metabolic network model for both FBA and 13C-MFA analyses. For 13C-MFA, include complete atom transition information for all reactions [57] [66]. The model should cover central carbon metabolism including glycolysis, pentose phosphate pathway, TCA cycle, and anaplerotic reactions.
Flue Estimation and Prediction: Perform 13C-MFA using specialized software (e.g., INCA, Metran, or Iso2Flux) to estimate intracellular fluxes by fitting the labeling data and external fluxes [62] [67]. Conduct FBA using the same metabolic network model, constraining the model with measured external fluxes and using appropriate objective functions (e.g., biomass maximization) [63] [65].
Statistical Analysis and Validation: For 13C-MFA, determine goodness-of-fit using χ²-test and calculate flux confidence intervals [66]. For FBA, perform flux variability analysis to characterize the range of possible optimal solutions [57] [63]. Compare fluxes at key metabolic nodes and calculate correlation metrics between FBA predictions and 13C-MFA estimates.
Protocol for Synergistic Application in Metabolic Engineering
The combination of FBA and 13C-MFA can be particularly powerful for metabolic engineering applications. The following protocol outlines their integrated use:
Initial Strain Design: Use FBA with genome-scale models to identify potential genetic modifications (gene knockouts, additions, or regulatory changes) that would enhance production of target compounds [59] [65]. Leverage algorithms such as OptKnock or similar approaches to couple growth with product formation.
Experimental Implementation: Construct engineered strains based on FBA predictions and cultivate them under production conditions.
Physiological Characterization: Perform 13C-MFA experiments with the engineered strains to quantify the actual metabolic flux distributions resulting from the genetic modifications [63] [66]. Compare these with the FBA predictions to identify discrepancies.
Model Refinement: Use the 13C-MFA results to refine the stoichiometric model and constraint sets used in FBA [63]. This may include updating reaction stoichiometry, adding missing transport steps, or incorporating regulatory constraints based on the experimental flux data.
Iterative Strain Improvement: Use the refined model to generate new strain design predictions, then experimentally implement and validate these designs using 13C-MFA [59]. This iterative cycle of prediction and experimental validation accelerates the development of high-performing production strains.
Table 3: Key Research Reagent Solutions for FBA and 13C-MFA Studies
| Category | Specific Items | Function/Application | Examples/Sources |
|---|---|---|---|
| Isotopic Tracers | [1-13C]Glucose, [U-13C]Glucose, [1,2-13C]Glucose, other position-specific labels | Create distinct labeling patterns for flux elucidation through specific metabolic pathways | Cambridge Isotope Laboratories, Sigma-Aldrich |
| Analytical Instruments | GC-MS, LC-MS, NMR systems | Measure isotopic labeling patterns in intracellular metabolites and extracellular compounds | Agilent, Thermo Fisher, Bruker, Waters |
| Metabolic Modeling Software | COBRA Toolbox, INCA, Metran, Iso2Flux, p13CMFA | Perform FBA simulations and 13C-MFA flux estimations | Various academic and open-source platforms |
| Stoichiometric Models | BiGG Database, ModelSeed, organism-specific GEMs | Provide curated metabolic networks for constraint-based modeling and flux analysis | BiGG Models, http://bigg.ucsd.edu/ |
| Cell Culture Components | Defined minimal media, serum-free formulations, custom supplements | Maintain consistent metabolic conditions and minimize unaccounted carbon sources | Custom formulations, commercial basal media |
The comparative analysis of FBA predictions and 13C-MFA estimated fluxes reveals these methods as complementary rather than competing approaches to metabolic flux determination [57] [63]. FBA provides a genome-scale perspective based on biochemical constraints and optimization principles, making it particularly valuable for hypothesis generation and initial strain design in metabolic engineering projects [59] [65]. Conversely, 13C-MFA delivers high-resolution quantification of fluxes in central carbon metabolism, serving as an essential validation tool and providing insights into pathway operations that cannot be obtained through constraint-based modeling alone [63] [62]. The integration of both approaches creates a powerful framework for understanding cellular metabolism.
Future methodological developments are likely to further enhance the synergy between these approaches. Bayesian statistical methods are emerging as promising frameworks for 13C-MFA, allowing more robust handling of model uncertainty and multi-model inference [54]. For FBA, approaches such as Comparative Flux Sampling Analysis (CFSA) enable identification of metabolic engineering targets through systematic comparison of flux spaces corresponding to different physiological states [59]. Additionally, parsimonious 13C-MFA (p13CMFA) incorporates flux minimization principles from FBA into the 13C-MFA framework, potentially improving flux resolution when working with large networks or limited measurement sets [67]. As these methodologies continue to evolve and integrate, they will further solidify the role of metabolic flux analysis as an indispensable tool for understanding and engineering cellular metabolism.
Quantifying Prediction Accuracy and Uncertainty in Flux Estimates
Flux estimation, the process of quantifying the flow of metabolites through biochemical reactions in living cells, is a cornerstone of systems biology and metabolic engineering. Its accuracy directly impacts advancements in drug discovery, microbial strain improvement, and the understanding of disease mechanisms [8] [10]. Predictive models in this domain are inherently underdetermined, meaning innumerable flux distributions can satisfy the basic stoichiometric constraints of a metabolic network [42] [11]. To resolve this, objective functions are employed as mathematical surrogates for cellular goals, such as maximizing biomass growth or the production of a specific metabolite [8]. The selection of an appropriate objective function is arguably the most critical, and often most uncertain, step in the predictive pipeline. An ill-suited objective can lead to significant deviations from true biological behavior, making the rigorous quantification of both prediction accuracy and associated uncertainty paramount for reliable biological inference. This guide provides a comparative analysis of contemporary methods, focusing on their experimental performance, underlying protocols, and their approach to managing this inherent uncertainty.
Comparative Analysis of Flux Estimation Methods
The field has moved beyond simple Flux Balance Analysis (FBA) towards more sophisticated frameworks that integrate diverse data types and explicitly account for uncertainty. The table below compares the core features and quantitative performance of several key methods.
Table 1: Comparison of Modern Flux Estimation and Uncertainty Quantification Frameworks
| Method Name | Core Approach | Ideal Use Case | Reported Performance & Accuracy | Uncertainty Handling |
|---|---|---|---|---|
| TIObjFind [8] [10] | Integrates FBA with Metabolic Pathway Analysis (MPA) to infer data-driven objective functions via Coefficients of Importance (CoIs). | Identifying context-specific metabolic objectives and shifting cellular priorities in response to environmental changes. | Demonstrates a strong match with experimental flux data and reduced prediction error in case studies on Clostridium species. | Quantifies reaction importance (CoIs); uncertainty is inferred from pathway usage and fit to data. |
| BayFlux [68] | Employs Bayesian inference to sample flux distributions compatible with experimental data for genome-scale models. | Quantifying full distributions of possible fluxes, especially when distinct flux regions fit data equally well (non-gaussianity). | Produces narrower, more precise flux distributions (reduced uncertainty) with genome-scale models vs. traditional core models. | Directly quantifies uncertainty via posterior flux distributions, revealing multiple plausible flux states. |
| Flux Cone Learning (FCL) [11] | Uses Monte Carlo sampling of the metabolic flux cone and machine learning to link flux space geometry to phenotypes. | Predicting gene deletion phenotypes (e.g., essentiality) and other fitness outcomes without a pre-defined objective function. | Best-in-class 95% accuracy predicting E. coli gene essentiality, outperforming FBA; effective in complex organisms. | Captures phenotypic uncertainty through variance in sampled flux cones and model predictions. |
| E-Flux2 & SPOT [69] | Integrates transcriptomic data with genome-scale models to infer flux distributions, with (E-Flux2) or without (SPOT) a known objective. | Predicting system-wide, condition-specific fluxes when 13C-MFA data is unavailable but gene expression data is. | Average correlation with measured fluxes: 0.59 - 0.87 (across E. coli & S. cerevisiae), outperforming other transcriptome-integration methods. | Uncertainty is implicit in the fit to transcriptomic data; method does not directly quantify flux uncertainty. |
| Validation-based MFA [70] | Selects the best 13C-MFA model using an independent validation dataset, not used during model fitting. | Robust model selection for 13C-MFA to prevent overfitting/underfitting, especially when measurement errors are uncertain. | Consistently selects the correct model in simulations, robust to errors in measurement uncertainty estimates. | Mitigates model structure uncertainty, a key source of error not addressed by other parameter-focused methods. |
A critical finding across studies is that the completeness of the underlying metabolic model significantly impacts the certainty of predictions. For instance, BayFlux demonstrated that using genome-scale models instead of smaller core models resulted in narrower flux distributions, directly reducing prediction uncertainty [68]. Furthermore, methods that avoid a single optimality assumption, like FCL and BayFlux, are particularly valuable for modeling complex systems such as human tissues or microbial communities, where a universal objective function is unknown [42] [11].
Experimental Protocols for Key Methods
Protocol for Topology-Informed Objective Find (TIObjFind)
The TIObjFind framework identifies metabolic objective functions that best align with experimental data through a multi-step process [8] [10].
- Single-Stage Optimization: The framework solves an optimization problem that minimizes the squared error between predicted fluxes (
v) and experimental flux data (v_exp), while simultaneously maximizing a hypothesized, distributed objective function (c_obj · v). This step identifies a feasible flux distribution (v*) that balances fit to data and metabolic objective. - Mass Flow Graph (MFG) Construction: The optimized flux distribution
v*is mapped onto a directed, weighted graph called the Mass Flow Graph. Nodes represent metabolic reactions, and edge weights represent the flux between them. - Metabolic Pathway Analysis (MPA) and Minimum Cut Sets: A path-finding algorithm (e.g., Boykov-Kolmogorov) is applied to the MFG to identify critical pathways between a source (e.g., glucose uptake) and a target (e.g., product secretion). This analysis calculates Coefficients of Importance (CoIs), which quantify each reaction's contribution to the overall objective.
- Iteration and Analysis: The CoIs inform the objective function for subsequent optimizations, and the process can be repeated. Comparing CoIs across different biological stages reveals shifts in metabolic priorities.
Protocol for Validation-Based 13C Metabolic Flux Analysis (MFA)
This protocol provides a robust alternative to traditional, informal model selection for 13C-MFA [70].
- Experimental Design and Data Collection: Conduct two separate isotopic tracer experiments. The first tracer provides the estimation data (
D_est), and the second, distinct tracer provides the validation data (D_val). - Model Candidate Development: Define a sequence of model candidates (
M1, M2, ... Mk) with increasing complexity (e.g., by adding or removing reactions or compartments). - Parameter Estimation: For each model candidate
Mk, perform parameter estimation (model fitting) using only the estimation dataD_est. - Validation-Based Model Selection: Using the parameters fit from
D_est, calculate the Sum of Squared Residuals (SSR) for each model against the independent validation dataD_val. - Model Choice: Select the model candidate
Mkthat achieves the smallest SSR with respect to the validation dataD_val. This model is chosen for final flux estimation, as it best predicts new, unseen data.
Signaling Pathways and Workflow Visualizations
The following diagrams illustrate the logical workflows of the compared methods and the key signaling concept of bi-directional flux in environmental exchange models.
Figure 1: The TIObjFind Workflow for Identifying Objective Functions
Figure 2: The Flux Cone Learning (FCL) Predictive Pipeline
Figure 3: Bi-Directional NH₃ Exchange Pathways in Land-Atmosphere Models
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful execution of the protocols above requires a suite of computational and experimental resources.
Table 2: Key Reagents and Tools for Flux Estimation Research
| Tool/Reagent | Category | Primary Function | Example Use Case |
|---|---|---|---|
| Genome-Scale Model (GEM) | Computational | A structured database (stoichiometric matrix S, flux bounds) defining all known metabolic reactions in an organism. | Serves as the core constraint system for FBA, TIObjFind, FCL, and BayFlux [8] [11]. |
| 13C-Labeled Substrates | Experimental | Tracer compounds (e.g., [1-13C]glucose) fed to cells to generate isotopic patterns (Mass Isotopomer Distributions). | Provides the experimental data (D_est, D_val) for 13C-MFA and validation-based model selection [70]. |
| Monte Carlo Sampler | Computational/Algorithm | A tool for randomly sampling the high-dimensional space of feasible fluxes defined by a GEM. | Generates the feature set for training predictive models in Flux Cone Learning [11]. |
| Mass Spectrometer | Experimental | Instrumentation to precisely measure the abundance of different mass isotopomers in metabolites. | Quantifies Mass Isotopomer Distributions (MIDs), the primary data for 13C-MFA [70]. |
| Transcriptomic Dataset | Experimental | Genome-wide measurements of gene expression levels (e.g., via RNA-seq). | Serves as input for E-Flux2 and SPOT to infer condition-specific flux distributions [69]. |
Flux prediction is a critical capability in fields ranging from systems biology to environmental science, enabling researchers to understand and optimize complex dynamic systems. The performance of these predictive models is benchmarked on three core metrics: accuracy in matching experimental observations, computational speed for practical feasibility, and robustness across diverse conditions. This guide provides a comparative analysis of prominent flux prediction methodologies, including traditional constraint-based models and modern machine learning approaches, to inform selection for scientific and industrial applications.
Comparative Performance Analysis of Flux Prediction Methods
The table below summarizes the key performance characteristics of different flux prediction methodologies, as reported in recent experimental studies.
Table 1: Performance Comparison of Flux Prediction Methods
| Methodology | Core Application | Reported Accuracy Metrics | Computational Speed & Scalability | Noted Robustness Features |
|---|---|---|---|---|
| TIObjFind Framework(FBA-MPA Hybrid) | Metabolic flux prediction in biological systems [8] | Reduces prediction error and improves alignment with experimental flux data [8] | Not explicitly quantified; involves solving an optimization problem and pathway analysis [8] | Captures adaptive metabolic shifts and pathway usage under different environmental conditions [8] |
| Topology-Based ML(Random Forest) | Predicting metabolic gene essentiality [71] | F1-Score: 0.400 (Precision: 0.412, Recall: 0.389) [71] | Not explicitly quantified; "structure-first" approach avoids complex simulations [71] | Superior handling of biological redundancy in metabolic networks compared to simulation [71] |
| Extreme Gradient Boosting (XGBoost) | Predicting ecosystem-scale CO₂ flux [72] | RMSE: 1.81 μmol m⁻² s⁻¹, R²: 0.86 [72] | Not explicitly quantified; enables gap-filling and upscaling of flux tower measurements [72] | Generalizes to ecologically similar sites; performance drops in unique ecosystems [72] |
| Extremely Randomized Trees (ERT) | Predicting permeate flux in membrane distillation [73] | R²: 0.905, MAE: 2.614, RMSE: 4.588 (test set) [73] | Not explicitly quantified; ensemble method [73] | Handles complex, nonlinear interactions among multiple operational parameters [73] |
| Natural Gradient Boosting (NGRB) | Predicting CO₂ flux in underground coal fire areas [74] | R²: 0.967, MAE: 0.234 [74] | Not explicitly quantified; reduces need for costly physical experiments [74] | Effective in a complex, challenging physical environment [74] |
| Long Short-Term Memory (LSTM) | Predicting reactivity and flux in pebble bed reactors [75] | R²: 0.9914 on testing set [75] | Not explicitly quantified; trained on data from zone-based simulator PEARLSim [75] | Capable of forecasting long-term reactivity responses to operational changes [75] |
Experimental Protocols and Methodologies
A critical factor in interpreting performance data is understanding the experimental protocols from which they were derived. The following section details the methodologies behind several key studies cited in this guide.
Topology-Informed Objective Find (TIObjFind)
The TIObjFind framework was developed to identify context-specific metabolic objective functions by integrating Flux Balance Analysis (FBA) with Metabolic Pathway Analysis (MPA) [8]. Its experimental protocol can be summarized in three key steps:
- Optimization Problem Formulation: The framework solves an optimization problem that minimizes the difference between model-predicted fluxes and experimentally observed flux data, while simultaneously maximizing an inferred metabolic objective [8].
- Mass Flow Graph (MFG) Construction: The FBA solutions are mapped onto a graph representation of the metabolic network, enabling a pathway-based interpretation of flux distributions [8].
- Pathway Analysis and Coefficient Assignment: A path-finding algorithm analyzes "Coefficients of Importance" (CoIs) between start (e.g., glucose uptake) and target reactions (e.g., product secretion). These coefficients quantify each reaction's contribution to the inferred cellular objective, moving beyond a single-reaction optimization goal like biomass maximization [8].
Topology-Based Machine Learning for Gene Essentiality
This study benchmarked a machine learning model against traditional FBA for predicting genes essential for metabolic function [71]. The experimental workflow was as follows:
- Feature Engineering: A reaction-reaction graph was constructed from the e_coli_core metabolic model. Graph-theoretic features, including betweenness centrality and PageRank, were calculated to describe the topological role of each gene within the network [71].
- Model Training and Benchmarking: A
RandomForestClassifierwas trained on these topological features. Its performance was rigorously evaluated against a standard FBA single-gene deletion analysis using a curated ground-truth dataset of known essential genes [71]. - Performance Evaluation: The model achieved an F1-Score of 0.400, significantly outperforming the standard FBA approach, which failed to identify any known essential genes under the test conditions (F1-Score: 0.000) [71].
Machine Learning for CO₂ Flux Prediction
The high-performance XGBoost model for ecosystem-scale CO₂ flux (FCO₂) prediction was developed through a detailed protocol [72]:
- Data Sourcing and Standardization: Data was sourced from the National Ecological Observatory Network (NEON), a subset of AmeriFlux sites chosen for its standardized measurement protocols, which mitigates inconsistencies common in broader network data [72].
- Predictor Selection: Thirty-five environmental drivers and site-specific variables were used as model predictors [72].
- Model Training and Comparison: Seven different machine learning algorithms were trained and compared. XGBoost consistently produced the most accurate predictions for half-hourly FCO₂ [72].
- Validation: Model performance was tested on 44 sites, showing excellent results in ecologically similar regions but poorer performance in unique ecosystems like the Pacific Northwest [72].
Figure 1: A decision workflow for selecting and evaluating flux prediction methodologies, comparing traditional and machine learning approaches.
Successful flux prediction research relies on a combination of computational tools, datasets, and biological resources. The following table outlines essential components of the research toolkit.
Table 2: Essential Research Reagents and Resources for Flux Prediction
| Tool/Resource Name | Type | Primary Function in Research |
|---|---|---|
| Genome-Scale Metabolic Models (GEMs) | Computational Model | Provide a stoichiometric matrix representing all known metabolic reactions in an organism, serving as the core constraint structure for FBA [8]. |
| FLUXNET / AmeriFlux / ICOS | Observational Data Network | Provide standardized, tower-based ecosystem-scale CO₂ flux measurements for training and validating environmental flux models [76] [72]. |
| PEARLSim (Zone-Based Simulator) | Computational Tool | Generates high-fidelity operational and flux data for pebble bed reactors by combining Monte Carlo transport with fuel inventory management; used to train LSTM models [75]. |
| KEGG / EcoCyc | Biological Database | Provide extensive, curated information on biological pathways, genomes, and metabolites, forming the foundational database for constructing metabolic networks [8]. |
| Extreme Gradient Boosting (XGBoost) | Machine Learning Algorithm | A powerful, scalable ensemble tree-based algorithm frequently identified as a top performer for regression tasks like environmental flux prediction [73] [72]. |
| SHAP (SHapley Additive exPlanations) | Interpretability Tool | A post-hoc analysis method used to interpret ML model predictions by quantifying the contribution of each input feature to the final output [73]. |
| Ceramic Membranes | Physical Material | High-stability membranes used in direct contact membrane distillation (DCMD); their performance (permeate flux) is a key target for predictive modeling [73]. |
| Eddy Covariance Method | Measurement Technique | The standard technique for measuring turbulent fluxes of CO₂, water vapor, and energy between the land surface and the atmosphere at ecosystem scales [72]. |
Model Selection Practices for Choosing the Most Statistically Justified Model
In the fields of systems biology and metabolic engineering, computational models such as 13C-Metabolic Flux Analysis (13C-MFA) and Flux Balance Analysis (FBA) are indispensable for predicting intracellular metabolic fluxes that cannot be directly measured. These constraint-based methods rely on metabolic network models operating at steady state, where reaction rates and metabolite levels are invariant. The accuracy of flux predictions, however, is highly dependent on selecting the most statistically justified model architecture and objective function. Model selection and validation are critical for ensuring these computational tools provide reliable insights into basic biology and effective metabolic engineering strategies [57].
Despite advances in quantifying flux uncertainty, validation and model selection methods have been historically underappreciated in metabolic modeling. The selection of an appropriate model directly influences the fidelity of model-derived fluxes to real in vivo conditions, impacting subsequent scientific conclusions and engineering applications. This guide provides a comparative analysis of contemporary model selection practices, focusing on statistical validation methods and emerging computational frameworks that enhance model robustness for research and drug development [57].
Comparative Analysis of Model Selection Frameworks
The statistical rigor of a metabolic model is evaluated through validation and model selection procedures. These practices determine how well a model's predictions align with experimental data and which model structure is most probable given the available evidence.
The Chi-Squared Test of Goodness-of-Fit in 13C-MFA
The most widely used quantitative validation and selection approach in 13C-MFA is the χ²-test of goodness-of-fit. This test evaluates whether the differences between the experimentally measured Mass Isotopomer Distribution (MID) values and those estimated by the model are statistically significant, helping researchers determine if their model provides an adequate fit to the isotopic labeling data [57].
However, this method has notable limitations. The standard χ²-test can be insufficient for comprehensively validating a model, as it may not fully account for all sources of uncertainty or model structural errors. Consequently, relying solely on this test is increasingly viewed as inadequate for robust model selection. Complementary and alternative forms of validation are often necessary to confirm model accuracy [57].
Emerging and Integrated Frameworks
Recent research has developed more sophisticated frameworks that integrate multiple data types and analytical techniques to improve model selection.
- Combined Model Validation Framework for 13C-MFA: A promising development incorporates metabolite pool size information into the validation process. This framework leverages new developments in the field, advocating for a multi-faceted approach that goes beyond the traditional goodness-of-fit test. By including pool size data, this method provides additional constraints and validation checkpoints, potentially leading to the selection of more biologically realistic models [57].
- TIObjFind Framework for FBA: For FBA models, selecting an appropriate objective function is a fundamental aspect of model selection. The novel TIObjFind (Topology-Informed Objective Find) framework integrates Metabolic Pathway Analysis (MPA) with FBA to systematically infer metabolic objectives from experimental data [8]. This framework determines Coefficients of Importance (CoIs), which quantify each reaction's contribution to a hypothesized objective function. By solving an optimization problem that minimizes the difference between predicted and experimental fluxes, TIObjFind identifies the objective function that best aligns with the observed metabolic phenotype. This data-driven approach helps select the most appropriate biological objective for an FBA model under specific conditions, moving beyond generic assumptions like biomass maximization [8].
- ObjFind Framework: A related approach, ObjFind, also infers objective functions by calculating coefficients that represent the relative importance of different reactions. A higher coefficient suggests that a reaction flux is operating near its maximum potential in the experimental data. While powerful, this method can be prone to overfitting to particular conditions if not carefully applied [8].
Table 1: Comparison of Model Selection Frameworks and Their Applications
| Framework/Method | Primary Modeling Context | Core Function | Key Inputs | Key Outputs/Measures |
|---|---|---|---|---|
| χ²-test of Goodness-of-Fit [57] | 13C-MFA | Validates model fit to labeling data | Measured vs. simulated Mass Isotopomer Distributions (MIDs) | Goodness-of-fit statistic (p-value) |
| Combined Validation with Pool Sizes [57] | 13C-MFA | Model validation and selection | Isotopic labeling data, metabolite pool sizes | Improved model discrimination and validation |
| TIObjFind [8] | FBA | Identifies context-specific objective functions | Stoichiometric network, experimental flux data | Coefficients of Importance (CoIs), optimized objective function |
| ObjFind [8] | FBA | Identifies objective function weights | Stoichiometric network, experimental flux data | Reaction weights/coefficients for the objective function |
| Random Forest Regression [77] | Empirical Flux Prediction | Predicts specific flux based on operational data | Historical flux and water quality data | Predicted future flux (R², Mean Square Error) |
Experimental Protocols for Model Validation
Implementing robust experimental protocols is essential for generating the data required for statistically sound model selection.
Protocol for 13C-MFA Model Validation
This protocol outlines the key steps for validating a 13C-Metabolic Flux Analysis model using the χ²-test and additional data.
- Experimental Design and Tracer Selection: Design a parallel labeling experiment. Utilize multiple 13C-labeled tracers (e.g., [1-13C]glucose, [U-13C]glucose) to introduce distinct isotopic patterns that collectively provide more comprehensive information for flux estimation [57].
- Data Acquisition:
- Isotopic Labeling: At metabolic steady state, quench the culture and extract metabolites. Measure the Mass Isotopomer Distribution (MID) of intracellular metabolites using Mass Spectrometry (MS) or NMR [57].
- Metabolite Pool Sizes: Quantify the concentrations of intracellular metabolites. This data is crucial for the advanced validation framework that incorporates pool size information [57].
- External Fluxes: Precisely measure uptake rates of substrates (e.g., glucose) and secretion rates of products (e.g., lactate, CO2) and biomass. These fluxes constrain the solution space of the model [57].
- Computational Fitting and Validation:
- Flux Estimation: Input the measured MIDs and external fluxes into the 13C-MFA software. Use an optimization algorithm to find the flux map that minimizes the difference between simulated and measured MIDs [57].
- Goodness-of-Fit Test: Perform the χ²-test to compare the simulated and experimental MIDs. A statistically non-significant result (typically p > 0.05) suggests the model is an acceptable fit for the data [57].
- Advanced Validation: Incorpo rate the measured metabolite pool sizes into the validation process. Evaluate if the model can simultaneously explain both the isotopic labeling and the metabolite concentrations to strengthen confidence in the selected model [57].
Protocol for FBA Objective Function Identification with TIObjFind
This protocol details the steps for using the TIObjFind framework to identify the most statistically justified objective function for an FBA model.
- Model and Data Preparation:
- Stoichiometric Model: Define a genome-scale stoichiometric model (GSSM) for the organism, including all known metabolic reactions, their stoichiometry, and bounds [57] [8].
- Experimental Flux Data: Acquire a set of measured internal or external fluxes under a specific condition. This data can come from 13C-MFA experiments or literature. This dataset is denoted as ( v^{\text{exp}} ) [8].
- Framework Application:
- Optimization Problem Setup: TIObjFind formulates an optimization problem that minimizes the difference between FBA-predicted fluxes and the experimental data ( v^{\text{exp}} ), while simultaneously maximizing an inferred, weighted-sum objective function [8].
- Mass Flow Graph (MFG) Construction: Map the FBA solutions onto a graph structure that represents the network topology and flux distributions [8].
- Path-Finding Algorithm: Apply a path-finding algorithm on the MFG to analyze the Coefficients of Importance (CoIs) between key start (e.g., substrate uptake) and target (e.g., product secretion) reactions [8].
- Model Selection and Interpretation:
- Coefficient Analysis: Examine the calculated CoIs. Reactions with higher coefficients are interpreted as being more critical to the cellular objective under the tested condition [8].
- Objective Function Selection: Use the pattern of CoIs to define or select a biologically relevant objective function (e.g., a weighted sum of key reaction fluxes) for future FBA simulations under similar conditions. This selected model is considered more statistically justified as its predictions are grounded in experimental flux data [8].
The following workflow diagram illustrates the key steps and decision points in the TIObjFind framework for FBA model selection.
Diagram 1: TIObjFind Model Selection Workflow for FBA.
The Scientist's Toolkit: Key Research Reagents and Materials
Successful execution of the experimental protocols requires specific tools and reagents. The following table details essential items for conducting flux analysis and model selection experiments.
Table 2: Key Research Reagents and Materials for Metabolic Flux Studies
| Item Name | Function/Application |
|---|---|
| 13C-Labeled Tracers (e.g., [1-13C]Glucose, [U-13C]Glucose) [57] | Substrates fed to biological systems to generate unique isotopic labeling patterns in metabolites, which are used for flux estimation in 13C-MFA. |
| Mass Spectrometer (MS) [57] | Analytical instrument used to precisely measure the Mass Isotopomer Distribution (MID) of metabolites from tracer experiments. |
| Stoichiometric Metabolic Model (e.g., from KEGG, EcoCyc) [8] | A computational network model containing all known metabolic reactions for an organism; the foundational structure for FBA and 13C-MFA. |
| Flux Analysis Software (e.g., for 13C-MFA or FBA) [57] | Computational tools that implement algorithms for estimating fluxes from labeling data (13C-MFA) or optimizing fluxes against an objective (FBA). |
| Experimental Flux Dataset [8] | A set of measured internal or external metabolic fluxes, often obtained via 13C-MFA, used as a benchmark for validating or inferring FBA objective functions. |
Selecting the most statistically justified model is a critical step in flux prediction research. While traditional methods like the χ²-test provide a foundational goodness-of-fit measure, they are no longer sufficient in isolation. The emerging generation of model selection practices, such as integrating metabolite pool sizes in 13C-MFA and employing data-driven frameworks like TIObjFind for FBA, represents a significant advancement. These approaches leverage multiple data types and network topology to infer biological objectives and select models that are more deeply grounded in experimental evidence.
For researchers and drug development professionals, adopting these robust validation and selection procedures is paramount. It enhances confidence in model-derived fluxes, which can inform metabolic engineering strategies and the identification of novel drug targets. As the field moves forward, the continued development and application of sophisticated model selection criteria will be essential for achieving a more accurate, predictive understanding of cellular metabolism.
Flux prediction is a cornerstone of systems biology, critical for understanding cellular metabolism and advancing metabolic engineering and drug development. For decades, Flux Balance Analysis (FBA) has been the predominant constraint-based method for predicting metabolic fluxes using genome-scale metabolic models (GEMs). However, traditional FBA faces inherent challenges, including its reliance on predefined biological objective functions and limited capacity to integrate multi-omics data. Recently, Machine Learning (ML) frameworks have emerged as powerful alternatives or complements to FBA. This guide provides an objective comparison of these approaches, evaluating their performance, methodologies, and applicability through experimental data and case studies.
Fundamental Principles and Methodologies
Traditional Flux Balance Analysis
FBA is a constraint-based approach that predicts metabolic flux distributions by assuming organisms operate at metabolic steady-state and optimize a defined cellular objective [65] [78]. The solution space is constrained by the stoichiometric matrix of the metabolic network and bounds on reaction fluxes.
- Core Assumption: Microorganisms maximize an objective function, typically biomass production for microbial systems [78].
- Common Variants: Parsimonious FBA (pFBA) minimizes total flux while achieving optimal objective value [40]; Dynamic FBA (dFBA) incorporates kinetic changes in extracellular metabolites [79]; and regulatory FBA (rFBA) integrates gene regulatory constraints [10].
Machine Learning Frameworks
ML approaches learn the relationship between input features (e.g., omics data, environmental conditions) and metabolic fluxes from experimental data, reducing dependence on prior assumptions about cellular objectives [40] [21].
- Supervised Learning: Directly predicts fluxes from features like transcriptomics or proteomics data [40].
- Topology-Based Models: Utilize graph-theoretic features (e.g., betweenness centrality) from metabolic network structure to predict gene essentiality or fluxes [71].
- Hybrid Neural-Mechanistic Models: Embed mechanistic constraints of FBA within trainable neural network architectures, such as Artificial Metabolic Networks (AMNs) [21].
Comparative Performance Analysis
The table below summarizes quantitative performance comparisons between traditional FBA and various ML frameworks across key predictive tasks, as reported in experimental studies.
Table 1: Quantitative Performance Comparison of FBA vs. Machine Learning Frameworks
| Predictive Task | Organism/System | Traditional FBA Performance | ML Framework Performance | Key Metric | Citation |
|---|---|---|---|---|---|
| Internal/External Flux Prediction | E. coli | Baseline (pFBA) | Smaller prediction errors vs pFBA | Mean Squared Error | [40] |
| Gene Essentiality Prediction | E. coli Core Model | F1-Score: 0.000 | F1-Score: 0.400 (Precision: 0.412, Recall: 0.389) | F1-Score | [71] |
| Quantitative Phenotype Prediction | E. coli, P. putida | Limited quantitative accuracy | Systematic outperformance of constraint-based models | Growth Rate Prediction Accuracy | [21] |
| Growth Rate Prediction in Communities | Human/Mouse Gut Bacteria | Low correlation with in vitro data (semi-curated GEMs) | Improved accuracy with curated models & ML integration | Correlation with Experimental Data | [41] |
Detailed Experimental Protocols and Case Studies
Case Study 1: Omics-Based Flux Prediction inE. coli
- Objective: Compare omics-based supervised ML with traditional pFBA for predicting metabolic fluxes under various conditions [40].
- Protocol:
- Input Data: Transcriptomics and/or proteomics data for E. coli across multiple conditions.
- ML Training: Supervised ML models were trained to map omics data to experimentally determined or FBA-simulated flux distributions.
- Benchmarking: ML predictions and pFBA predictions were compared against a reference set of fluxes.
- Outcome: The omics-based ML approach demonstrated a consistent advantage, achieving smaller prediction errors for both internal and external metabolic fluxes compared to the pFBA baseline [40].
Case Study 2: Topology-Based ML for Gene Essentiality
- Objective: Predict essential metabolic genes using network topology, benchmarking against FBA single-gene deletion analysis [71].
- Protocol:
- Feature Engineering: A reaction-reaction graph was constructed from the ecolicore model. Graph-theoretic features (e.g., betweenness centrality, PageRank) were computed for each gene.
- Model Training: A
RandomForestClassifierwas trained on these topological features using a curated ground-truth dataset of essential genes. - Validation: Model performance (Precision, Recall, F1-Score) was rigorously evaluated against a standard FBA deletion analysis.
- Outcome: The topology-based ML model achieved an F1-Score of 0.400, decisively outperforming the standard FBA approach, which failed to identify any known essential genes (F1-Score: 0.000) [71].
Case Study 3: Neural-Mechanistic Hybrid Modeling
- Objective: Improve quantitative growth rate and phenotype predictions by embedding FBA constraints into a neural network [21].
- Protocol:
- Model Architecture: A hybrid AMN model was developed, featuring a trainable neural layer that processes inputs (e.g., medium composition) followed by a mechanistic layer that enforces FBA-derived steady-state constraints.
- Training: The model was trained on sets of flux distributions from FBA simulations or experimental data.
- Testing: The trained model was used to predict phenotypes of E. coli and Pseudomonas putida in different media and with gene knock-outs.
- Outcome: The hybrid models systematically outperformed traditional constraint-based models and required training data orders of magnitude smaller than classical ML methods [21].
Workflow and Pathway Visualization
The following diagrams illustrate the fundamental workflows of the traditional FBA and a generalized ML-based framework for flux prediction.
Traditional FBA Workflow
Diagram 1: Traditional FBA relies on a GEM, constraints, and a pre-defined objective function to compute an optimal flux distribution via linear programming.
Machine Learning Framework for Flux Prediction
Diagram 2: ML frameworks learn a mapping from input features to flux distributions using experimental data for training, avoiding the need for an explicit objective function.
The Scientist's Toolkit: Essential Research Reagents and Tools
The table below details key software, databases, and computational tools essential for conducting research in metabolic flux prediction.
Table 2: Key Research Reagent Solutions for Flux Prediction Research
| Tool/Resource Name | Type | Primary Function | Relevance |
|---|---|---|---|
| COBRA Toolbox / cobrapy | Software Toolbox | Perform FBA and related constraint-based analyses [21] [65]. | Standard ecosystem for building, simulating, and analyzing GEMs. |
| AGORA | Model Database | Repository of semi-curated GEMs for gut bacteria [41]. | Provides starting point for modeling microbial communities; quality varies. |
| MEMOTE | Quality Control Tool | Suite for testing and ensuring GEM quality and consistency [65] [41]. | Essential for validating model functionality before FBA/ML use. |
| TIObjFind | Framework | Infers metabolic objective functions from data using topology-informed optimization [10]. | Enhances FBA interpretability and alignment with experimental fluxes. |
| COMETS | Simulation Tool | Performs dynamic FBA simulations in spatial and temporal contexts [78] [41]. | Models complex community dynamics and batch processes. |
| MICOM | Software Tool | Models microbial communities using FBA with abundance constraints [41]. | Predicts growth and interactions in multi-species consortia. |
| Artificial Metabolic Networks (AMNs) | Hybrid Model | Embeds FBA constraints within neural networks for phenotype prediction [21]. | Exemplifies the neural-mechanistic hybrid approach. |
The comparative analysis reveals a nuanced landscape. Traditional FBA remains a powerful, knowledge-driven tool for exploring metabolic capabilities and generating testable hypotheses, especially when a relevant objective function is known. Its main strengths are interpretability and a strong foundation in biochemical networks.
However, evidence shows that ML frameworks can achieve superior predictive accuracy in specific tasks, such as quantitative flux prediction [40] and gene essentiality identification [71]. Their key advantage is the ability to learn complex, condition-specific relationships from high-dimensional data without relying on a pre-defined objective function.
The emerging paradigm of hybrid modeling, which embeds mechanistic constraints into ML architectures [21], is particularly promising. This approach leverages the predictive power of ML while adhering to biochemical laws, resulting in models that are both accurate and physiologically plausible. For researchers, the choice between FBA and ML is not binary but strategic. FBA is ideal for foundational network analysis, while ML and hybrid models offer enhanced precision for quantitative phenotype prediction, especially when integrating multi-omics data or tackling problems where cellular objectives are unclear.
Conclusion
The comparative analysis confirms that no single, consensus objective function exists for all flux prediction scenarios. The choice is highly condition-dependent and must be carefully validated against experimental data. While traditional FBA with objectives like maximal biomass production remains a cornerstone, parsimonious solutions and multi-objective optimizations often yield more realistic predictions for complex phenotypes like ageing. The emergence of machine learning frameworks, such as ML-Flux, represents a paradigm shift, offering superior computational speed and accuracy by directly mapping isotope patterns to fluxes. For future research, the integration of these data-driven methods with robust biochemical networks promises to democratize quantitative metabolic profiling. This will significantly accelerate therapeutic development and synthetic biology by providing a more dynamic and reliable readout of cellular states in health and disease.
References
- 1. Introduction to flux balance analysis (FBA) [https://www.protocols.io/view/introduction-to-flux-balance-analysis-fba-daz22f8e.html]
- 2. What is flux balance analysis? - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3108565/]
- 3. The choice of the objective function in flux balance analysis is ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9560524/]
- 4. Comparison and analysis of objective functions in flux ... [https://pubmed.ncbi.nlm.nih.gov/25044958/]
- 5. The Biomass Objective Function - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC2912156/]
- 6. Enhanced flux prediction by integrating relative expression ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6532942/]
- 7. Metabolic Objectives and Trade-Offs: Inference and Applications [https://pmc.ncbi.nlm.nih.gov/articles/PMC11857637/]
- 8. Optimization-based framework with flux balance analysis (FBA ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1013635]
- 9. Introduction to BioCyc and Pathway Tools [https://biocyc.org/intro.shtml]
- 10. Optimization-based framework with flux balance analysis (FBA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12578352/]
- 11. Accurate prediction of gene deletion phenotypes with Flux ... [https://www.nature.com/articles/s41467-025-63436-9]
- 12. An evaluation of machine-learning for predicting phenotype [https://pmc.ncbi.nlm.nih.gov/articles/PMC7048706/]
- 13. Benchmarking machine learning and parametric methods ... [https://www.nature.com/articles/s41598-024-57234-4]
- 14. Flux sampling is a powerful tool to study metabolism under ... [https://www.nature.com/articles/s41540-019-0109-0]
- 15. Current state and challenges for dynamic metabolic modeling [https://www.sciencedirect.com/science/article/pii/S1369527416300960]
- 16. Enhanced flux potential analysis links changes in enzyme ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11965317/]
- 17. The choice of the objective function in flux balance analysis is ... [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0276112]
- 18. Clostridium butyricum maximizes growth while minimizing enzyme usage and ATP production: metabolic flux distribution of a strain cultured in glycerol | BMC Systems Biology | Full Text [https://bmcsystbiol.biomedcentral.com/articles/10.1186/s12918-017-0434-0]
- 19. An unconventional uptake rate objective function approach enhances applicability of genome-scale models for mammalian cells | npj Systems Biology and Applications [https://www.nature.com/articles/s41540-019-0103-6]
- 20. ThermOptCobra: Thermodynamically optimal construction and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12329557/]
- 21. A neural-mechanistic hybrid approach improving the ... [https://www.nature.com/articles/s41467-023-40380-0]
- 22. Multi-objective optimization model and algorithm ... [https://www.sciencedirect.com/science/article/pii/S2666519025000676]
- 23. Multi-Objective Drug Molecule Optimization Based on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12389091/]
- 24. Multi-and many-objective optimization: present and future ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2023.1288626/full]
- 25. Optimization-based framework with flux balance analysis ... [https://pubmed.ncbi.nlm.nih.gov/41144574/]
- 26. Deep learning with improved hybrid neuro-turning for ... [https://www.sciencedirect.com/science/article/abs/pii/S2214714424010675]
- 27. An Artificial Neural Network (ANN) Model to Predict Critical ... [https://www.sciencedirect.com/science/article/pii/S0029549324008367]
- 28. ReactorNet based on machine learning framework to ... [https://www.nature.com/articles/s41598-025-13794-7]
- 29. Accurate and rapid determination of metabolic flux by deep ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10659294/]
- 30. Comparison of Standalone and Hybrid Machine Learning ... [https://www.mdpi.com/1996-1073/16/7/3182]
- 31. Optimization-based framework with flux balance analysis ... [https://journals.plos.org/ploscompbiol/article/figures?id=10.1371/journal.pcbi.1013635]
- 32. Comparative Analysis of Machine Learning Models for ... [https://www.mdpi.com/2076-3417/15/7/3636]
- 33. Overexpression of Ssd1 and calorie restriction extend ... [https://elifesciences.org/reviewed-preprints/108892v2]
- 34. Sis2 regulates yeast replicative lifespan in a dose- ... [https://www.nature.com/articles/s41467-023-43233-y]
- 35. Flux-P: Automating Metabolic Flux Analysis - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC3901227/]
- 36. Metabolic flux analysis: a comprehensive review on sample ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9449821/]
- 37. SIMPEL: using stable isotopes to elucidate dynamics of ... [https://www.nature.com/articles/s42003-024-05844-z]
- 38. Systematic evaluation of objective functions for predicting ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1949037/]
- 39. NEXT-FBA: A hybrid stoichiometric/data-driven approach to ... [https://www.sciencedirect.com/science/article/pii/S1096717625000461]
- 40. Predicting metabolic fluxes from omics data via machine ... [https://www.sciencedirect.com/science/article/pii/S2001037023003586]
- 41. Predicting microbial interactions with approaches based on ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-024-05651-7]
- 42. Flux sampling and context-specific genome-scale ... [https://www.sciencedirect.com/science/article/abs/pii/S0167779925002719]
- 43. Predicting metabolic fluxes from omics data via machine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10590844/]
- 44. Flux Balance Analysis | Virginia - iGEM 2025 [https://2025.igem.wiki/virginia/fba]
- 45. MINN: A metabolic-informed neural network for integrating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12359237/]
- 46. Algorithms and tools for data-driven omics integration to ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06446-x]
- 47. Lexicographic Optimization Strategy [https://www.emergentmind.com/topics/lexicographic-optimization-strategy]
- 48. A two-stage Multi-Criteria Optimization method for service ... [https://www.sciencedirect.com/science/article/abs/pii/S0167739X21000935]
- 49. An Efficient Linearized Optimization Framework for ... [https://educationaldatamining.org/EDM2025/proceedings/2025.EDM.long-papers.31/index.html]
- 50. A column generation approach for the lexicographic ... [https://link.springer.com/article/10.1007/s00291-023-00741-z]
- 51. [1805.03437] Exact Lexicographic Scheduling and ... [https://arxiv.org/abs/1805.03437]
- 52. An improved algorithm for flux variability analysis [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-05089-9]
- 53. 13CFLUX - Third-generation high-performance engine for ... [https://arxiv.org/html/2509.23847]
- 54. Rethinking 13C-metabolic flux analysis – The Bayesian ... [https://www.sciencedirect.com/science/article/pii/S1096717624000508]
- 55. Quantum Algorithm Targets Metabolic Networks in Step ... [https://thequantuminsider.com/2025/11/25/quantum-algorithm-targets-metabolic-networks-in-step-toward-biological-modeling/]
- 56. Flux-sum coupling analysis of metabolic network models [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1012972]
- 57. Model Validation and Selection in Metabolic Flux Analysis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10922127/]
- 58. Model Validation and Selection in Metabolic Flux Analysis ... [https://arxiv.org/abs/2303.12651]
- 59. CFSA: Comparative flux sampling analysis as a guide for ... [https://www.sciencedirect.com/science/article/pii/S2214030124000130]
- 60. Goodness of Fit: Definition & Tests [https://statisticsbyjim.com/basics/goodness-of-fit/]
- 61. Goodness of fit [https://en.wikipedia.org/wiki/Goodness_of_fit]
- 62. A guide to 13C metabolic flux analysis for the cancer biologist [https://www.nature.com/articles/s12276-018-0060-y]
- 63. Synergy between 13 C-metabolic flux analysis and ... [https://www.sciencedirect.com/science/article/abs/pii/S1096717610001023]
- 64. 13C metabolic flux analysis - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC9493264/]
- 65. Model Validation and Selection in Metabolic Flux Analysis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10055486/]
- 66. Publishing 13C metabolic flux analysis studies: A review and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5866723/]
- 67. p13CMFA: Parsimonious 13C metabolic flux analysis [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1007310]
- 68. BayFlux: A Bayesian method to quantify metabolic Fluxes and their... [https://pubmed.ncbi.nlm.nih.gov/37948450/]
- 69. E- Flux 2 and SPOT: Validated Methods for Inferring... | PLOS One [https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0157101]
- 70. Validation-based model selection for 13C metabolic flux analysis with... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9022838/]
- 71. A Topology-Based Machine Learning Model Decisively ... [https://arxiv.org/abs/2507.20406]
- 72. Machine Learning-Based Prediction of Ecosystem-Scale ... [https://www.mdpi.com/2073-445X/14/1/124]
- 73. Prediction of permeate flux of ceramic membrane-based ... [https://www.sciencedirect.com/science/article/pii/S0011916425010665]
- 74. Advanced machine learning schemes for prediction CO2 ... [https://www.sciencedirect.com/science/article/pii/S2090123224004880]
- 75. Predicting and forecasting reactivity and flux using long ... [https://arxiv.org/html/2511.05118v1]
- 76. Global carbon flux dataset generated by fusing remote ... [https://www.nature.com/articles/s41597-025-05672-8]
- 77. A Random Forest Regression Approach for Prediction of Specific Flux for Ultrafiltration Membranes [https://scholars.unh.edu/thesis/1981/]
- 78. Advances in flux balance analysis by integrating machine ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8382995/]
- 79. Modeling host–pathway dynamics at the genome scale ... [https://www.sciencedirect.com/science/article/pii/S1096717625000849]