Engineering Yeast Cell Factories: A Comprehensive Guide to Heterologous Pathway Expression for Drug Development
This article provides researchers, scientists, and drug development professionals with a systematic framework for harnessing yeast cell factories for heterologous pathway expression.
Engineering Yeast Cell Factories: A Comprehensive Guide to Heterologous Pathway Expression for Drug Development
Abstract
This article provides researchers, scientists, and drug development professionals with a systematic framework for harnessing yeast cell factories for heterologous pathway expression. It explores the foundational principles of yeast host systems, details advanced methodological tools for pathway engineering, addresses common troubleshooting and optimization challenges, and offers validation strategies for comparative analysis. By integrating the latest research on promoter engineering, cellular lifespan extension, and multi-omics validation, this guide serves as a critical resource for advancing the production of therapeutic compounds, including complex plant-derived natural products and membrane proteins, in yeast-based platforms.
Yeast Host Systems and Expression Fundamentals: Building Your Genetic Toolbox
Comparative Advantages of Yeast Systems for Eukaryotic Protein Production
The development of efficient and robust systems for eukaryotic protein production is a cornerstone of modern biotechnology, supporting the manufacture of biologics, industrial enzymes, and research reagents. Among available platforms, yeast-based expression systems offer a powerful combination of eukaryotic processing capabilities, rapid growth, and scalability. This application note delineates the comparative advantages of major yeast expression systems within the context of heterologous pathway expression in yeast cell factories. We provide a structured quantitative comparison, detailed experimental methodologies for core protocols, and visualization of critical pathways to guide researchers in selecting and implementing the optimal yeast platform for their specific protein production needs.
Comparative Analysis of Major Yeast Expression Systems
The selection of an appropriate yeast host is critical for the successful production of a recombinant eukaryotic protein. The most commonly utilized systems each possess distinct profiles of advantages and limitations, making them suited to different applications.
Table 1: Key Characteristics of Major Yeast Production Systems
| Feature | Saccharomyces cerevisiae | Komagataella phaffii | Ogataea polymorpha |
|---|---|---|---|
| Typical Application | Traditional cell factory; well-studied model organism [1] | High-yield production of biopharmaceuticals and industrial enzymes [2] | Production of therapeutics and antivirals; thermotolerant processes [2] |
| Key Strength | Extensive genetic tools; GRAS status; rapid growth [3] [1] | Very high cell-density fermentation; strong, regulated promoters; superior secretion [2] | Thermotolerance (30-50°C); low hyper-mannosylation; multi-carbon source use [2] |
| Promoter Example | GAL1, GAL10 [3] | Methanol-inducible AOX1 [3] [2] | Methanol-inducible promoters [2] |
| Post-Translational Modification | Hyper-mannosylation of N-glycans can occur | Capable of human-like glycosylation patterns [4] | Human-like glycoproteins; relatively low hyper-mannosylation [2] |
| Genetic Engineering | Highly advanced (CRISPR, synthetic biology) [1] [5] | Advanced (CRISPR/Cas9 available) [2] | Well-developed genetic tools [2] |
| Regulatory Status | GRAS (Generally Recognized as Safe) [3] | GRAS status for specific products [2] | GRAS status [2] |
Table 2: Quantitative Performance Metrics for Yeast Systems
| Metric | S. cerevisiae | K. phaffii | O. polymorpha |
|---|---|---|---|
| Growth Temperature Range | 28-30°C (typical) | 28-30°C | 30-50°C [2] |
| Fermentation Cell Density | High | Very High [2] | High [2] |
| Representative Protein Yield | Moderate | High (e.g., >g/L scale reported) [2] | High [2] |
| Market Impact (Cell Wall Extract Market 2021) | ~$2.09 Billion (as part of global market) [6] | Significant (as a key contributor) | Growing |
The data above demonstrate that while S. cerevisiae remains a versatile and genetically tractable workhorse, methylotrophic yeasts like K. phaffii and O. polymorpha offer distinct advantages for demanding industrial applications, particularly where high cell density, strict promoter regulation, or thermotolerance are required.
Essential Experimental Protocols
Basic Protocol 1: Target Optimization and Construct Design
The initial bioinformatic analysis of the target protein is crucial for maximizing the likelihood of high expression and solubility [7]. This protocol outlines a computational workflow for target optimization prior to gene synthesis.
Materials & Reagents
- Hardware: Computer with internet access.
- Software: NCBI BLAST, ColabFold (AlphaFold2 server), XtalPred.
- Files: Target protein sequence(s) in FASTA format.
Procedure
- pBLAST against PDB Database: Navigate to the NCBI BLAST website. Select "Protein BLAST" and input your target protein sequence in FASTA format. Set the database to "Protein Data Bank proteins (pdb)" and select the "PSI-BLAST" algorithm. Run the search using default parameters. Identify homologous structures with ≥40% sequence identity and 75-80% query coverage. Use these alignments to inform the design of your expression construct, prioritizing domains from homologs with high query coverage [7].
- Modeling with AlphaFold: For targets lacking close PDB homologs, use the ColabFold: AlphaFold2 server. Input the target sequence and run the analysis with default parameters. The tool will generate five models, with each residue colored by its predicted Local Distance Difference Test (pLDDT) score. A high pLDDT score indicates confidence in the local structure prediction. Use this model to identify and potentially exclude poorly structured, disordered regions from your final construct to enhance the probability of soluble expression [7].
- Codon Optimization: Upon finalizing the target protein sequence, utilize commercial gene synthesis services that offer host-specific codon optimization (e.g., for K. phaffii or S. cerevisiae). This step is critical for maximizing translational efficiency and protein yield [7].
Basic Protocol 2: High-Throughput Transformation and Screening
This protocol describes a high-throughput (HTP) method for transforming and screening multiple expression constructs in a 96-well format, adapted from established pipelines [7].
Materials & Reagents
- Expression Vectors: Codon-optimized, synthetically derived genes cloned into an appropriate yeast expression vector (e.g., pMCSG53 for E. coli, or a methanol-inducible vector for K. phaffii).
- Yeast Strains: Chemically competent cells of your chosen yeast system (e.g., S. cerevisiae, K. phaffii).
- Media: Appropriate selective medium (e.g., SD -Ura for S. cerevisiae).
- Equipment: 96-well deep-well plates, microplate shaker incubator, liquid handling robot (optional, for automation).
Procedure
- Transformation: In a 96-well plate, transform the expression constructs into your competent yeast strain using a high-efficiency protocol (e.g., lithium acetate method for S. cerevisiae). Plate the transformation mixtures onto solid selective medium and incubate until colonies form [7].
- Inoculation and Growth: Pick individual colonies into a 96-deep-well plate containing liquid selective medium. Seal the plate with a breathable seal and incubate with shaking (e.g., 220 rpm) at the standard growth temperature (e.g., 30°C) until the cultures reach mid-log phase.
- Protein Expression Induction: For inducible systems like AOX1 in K. phaffii, add the inducer (e.g., methanol to a final concentration of 0.5-1%) to the culture. For S. cerevisiae GAL promoters, induce by adding galactose. Continue incubation with shaking for a defined period (e.g., 24-48 hours) [7].
- Solubility Screening: Harvest cells by centrifugation. Lyse the cell pellets using chemical lysis or enzymatic digestion (e.g., lyticase). Centrifuge the lysates to separate soluble and insoluble fractions. Analyze the soluble fraction for the presence of the target protein, typically via SDS-PAGE or western blot [7].
Basic Protocol 3: Engineering Robust Yeast Cell Factories
Enhancing the cellular robustness of yeast can significantly extend its operational lifespan and improve recombinant protein yield under industrial fermentation conditions [5]. This protocol outlines a genetic strategy to improve chronological lifespan.
Materials & Reagents
- Strains & Plasmids: S. cerevisiae background strain, CRISPR/Cas9 plasmid system, donor DNA for gene editing.
- Media: YPD medium, synthetic dropout media as needed.
- Molecular Biology Reagents: DNA polymerases, restriction enzymes, cloning kits, primers.
Procedure
- Genetic Modifications: To enhance robustness, engineer the yeast chassis by:
- Downregulating TOR1: Use CRISPR/Cas9 to replace the native promoter of the TOR1 gene with a weaker promoter to downregulate its expression.
- Deleting HDA1: Design a gRNA and a donor DNA template to completely knock out the histone deacetylase gene HDA1 [5].
- Strain Validation: Transform the genetic constructs into the host yeast strain. Select positive clones and verify the genetic modifications via PCR and DNA sequencing.
- Phenotypic Analysis:
- Chronological Lifespan (CLS) Assay: Measure the viability of the engineered strains over time in stationary phase culture compared to the wild-type control. The robust strains should show a significantly extended CLS [5].
- Production Assay: Cultivate the validated strains in shake-flask batch fermentations and measure the titer of the target product (e.g., fatty alcohols, a recombinant protein). Successful engineering should result in a substantial increase in yield (e.g., up to 56% for fatty alcohols) [5].
Signaling Pathways and Workflows
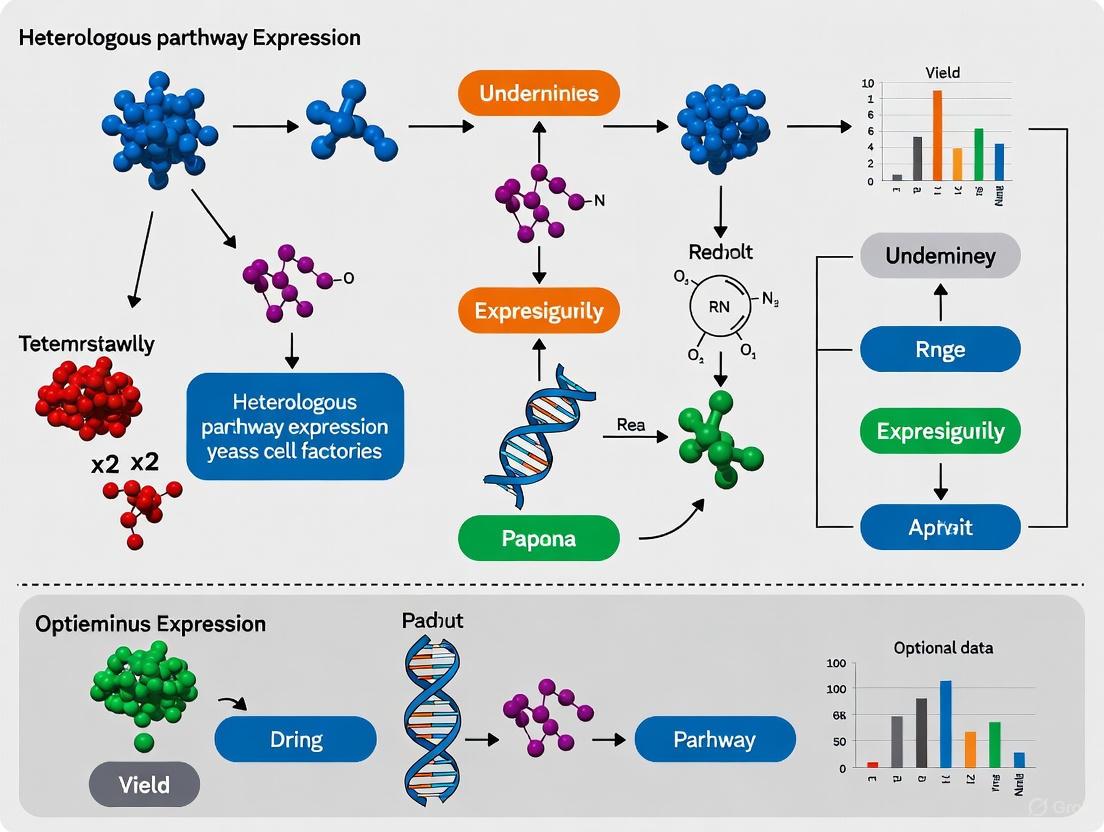
Methanol Induction Pathway in Methylotrophic Yeast
The high productivity of methylotrophic yeast systems is driven by the tightly regulated methanol utilization pathway. The following diagram illustrates the core logic of gene induction, centered on the AOX1 promoter, which is a key tool for recombinant protein production.
High-Throughput Protein Screening Pipeline
The integration of bioinformatic design with high-throughput experimental screening creates an efficient pipeline for identifying successful expression constructs, as summarized in the workflow below.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents and Tools for Yeast Protein Production
| Item | Function/Application | Example Use-Case |
|---|---|---|
| Methanol-Inducible Promoters (e.g., AOX1) | Strong, tightly regulated promoter for high-level expression in methylotrophic yeasts [2]. | Inducing recombinant protein expression in Komagataella phaffii by adding methanol to the culture medium [2]. |
| CRISPR/Cas9 Systems | Precision genome editing for strain engineering (e.g., gene knock-outs, promoter swaps) [2] [5]. | Knocking out the HDA1 gene in S. cerevisiae to enhance cellular robustness and extend chronological lifespan [5]. |
| Bioinformatics Software (BLAST, ColabFold) | Computational analysis of target proteins to predict structure, identify domains, and optimize construct design [7]. | Identifying and truncating intrinsically disordered regions of a target protein to improve its solubility during recombinant expression [7]. |
| High-Throughput Screening Systems | Automated liquid handling and analysis for parallel testing of many constructs or conditions in microplates [7]. | Rapidly screening hundreds of different protein constructs for solubility and expression level in a 96-well format [7]. |
| Synthetic DNA & Cloning Services | Provides codon-optimized, sequence-verified genes cloned into expression vectors, accelerating the construct generation process [7]. | Sourcing a library of codon-optimized genes for a structural genomics project, delivered pre-cloned in an expression vector [7]. |
The selection of an appropriate microbial host is a foundational step in the construction of efficient yeast cell factories for heterologous pathway expression. This decision critically influences the yield, functionality, and scalability of target recombinant proteins and metabolites. While prokaryotic systems like E. coli offer simplicity and high growth rates, they often lack the eukaryotic machinery necessary for proper protein folding, complex post-translational modifications, and secretion of sophisticated biologics [8]. Among eukaryotic hosts, yeast systems strike a balance between the cellular complexity of mammalian cells and the ease of use of prokaryotes. For researchers and drug development professionals, the choice frequently narrows to two principal workhorses: the conventional, well-characterized Saccharomyces cerevisiae and the robust, high-yield Komagataella phaffii (formerly Pichia pastoris) [9] [10]. This application note provides a structured comparison of these systems and outlines detailed protocols to guide host selection and engineering within the context of advanced heterologous pathway research.
Host System Comparison:S. cerevisiaevs.K. phaffii
The optimal host varies significantly with the specific application. The following tables summarize the core characteristics, advantages, and challenges of each system to inform the selection process.
Table 1: Fundamental Characteristics of Yeast Expression Systems
| Characteristic | Saccharomyces cerevisiae | Komagataella phaffii |
|---|---|---|
| Doubling Time | ~90 minutes [8] | 60–120 minutes [8] |
| Genetic Tools | Extensive, well-developed [1] [11] | Advanced, rapidly expanding [12] [13] |
| Secretory Capacity | High [10] | Very High; limited endogenous secretion simplifies purification [8] [9] |
| Post-Translational Modifications | N- & O-hyperglycosylation [8] | Human-like glycosylation (shorter high-mannose chains) [8] [10] |
| Typical Glycosylation Pattern | Hypermannosylation (50-150 mannose residues) [8] | Shorter high-mannose chains (~20 mannose residues) [10] |
| Inducible Promoter Example | Galactose-inducible (GAL1, GAL10) | Methanol-inducible (PAOX1) [8] [12] |
| Constitutive Promoter Example | Phosphoglycerate kinase (PGK1) | Glyceraldehyde-3-phosphate dehydrogenase (PGAP) [12] |
| Carbon Source | Glucose, Sucrose, Galactose [14] | Glycerol, Methanol, Glucose [8] [12] |
Table 2: Applied Considerations for Host Selection
| Parameter | Saccharomyces cerevisiae | Komagataella phaffii |
|---|---|---|
| Key Advantages | • GRAS status• Extensive synthetic biology toolkit [1] [11]• Fast growth• Known physiology | • Extremely high cell densities in bioreactors• Strong, tightly regulated promoters [8] [12]• Low secretion of endogenous proteins [8]• Suitable for membrane protein production [8] |
| Common Challenges | • Hyperglycosylation which can be immunogenic [8]• Metabolic burden at scale | • Methanol metabolism requires specific safety protocols• Codon bias may require optimization [9]• Process optimization is often protein-specific [8] |
| Ideal Applications | • Production of ethanol, flavors, and metabolites [14] [5]• Expression of intracellular proteins• Pathway prototyping and screening | • Production of subunit vaccines and therapeutic proteins [8] [9]• High-level secretory production of enzymes [12] [9]• Expression of complex proteins requiring minimal glycosylation |
Experimental Protocols for Host Engineering and Analysis
Protocol: Engineering aK. phaffiiMutS Strain for Enhanced Protein Secretion
Background: The K. phaffii MutS (Methanol utilization slow) phenotype, resulting from the disruption of the AOX1 gene, slows methanol metabolism. This can be beneficial for certain proteins by reducing metabolic burden and heat generation during induction, potentially leading to higher yields of functional protein [8] [12]. This protocol leverages CRISPR-Cas9 for precise genome editing.
Materials:
- K. phaffii host strain (e.g., GS115, X-33)
- CRISPR-Cas9 plasmid system for K. phaffii
- Donor DNA fragment for gene knockout (designed below)
- Solutions for yeast transformation (e.g., lithium acetate/PEG)
- YPD media, Minimal Dextrose (MD) plates, Minimal Methanol (MM) plates
- Synthetic oligonucleotides for PCR and sequencing
Procedure:
- gRNA Design and Plasmid Construction: Design a gRNA with a 20-nucleotide sequence targeting the AOX1 gene coding region. Clone this gRNA expression cassette into a K. phaffii-specific CRISPR-Cas9 plasmid [12].
- Donor DNA Preparation: Design a linear donor DNA fragment containing a selectable marker (e.g., ARG4) flanked by ~500 bp homology arms upstream and downstream of the AOX1 start and stop codons, respectively. Amplify this fragment via PCR.
- Transformation: Co-transform the K. phaffii host strain with the CRISPR-Cas9 plasmid and the linear donor DNA fragment using a standard lithium acetate method.
- Selection and Screening: Plate transformed cells onto MD plates without the appropriate amino acid to select for transformants that have integrated the marker. Isolate individual colonies.
- Phenotype Confirmation (MutS): Streak potential mutants on MM plates. A MutS strain will show significantly slower growth compared to a wild-type (Mut+) control strain over 2-3 days.
- Genotypic Verification: Verify the AOX1 gene replacement by colony PCR using primers flanking the integration site and subsequent DNA sequencing.
Protocol: Analyzing Secretion Efficiency in Yeast Using a β-Glucosidase Reporter
Background: Quantifying secretion efficiency is vital for host screening and engineering. This protocol uses a secreted β-glucosidase (BGL) as a reporter, adaptable for both S. cerevisiae and K. phaffii [15].
Materials:
- Yeast strain with integrated gene for secreted BGL (e.g., with S. cerevisiae α-mating factor signal peptide)
- Deep-well plates or shake flasks
- Appropriate induction media
- Substrate: p-Nitrophenyl β-D-glucopyranoside (pNPG) in buffer
- Stop solution: 1 M Na2CO3
- Microplate reader
Procedure:
- Culture and Induction: Inoculate strains in deep-well plates containing selective media. Grow to mid-log phase, then induce expression (e.g., with methanol for K. phaffii PAOX1, or galactose for S. cerevisiae GAL promoters).
- Sample Collection: At regular intervals post-induction (e.g., 24, 48, 72 hours), centrifuge culture aliquots to separate cells from supernatant.
- Secreted Enzyme Assay: Incubate the cell-free supernatant with pNPG substrate. The enzymatic reaction releases yellow p-nitrophenol.
- Reaction Termination and Quantification: After a fixed time, add stop solution (Na2CO3) and measure the absorbance at 405 nm using a microplate reader.
- Data Analysis: Calculate BGL activity from a p-nitrophenol standard curve. Normalize activity to cell density (OD600) or total cell protein to determine specific secretion efficiency.
Workflow Visualization
The following diagram illustrates the logical workflow for selecting and engineering a yeast host for a heterologous protein production project, integrating the protocols described above.
The Scientist's Toolkit: Key Research Reagents
Success in yeast metabolic engineering relies on a suite of specialized reagents and genetic tools. The following table details essential components for constructing and analyzing yeast cell factories.
Table 3: Essential Reagents for Yeast Cell Factory Engineering
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| CRISPR-Cas9 System | Enables precise gene knockouts, integrations, and edits [5]. | Disrupting HOC1 in K. phaffii to increase cell wall permeability and protein secretion [15]. |
| AOX1 Promoter (PAOX1) | Strong, methanol-inducible promoter in K. phaffii for tight regulation and high expression levels [8] [12]. | Controlling the expression of a therapeutic antibody fragment, inducing with methanol in a bioreactor. |
| S. cerevisiae GAL Promoters | Strong, tightly glucose-repressed and galactose-induced promoters for controlled expression [11]. | Regulating a heterologous metabolic pathway to prevent burden during the growth phase. |
| α-Mating Factor (MFα) Signal Peptide | Directs the secretion of recombinant proteins into the culture medium in both S. cerevisiae and K. phaffii [10] [15]. | Secretion of β-glucosidase (BGL) reporter enzyme for quantifying secretion efficiency. |
| 2µ Plasmid Origin | High-copy-number origin for episomal plasmid maintenance in S. cerevisiae [14]. | Rapid pathway prototyping and screening of enzyme variant libraries. |
| Zeocin / Antibiotic Markers | Selective agents for maintaining plasmids and selecting for transformants in various yeast strains [12]. | Maintaining expression vectors in K. phaffii strains like X-33 and SMD1168H. |
The strategic choice between S. cerevisiae and K. phaffii is not a matter of overall superiority, but of aligning host biology with project objectives. S. cerevisiae remains an unparalleled platform for fundamental research, pathway prototyping, and the production of a wide range of metabolites and intracellular proteins, thanks to its extensive genetic toolbox and well-understood physiology [1] [11]. In contrast, K. phaffii excels in high-density fermentations and the secretory production of complex proteins where its strong, regulated promoters, high secretion capacity, and more human-like glycosylation are decisive advantages [8] [12] [9]. The ongoing development of synthetic biology tools is progressively blurring the historical limitations of each system, enabling researchers to custom-engineer both conventional and non-conventional yeasts into highly efficient, task-specific cell factories for the next generation of biologics and bio-based chemicals [13].
The engineering of yeast cell factories for heterologous pathway expression represents a cornerstone of modern industrial biotechnology, enabling the production of high-value therapeutics, enzymes, and biofuels. The efficiency of these cell factories is fundamentally governed by the precise regulation of key genetic elements: promoters, terminators, and selection markers. These components collectively control transcriptional initiation, transcriptional termination, and the selective pressure necessary for strain construction [16]. Recent advances in synthetic biology, CRISPR/Cas9 genome editing, and artificial intelligence (AI)-assisted design have dramatically accelerated the optimization of these elements, leading to substantial improvements in protein folding, pathway flux, and最终产量 [17] [18] [16]. This Application Note details the properties, quantitative performance, and practical protocols for implementing these genetic elements within heterologous expression systems in yeast, providing a structured framework for researchers and drug development professionals.
Quantitative Comparison of Key Genetic Elements
Promoter Performance in Yeast Systems
Table 1: Characteristics and performance of common and engineered promoters in yeast.
| Host Organism | Promoter | Type | Strength/Performance | Application Context |
|---|---|---|---|---|
| Saccharomyces cerevisiae | TDH3P | Constitutive | One of the highest-performing native promoters [19] | Enhanced heterologous xylanase activity; cultivation on glucose and xylose [19] |
| Saccharomyces cerevisiae | SED1P | Constitutive | Moderate to high expression under stress [19] | Effective for heterologous cellulase and xylanase expression [19] |
| Komagataella phaffii | AOX1 | Inducible (Methanol) | Very strong induction | Common in classical integrative systems [16] |
| Komagataella phaffii | GAP | Constitutive | Strong | High-level expression without methanol induction [16] |
| Aspergillus niger | AAmy | Constitutive | High | Used in a modular platform for expressing diverse proteins [17] |
| Engineered (AI-Designed) | DOSDiff | Constitutive | Up to 1.70-fold expression gain [18] | Demonstrated cross-species functionality in P. pastoris [18] |
Terminator and Selection Marker Efficiency
Table 2: Functional characteristics of terminators and selection markers.
| Element Type | Specific Name | Host | Key Feature/Function | Experimental Note |
|---|---|---|---|---|
| Terminator | DIT1T | S. cerevisiae | Outperformed benchmark ENO1P/T terminator [19] | Used effectively in combination with SED1P and TDH3P [19] |
| Terminator | AnGlaA | A. niger | Native terminator used in a heterologous expression platform [17] | Part of a cassette with the AAmy promoter for high-yield production [17] |
| Selection Marker | Geneticin (G418) | K. phaffii | Antibiotic resistance marker | Used for selection in CRISPR/Cas9 systems with episomal Cas9/sgRNA plasmids [20] |
| Selection Marker | Kl.URA3 | K. phaffii | Auxotrophic marker | Enables selection in chaperone library and mating protocols [21] |
| Selection Marker | kanMX (GAL10) | S. cerevisiae | Antibiotic resistance with inducible promoter | Allows selection on G418; induced by galactose in query strains [21] |
Experimental Protocols
Protocol 1: CRISPR/Cas9-Mediated Markerless Integration inKomagataella phaffii
This protocol facilitates the seamless integration of expression cassettes into specific genomic loci of K. phaffii without the need for antibiotic resistance markers, ideal for food-grade protein production [20].
Materials and Reagents:
- Cas9/sgRNA plasmid (e.g., CRISPi04576, CRISPiPFK1, CRISPi_ROX1)
- Donor helper plasmid (e.g., crBB3_14 for one transcription unit)
- K. phaffii host strain
- Restriction enzymes (BsaI, BpiI)
- T4 DNA Ligase
- PEG/LiAc transformation mix
- Solid and liquid media (YPG, SC)
Methodology:
- sgRNA and Donor Construction: Clone the sgRNA sequence targeting a specific neutral locus (e.g., 04576, PFK1, ROX1) into a Cas9/sgRNA plasmid. Assemble the heterologous gene expression cassette (promoter-gene-terminator) into the corresponding donor helper plasmid via Golden Gate assembly using BsaI or BpiI [20].
- Co-transformation: Co-transform approximately 1 µg of the linearized donor DNA fragment and the Cas9/sgRNA plasmid into competent K. phaffii cells using a standard PEG/LiAc method [20].
- Screening and Validation: Plate cells on non-selective medium. After 2-3 days, screen individual colonies for correct integration using colony PCR. Verify protein expression via fluorescence (for eGFP) or enzymatic assays [20].
Protocol 2: Evaluating Promoter Strength inSaccharomyces cerevisiae
This protocol outlines a procedure for comparing the performance of different promoters under specific cultivation conditions, as their activity can be unpredictable and context-dependent [19].
Materials and Reagents:
- S. cerevisiae strains with reporter genes (e.g., xylanase, xylosidase) under test promoters (e.g., TDH3P, SED1P)
- Cultivation media: Glucose (aerobic/micro-aerobic), Xylose, Beechwood xylan
- Lignocellulosic biomass hydrolysate
Methodology:
- Strain Cultivation: Inoculate strains in triplicate into media containing different carbon sources (e.g., glucose, xylose, beechwood xylan). Cultivate under both aerobic and micro-aerobic conditions as required [19].
- Sample Harvesting: Harvest cells at the mid-exponential phase. Collect supernatant for secreted enzyme assays and cell pellet for intracellular analysis.
- Activity Assay: Perform enzymatic assays (e.g., on xylanase or xylosidase) tailored to the reporter gene. Measure product formation spectrophotometrically. Normalize activity to cell density or total protein content [19].
- Data Analysis: Compare specific enzyme activities across different promoter constructs and growth conditions to identify the best-performing promoter for the intended application.
Protocol 3: Chaperone Co-expression Screening for Enhanced Production
This protocol uses a mating-based strategy to identify cytosolic chaperones that improve the folding and production of heterologous small molecules or proteins in S. cerevisiae [21].
Materials and Reagents:
- Haploid chaperone overexpression library (MATa, 68 strains) [21]
- Haploid query strain (MATα) expressing the heterologous pathway (e.g., aspulvinone E)
- Solid YPG media with galactose
- Selective plates (SC-Ura + Gal + G418)
Methodology:
- Mating: Replica-pin the arrayed chaperone library strains and the query strain together onto solid YPG media with galactose as the carbon source. Incubate to allow mating [21].
- Diploid Selection: Transfer the mated colonies to a solid medium that selects for diploid cells (e.g., SC-Ura + Gal + G418).
- Production Screening: Transfer the array of diploid strains to a production medium. Assess the production of the target compound (e.g., measure aspulvinone E fluorescence or via UHPLC-DAD-TOFMS) [21].
- Validation: The best-performing chaperone hits (e.g., combined overexpression of YDJ1 and SSA1) should be validated in liquid batch fermentations to quantify the percentage yield improvement [21].
Visualization of Experimental Workflows
Chaperone Screening Workflow
CRISPR/Cas9 Workflow in K. phaffii
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential research reagents and their applications in yeast genetic engineering.
| Reagent/Tool | Function/Description | Application Example |
|---|---|---|
| GoldenPiCS Toolkit [20] | A modular cloning system based on Golden Gate assembly for building expression cassettes. | Assembling promoter, gene, and terminator modules for integration in K. phaffii. |
| CRISPR/Cas9 System [17] [20] | Enables precise double-strand breaks and markerless genomic integration via HDR. | Deleting native glucoamylase genes in A. niger; integrating expression cassettes in K. phaffii. |
| AI-Based Design Tool (DOSDiff) [18] | A deep learning framework (D3PM) for de novo design and optimization of promoter sequences. | Generating highly efficient synthetic promoters for S. cerevisiae and P. pastoris. |
| Chaperone Overexpression Library [21] | An arrayed library of S. cerevisiae strains overexpressing one or two cytosolic chaperones. | Identifying chaperones (e.g., Ydj1, Ssa1) that improve folding and yield of heterologous pathways. |
| Neutral Genomic Integration Sites [20] | Specific loci (e.g., 04576, PFK1, ROX1) that allow robust expression without affecting cell fitness. | Targeted, stable integration of heterologous genes in K. phaffii for consistent expression. |
The engineering of microbial cell factories, particularly using the yeast Saccharomyces cerevisiae, is a cornerstone of industrial biotechnology for the sustainable production of biofuels, bioplastics, pharmaceuticals, and high-value compounds [22] [23]. A critical enabling technology for this endeavor is the controlled heterologous expression of metabolic pathways, which is primarily facilitated by specialized plasmid vectors [24]. These vectors allow researchers to introduce, modify, and optimize genes from other organisms into the yeast host. The four main types of S. cerevisiae shuttle vectors—YIp (Yeast Integrating), YEp (Yeast Episomal), YCp (Yeast Centromeric), and YRp (Yeast Replicating)—each possess distinct replication and maintenance strategies, leading to characteristic stability, copy number, and gene expression levels [25] [24] [26]. The rational selection and application of these vectors are fundamental to achieving predictable control of gene expression, which is necessary for balancing metabolic flux, maximizing product titers, minimizing metabolic burden, and ultimately designing efficient yeast cell factories [27] [28] [22]. These vectors are shuttle plasmids, containing components that allow for propagation and selection in both E. coli (for convenient cloning) and S. cerevisiae (for functional expression) [24].
Plasmid Characteristics and Comparative Analysis
The functional differences between YIp, YEp, YCp, and YRp plasmids stem from their unique origins of replication and associated DNA elements, which directly influence their copy number, stability, and suitability for different applications in metabolic engineering [25] [26].
YEp (Yeast Episomal Plasmids) contain a fragment from the native yeast 2µ plasmid circle. This origin allows them to replicate independently of the chromosome as episomes and be maintained at high copy numbers, typically 50+ copies per cell [25]. This high copy number makes them ideal for applications requiring high-level protein expression. However, without selective pressure, they can be somewhat unstable due to uneven segregation during cell division [25].
YCp (Yeast Centromeric Plasmids) incorporate an Autonomously Replicating Sequence (ARS) along with a centromere (CEN) sequence. The centromere ensures proper segregation during mitosis, much like a natural chromosome. Consequently, YCp plasmids are very stable but are maintained at a low copy number, typically 1-2 copies per cell [25]. They are excellent for expressing genes where moderate, stable expression is required, or when the gene product is toxic at high levels.
YRp (Yeast Replicating Plasmids) contain an ARS but lack a centromere. While they can replicate independently and achieve a moderate to high copy number, they are highly unstable and tend to be lost rapidly during mitotic growth in non-selective conditions because they do not segregate efficiently to daughter cells [25] [26].
YIp (Yeast Integrating Plasmids) lack a yeast origin of replication entirely. To be maintained, they must be integrated directly into the host chromosome via homologous recombination [25] [24]. This results in very high stability, as the DNA is passed on like a native gene, but the copy number is typically low (1-2), unless targeted to multi-copy genomic loci. They are used when permanent, stable genetic modification is desired.
Table 1: Characteristics of Primary Yeast Plasmid Vector Systems
| Plasmid Type | Key Components | Copy Number | Stability | Primary Applications |
|---|---|---|---|---|
| YEp (Episomal) | 2µ plasmid origin | High (50+) [25] | Moderate [25] | High-level protein expression; transient overexpression |
| YCp (Centromeric) | ARS, CEN | Low (1-2) [25] | High [25] | Stable, moderate expression; expressing toxic proteins; genomic library construction |
| YRp (Replicating) | ARS | Moderate to High [26] | Low [25] | Rapid, transient gene expression; library screening where high copy number is initially beneficial |
| YIp (Integrating) | Homology arm(s) for recombination | Low (1-2), can be higher if targeted to rDNA [24] | Very High [24] | Stable, permanent gene integration; pathway assembly in chassis strains |
The following diagram illustrates the core replication and inheritance mechanisms of these four plasmid types, which underpin their characteristics described in Table 1.
Quantitative Data for Plasmid Selection
Beyond the basic characteristics, selecting a plasmid system for metabolic engineering requires consideration of quantitative data on plasmid burden and its impact on cell growth. The burden, manifested as reduced growth rates, arises from both the metabolic load of maintaining and replicating plasmid DNA and the expression of the selection marker itself [28].
Table 2: Impact of Plasmid Features on Cellular Burden and Performance
| Feature | Impact on Plasmid Burden/Copy Number | Experimental Consideration |
|---|---|---|
| Selection Marker | Auxotrophic markers (e.g., LEU2, URA3) can impose a significant metabolic burden, reducing growth rate more than the physical load of plasmid replication [28]. Dominant markers (e.g., KanMX) can alleviate this in non-auxotrophic strains. | Marker choice is critical. Auxotrophic selection requires specific host strains but is cost-effective. Antibiotic resistance allows for use in any strain but adds cost [25] [28]. |
| Plasmid Load | The physical DNA load has a minor impact on growth in haploid strains but a more significant effect in diploid strains [28]. | For high-copy YEp plasmids in diploids, the burden is more pronounced. Consider lower-copy vectors or genomic integration for complex pathway expression in diploids. |
| Promoter Strength | Strong promoters (e.g., PTDH3, PGK1) can enhance product formation but may increase burden if protein overexpression is toxic or drains cellular resources [27] [28]. | Weaker or inducible promoters (e.g., GAL1, CUP1) can be used to control expression timing and reduce burden during growth phases [27]. |
| Strain Ploidy | Plasmid burden is generally more pronounced in diploid strains compared to haploid strains [28]. | Haploid strains are often preferred for initial engineering to minimize unintended burden effects. |
Application Protocols for Heterologous Pathway Expression
This section provides detailed methodologies for employing yeast plasmid vectors in the context of optimizing heterologous biosynthetic pathways, a common task in developing yeast cell factories.
Protocol: Modular Pathway Optimization Using Plasmid Libraries
Objective: To generate and screen a combinatorial library of a heterologous biosynthetic pathway by varying the expression levels of individual genes using different plasmid systems. This is a powerful approach to balance metabolic flux [22].
Materials:
- Research Reagent Solutions: See Table 3 for a complete list.
- Host Strain: S. cerevisiae haploid strain with relevant auxotrophies (e.g., BY4741: his3Δ1, leu2Δ0, met15Δ0, ura3Δ0) [28].
- Plasmids: A set of YEp and YCp vectors with compatible selection markers (e.g., pRS42X series [HIS3] and pRS41X series [LEU2]) and a range of promoters (e.g., strong: PTDH3, intermediate: TEF1, weak: CYC1) [27] [28].
- Gene of Interest (GOI): Codon-optimized genes for your target pathway.
Table 3: Research Reagent Solutions for Plasmid-Based Pathway Engineering
| Reagent / Material | Function / Description | Example / Specifics |
|---|---|---|
| Shuttle Vectors (pRS series) | E. coli/S. cerevisiae plasmids with standardized selection (e.g., HIS3, LEU2) and origins (2μ, CEN/ARS) for flexible cloning and expression [28]. | pRS425 (2μ, LEU2), pRS414 (CEN/ARS, TRP1) |
| Promoter & Terminator Libraries | Modular DNA parts to tune transcription initiation and termination, creating a range of expression strengths for each pathway gene [27] [22]. | Promoters: PTDH3 (strong), TEF1 (medium), PSSA1 (induced post-diauxic) [27]. |
| Auxotrophic Drop-out Media | Selective media lacking specific amino acids or nucleotides to maintain plasmid pressure and ensure plasmid retention in transformed cells [25] [28]. | Synthetic Complete (SC) media lacking leucine (-Leu), uracil (-Ura), etc. |
| Enzymatic Assembly Mix | For seamless and efficient Golden Gate or Gibson assembly of multiple DNA fragments (promoter, GOI, terminator) into a plasmid backbone in a single reaction. | Commercial Gibson Assembly Master Mix or similar. |
| Yeast Transformation Kit | Chemical (LiAc) or electroporation-based reagents for high-efficiency introduction of plasmid DNA into competent yeast cells. | Lithium Acetate (LiAc)/Single-Stranded Carrier DNA/Polyethylene Glycol (PEG) method. |
Procedure:
- Modular Plasmid Construction: For each gene in the pathway, clone the coding sequence into multiple plasmid backbones. This creates a set of vectors where the gene is under the control of different promoters (strong, medium, weak) and maintained at different copy numbers (YEp for high, YCp for low). Use different selection markers for each plasmid to allow for co-transformation.
- Combinatorial Co-transformation: Co-transform the host yeast strain with different combinations of the pathway gene plasmids. For a 3-gene pathway with 3 expression levels each, this will generate a theoretical library size of 3^3 = 27 different strain combinations.
- Library Screening: Plate the transformed yeast cells on appropriate selective media (e.g., SC -Leu -Trp -Ura) to ensure all plasmids are maintained. Pick individual colonies into deep-well plates containing liquid selective media for micro-cultivation.
- Product Titer Analysis: After a suitable fermentation period (e.g., 72-96 hours, capturing both exponential and stationary phases), quantify the titer of the desired metabolic product using HPLC or LC-MS.
- Hit Validation: Re-culture the top-performing strains from the primary screen and re-evaluate their performance in small-scale flask fermentations to validate the increase in product titer and yield.
The workflow for this combinatorial approach, from plasmid construction to screening, is outlined below.
Protocol: Stable Pathway Integration Using YIp Vectors
Objective: To stably integrate a heterologous biosynthetic pathway into the yeast genome for long-term, selection-free expression, which is critical for industrial-scale fermentation.
Materials:
- Host Strain: S. cerevisiae strain with a defined auxotrophy (e.g., ura3Δ0).
- YIp Vector: Contains a selection marker (e.g., URA3) and the GOI, flanked by homology arms targeting a specific genomic locus (e.g., the rDNA cluster for multi-copy integration) [24].
- Restriction Enzymes: For linearizing the plasmid within the homology arm to stimulate homologous recombination.
Procedure:
- Vector Linearization: Digest the YIp plasmid with a restriction enzyme that cuts within the genomic homology arm sequence. This creates a linear DNA fragment with the GOI and marker flanked by homologous ends, dramatically increasing the efficiency of genomic integration.
- Yeast Transformation: Transform the linearized DNA fragment into the host yeast strain using a standard LiAc protocol.
- Selection and PCR Verification: Plate transformed cells on selective media (e.g., SC -Ura) to select for successful integrants. Screen resulting colonies by colony PCR using primers that span the integration junction to verify correct chromosomal integration.
- Curing (Optional): For markers like URA3 that allow for counterselection, the marker can be excised in a subsequent step by plating on 5-Fluoroorotic Acid (5-FOA) to select for cells that have lost the URA3 gene, allowing for iterative engineering [25].
Advanced and Emerging Methodologies
As the field of metabolic engineering advances, so do the tools for gene expression control. Beyond the classic plasmid systems, new technologies enable more dynamic and high-throughput optimization.
Advanced In Vivo Shuffling (GEMbLeR): GEMbLeR (Gene Expression Modification by LoxPsym-Cre Recombination) is an advanced method that allows for the generation of vast diversity in promoter and terminator combinations directly in the yeast genome [22]. It involves replacing a gene's native regulatory elements with pre-assembled arrays of different promoters and terminators, all flanked by orthogonal LoxPsym sites. Induction of Cre recombinase then shuffles these modules in vivo, creating a massive library of expression variants for a pathway without the need for re-cloning, enabling rapid strain optimization [22].
Considerations for Carbon Source and Fermentation Phase: The choice of promoter and plasmid must account for the fermentation conditions. So-called "constitutive" promoters (e.g., PTEF1, PTDH3) often show sharply decreased activity after glucose depletion (diauxic shift) [27]. For sustained production in industrial batch fermentations, promoters that are induced at low glucose levels (e.g., PADH2, PHXT7) or in the post-diauxic phase (e.g., PSSA1) can be more effective, ensuring pathway expression throughout the fermentation process [27].
Application Notes
Within the context of engineering yeast cell factories for heterologous pathway expression, the precise control of cellular processes is paramount. Post-translational modifications (PTMs) and accurate protein targeting are not merely fundamental biological phenomena; they are critical engineering parameters that directly influence the yield, stability, and functionality of recombinant proteins and metabolites [29] [30] [31]. Mastering these processes is essential for constructing efficient and robust microbial production platforms.
The Role of PTMs in Regulating Protein Stability and Function
PTMs are covalent processing events that rapidly and reversibly alter the properties of a protein, enabling cells to respond dynamically to environmental and metabolic cues. In metabolic engineering, understanding and harnessing these modifications allows for the fine-tuning of pathway enzyme activity and stability.
Ubiquitination and the Control of Metabolic Flux: Ubiquitination primarily serves as a signal for proteasomal degradation via the ubiquitin-proteasomal system (UPS) [30]. This modification is a key mechanism for controlling the concentration of regulatory proteins, such as cell cycle regulators and signaling molecules [29]. For a metabolic engineer, the UPS represents a tool for degrading rate-limiting or undesired enzymes. PTMs like phosphorylation often act as upstream signals that trigger ubiquitination, providing a layer of rapid, reversible regulation before the irreversible commitment to degradation [30]. This cross-talk creates a "PTM-activated degron," where a phosphorylation event can mark a protein for destruction, a mechanism that could be engineered to dynamically control metabolic pathway fluxes.
Methylation as a Regulatory Signal: Beyond its well-known role in histones, protein methylation is a crucial regulator of protein stability. For instance, the methyltransferase SETD7 methylates the NF-κB subunit RELA and hypoxia-inducible factor α (HIF-1α), creating a "methyl-activated degron" that targets these proteins for ubiquitination and degradation [30]. This demonstrates how a single PTM can directly influence the half-life of central regulatory proteins. Engineering the recognition of such degrons could be a strategy to stabilize key enzymes in a heterologous pathway.
Glycosylation for Robustness and Secretion: Glycosylation is vital for the structural integrity of fungal cell walls and the proper folding and secretion of proteins [29] [31].
N-Glycosylation andO-mannosylation are essential for virulence in pathogenic fungi, and their disruption leads to severe cell wall defects [29]. In yeast cell factories, hyperglycosylation of recombinant proteins is a common issue that can impair function [31]. Therefore, engineering the glycosylation pathway towards humanized patterns is a major focus for producing therapeutic proteins like antibodies [31].
Table 1: Key Post-Translational Modifications and Their Impact on Yeast Cell Factories
| PTM | Key Function | Impact on Heterologous Expression | Example in Yeast |
|---|---|---|---|
| Phosphorylation [29] | Signal transduction, enzyme regulation | Modulates activity of pathway enzymes; can trigger degradation | MAPK cascades regulating stress response and morphogenesis [29] |
| Ubiquitination [29] [30] | Targets proteins for degradation (K48-linkage); non-proteolytic signaling | Controls turnover of metabolic and regulatory proteins | Regulation of cyclins during cell cycle [29] |
| Sumoylation [29] | Transcriptional regulation, stress response, cell cycle | Can stabilize proteins or alter localization | Modification of transcription factors and heat shock proteins [29] |
| Glycosylation [29] [31] | Protein folding, stability, secretion, cell wall integrity | Affects secretion efficiency, activity, and immunogenicity of therapeutics | N- and O-glycosylation of cell wall mannoproteins and secreted enzymes [29] |
| Methylation [30] [32] | Gene regulation, protein stability | Can create degrons to destabilize specific proteins | Methylation of histones by Set1p, Set2p, Set5p, Dot1p [32] |
| Palmitoylation / Farnesylation [29] | Membrane anchoring, protein-protein interactions | Critical for localization of signaling proteins (e.g., Ras) | Ras1 localization to plasma membrane in C. albicans [29] |
Protein Targeting and Secretion for Efficient Production
For a yeast cell factory, the ultimate goal is often the high-yield production and secretion of a target compound. Protein targeting to organelles or the secretory pathway is therefore a critical engineering lever.
The Secretory Pathway as an Engineering Bottleneck: S. cerevisiae possesses a sophisticated eukaryotic secretory pathway, which is advantageous for producing correctly folded proteins with disulfide bonds [31]. However, the secretory transport process can be inefficient, creating a major bottleneck. Engineering strategies focus on overexpressing key components of this pathway, such as chaperones that aid folding and proteins involved in vesicle trafficking, to enhance the overall flux of heterologous proteins through the secretory system [31].
GPI Anchors and Cell Surface Display: Glycosylphosphatidylinositol (GPI) anchors are PTMs that tether proteins to the cell surface [29]. In yeast, these anchors can be exploited to display enzymes or binding proteins on the cell wall, a strategy useful for whole-cell biocatalysis or biosensor development [29] [31].
Subcellular Compartmentalization of Pathways: Rerouting heterologous metabolic pathways to specific subcellular compartments, such as the mitochondria or peroxisomes, can concentrate substrates, isolate toxic intermediates, and leverage unique cofactor pools [33]. This spatial reconfiguration is a powerful strategy to enhance pathway efficiency and yield.
Experimental Protocols
Protocol 1: Analyzing PTMs via Quantitative Mass Spectrometry
This protocol outlines a method for comparative analysis of protein localization and PTMs using quantitative mass spectrometry (MS) of subcellular fractions, adapted from studies in rat liver [34].
1. Principle: Subcellular fractionation is combined with quantitative MS to assign proteins and their modifications to specific organelles. This allows for the creation of a global cellular map and the study of how PTMs influence protein localization and stability.
2. Research Reagent Solutions:
- Lysis/Buffering: Amicon Ultra 0.5 ml 30KDa cellulose filters, 100 mM Tris pH 7.6, 1% lithium dodecyl sulfate, 50 mM ammonium bicarbonate.
- Reduction/Alkylation: 100 mM DTT, 22.5 mM iodoacetamide in urea buffer (100 mM Tris-HCL, pH 8.5, 8M urea).
- Digestion: Sequencing-grade trypsin, endoproteinase Lys-C.
- Quantification: TMT10 isobaric labeling kit.
- Special Equipment: Ultracentrifuge, SW55Ti rotor, LC-MS/MS system.
3. Step-by-Step Procedure:
A. Subcellular Fractionation: i. Prepare a post-nuclear supernatant (fraction E) from a homogenized cell culture. ii. Subject fraction E to differential centrifugation to isolate nuclear (N), heavy mitochondrial (M), light mitochondrial (L), microsomal (P), and cytosolic (S) fractions [34]. iii. Further resolve specific organellar fractions (e.g., L1) by density gradient centrifugation using a Nycodenz or sucrose step gradient.
B. Protein Digestion and TMT Labeling: i. For each fraction, reduce 20 μg of protein with DTT and alkylate with iodoacetamide. ii. Digest proteins using a sequential trypsin/Lys-C digestion protocol (16 hours trypsin, followed by 4 hours Lys-C) [34]. iii. Label the resulting peptides from each fraction with a different TMT reporter ion tag according to the manufacturer's instructions. Pool the labeled samples.
C. Mass Spectrometric Analysis: i. Analyze the pooled TMT-labeled peptide sample by LC-MS/MS. ii. For quantification, compare two primary methods: - TMT-MS2: Provides greatest proteome coverage but suffers from ratio compression, narrowing the accurate dynamic range [34]. - TMT-MS3: Uses synchronous precursor selection to diminish ratio compression, resulting in more accurate quantification but potentially lower coverage [34]. iii. Acquire data in a manner that allows for both protein identification and quantification of the TMT reporter ions.
D. Data Analysis: i. Identify proteins and map PTM sites using database search engines. ii. Quantify protein abundance across fractions based on TMT reporter ion intensities. iii. Use clustering algorithms to assign proteins to subcellular compartments based on their abundance profiles across the fractionation series, using a set of well-known marker proteins for validation [34].
Protocol 2: Engineering Yeast for Enhanced Heterologous Protein Secretion
This protocol details strategies to construct S. cerevisiae strains optimized for the high-level secretion of heterologous proteins, a critical goal for efficient downstream processing [31].
1. Principle: Enhance protein secretion by engineering multiple levels of the process: hyperexpression of the gene of interest, optimization of the secretory pathway, and humanization of glycosylation patterns.
2. Research Reagent Solutions:
- Genetic Tools: Codon-optimized synthetic genes, integration plasmids (YIp), episomal plasmids (YEp), CRISPR/Cas9 system for genome editing.
- Promoters/Terminators: Constitutive (e.g., PGK1, ADH1) and inducible (e.g., GAL1) promoters of varying strengths; optimized terminators.
- Culture Media: Selective media (e.g., SC dropout), induction media (e.g., containing galactose).
3. Step-by-Step Procedure:
A. Construct a Hyperexpression System: i. Codon Optimization: Synthesize the heterologous gene with codons optimized for S. cerevisiae to improve translational efficiency. Avoid rare codons, adjust GC content, and remove cryptic regulatory sequences [31]. ii. Select an Expression Vector: Choose a high-copy-number episomal plasmid (YEp) for high gene dosage or an integration plasmid (YIp) for stable, single-copy chromosomal insertion [31]. iii. Engineer Transcriptional Control: Clone the codon-optimized gene under the control of a strong, tunable promoter (e.g., inducible GAL1 or constitutive TEF1). Pair with a strong terminator to ensure efficient transcription termination [31].
B. Engineer the Secretory Pathway: i. Overexpress Chaperones: Co-express endoplasmic reticulum (ER) chaperones like BiP/Kar2p and protein disulfide isomerase (PDI) to facilitate proper folding and prevent ER stress [31]. ii. Modulate Vesicle Trafficking: Overexpression of proteins involved in vesicle formation (e.g., the small GTPase Sar1p) and fusion (e.g., the v-SNARE Sec22p) can enhance the flux of proteins from the ER to the Golgi and onward to the plasma membrane [31].
C. Perform Glycosylation Pathway Engineering:
i. Disable Hypermannosylation: Knock out genes responsible for adding elongated mannose chains (e.g., och1Δ) to reduce immunogenic hyperglycosylation [29] [31].
ii. Create Humanized Glycosylation Strains: Introduce pathways for the synthesis of complex human-type N-glycans. This typically involves the knock-in of multiple enzymes, such as mannosidases I and II, N-acetylglucosaminyltransferases I and II, and others, into an och1Δ background [31].
D. Analysis of Success: i. Measure protein yield in the culture supernatant using assays like SDS-PAGE, Western blot, or enzyme activity assays. ii. Analyze glycosylation patterns of the secreted protein using techniques such as lectin blotting or MS.
Table 2: Key Research Reagents for Yeast Metabolic Engineering
| Reagent / Tool | Function | Example Use Case |
|---|---|---|
| CRISPR/Cas9 System [31] | Precise genome editing | Knocking out glycosylation genes (e.g., OCH1); integrating heterologous pathways. |
| Codon-Optimized Genes [31] | Enhances translational efficiency and protein yield | Optimizing heterologous enzyme genes for expression in S. cerevisiae. |
| Inducible Promoters (e.g., GAL1) [31] | Provides temporal control over gene expression | Decoupling cell growth from product formation to avoid metabolic burden. |
| TMT Isobaric Labeling Kits [34] | Multiplexed quantitative proteomics | Simultaneously comparing protein abundance and PTMs across 10+ subcellular fractions. |
| Episomal Plasmids (YEp) [31] | High-copy-number gene expression | Rapid testing and high-level expression of heterologous pathway enzymes. |
Advanced Tools and Workflows for Pathway Design and Implementation
The construction of recombinant DNA molecules is a foundational technology in molecular biology and synthetic biology, enabling the study of gene function and the engineering of microbial cell factories. For researchers focusing on heterologous pathway expression in yeast, the choice of DNA assembly method is critical for efficiently building complex genetic constructs such as multi-gene pathways, CRISPR-Cas9 systems, and protein expression vectors [35]. The evolution from traditional restriction/ligation cloning to modern, seamless techniques has significantly accelerated the pace of metabolic engineering in yeast systems like Saccharomyces cerevisiae and Pichia pastoris [36] [37]. These advanced methods facilitate the precise assembly of multiple DNA fragments without leaving unwanted "scar" sequences, enabling the creation of optimized pathways for producing valuable compounds such as terpenoids, biofuels, and therapeutic proteins [36] [38]. This article provides a comprehensive overview of key DNA assembly strategies, with detailed protocols and applications specifically framed within the context of yeast cell factory engineering.
Molecular cloning has undergone a remarkable transformation since the pioneering work of Cohen and Boyer in the 1970s [35]. The early methods relied on restriction enzymes and DNA ligase to cut and paste DNA fragments into plasmid vectors. While these techniques revolutionized biological research, their limitations—including dependence on specific restriction sites and the potential for unwanted scar sequences—spurred the development of more sophisticated, seamless assembly methods [35]. The timeline below visualizes the evolution of these key cloning technologies, highlighting their development and relationships.
The contemporary molecular biology laboratory now has access to multiple advanced cloning techniques, each with distinct mechanisms and optimal use cases. The following table provides a quantitative comparison of the most widely used DNA assembly methods, highlighting key performance characteristics relevant to yeast metabolic engineering.
Table 1: Quantitative Comparison of DNA Assembly Methods
| Method | Mechanism | Typical Fragment Limit | Seamless? | Key Enzymes | Optimal Use Cases |
|---|---|---|---|---|---|
| Restriction Enzyme Ligation | Restriction digestion and ligation | 1-2 fragments | No | Type II RE, DNA Ligase | Simple inserts with compatible sites [39] |
| Gateway Cloning | Site-specific recombination | 1 fragment | No | Integrase, Excisionase | High-throughput transfer to multiple vectors [39] [35] |
| Gibson Assembly | Homologous recombination | 5-15 fragments | Yes | Exonuclease, Polymerase, Ligase | Modular pathway assembly [39] [40] |
| Golden Gate Assembly | Type IIS restriction-ligation | 30+ fragments | Yes | Type IIS RE, DNA Ligase | Combinatorial library construction [39] [40] |
| TOPO-TA Cloning | Topoisomerase-mediated | 1 fragment | No | Topoisomerase I | Rapid cloning of PCR products [39] |
Detailed Methodologies and Protocols
Restriction Enzyme Ligation Cloning
The traditional restriction enzyme ligation method remains a fundamental technique for simple cloning applications. The step-by-step workflow for this method is illustrated below.
Protocol: Restriction Enzyme Ligation Cloning
Step 1: Vector and Insert Preparation
- Digest 1-2 µg of vector and insert DNA with selected restriction enzymes in appropriate buffer. Incubate for 1-2 hours at enzyme-specific temperatures [39].
- Use a 3:1 molar ratio of insert to vector to enhance ligation efficiency.
Step 2: Gel Purification
- Run digested products on an agarose gel and excise bands of interest.
- Purify DNA using gel extraction kits to remove enzymes and buffers.
Step 3: Ligation
- Set up ligation reaction with T4 DNA ligase, using purified vector and insert fragments.
- Incubate at room temperature for 1 hour or 16°C overnight [35].
Step 4: Transformation and Screening
- Transform ligation reaction into competent E. coli cells.
- Screen colonies by colony PCR or restriction digest to identify correct clones.
Application Note for Yeast Engineering: While largely superseded by newer methods, restriction enzyme cloning remains useful for constructing basic expression cassettes with yeast-specific promoters such as TDH3P (glyceraldehyde-3-phosphate dehydrogenase) and SED1P, which have been shown to drive high-level expression of heterologous enzymes in S. cerevisiae [38].
Gibson Assembly
Gibson Assembly, developed by Daniel Gibson in 2009, allows for seamless assembly of multiple DNA fragments in a single isothermal reaction [40]. The mechanism employs a three-enzyme cocktail that simultaneously executes exonuclease activity, polymerase extension, and DNA ligation.
Protocol: Gibson Assembly
Step 1: Fragment Preparation with Homology Arms
- Amplify DNA fragments by PCR with 20-40 bp overlapping homologous sequences at their ends [40].
- Gel-purify PCR products to remove primers and non-specific amplification.
Step 2: Assembly Reaction
- Combine DNA fragments with Gibson Assembly master mix containing:
- T5 exonuclease: chews back 5' ends to create single-stranded overhangs
- Phusion DNA polymerase: fills in gaps in the annealed fragments
- Taq DNA ligase: seals nicks in the assembled DNA [40]
- Use approximately 100 ng of total DNA with equimolar ratios of each fragment.
- Combine DNA fragments with Gibson Assembly master mix containing:
Step 3: Incubation and Transformation
- Incubate reaction at 50°C for 30-60 minutes.
- Transform 2-5 µL of reaction into competent E. coli cells [40].
Step 4: Screening
- Screen colonies by analytical PCR or restriction digest.
Application Note for Yeast Engineering: Gibson Assembly is particularly valuable for assembling entire biosynthetic pathways—such as the mevalonate (MVA) pathway for terpenoid production—into yeast expression vectors in a single reaction [36]. Its ability to join up to 15 fragments makes it ideal for constructing multi-gene pathways without introducing scar sequences between genes.
Golden Gate Assembly
Golden Gate Assembly utilizes Type IIS restriction enzymes that cut outside their recognition sequences, enabling seamless assembly of multiple DNA fragments with unique overhangs in a single reaction [39] [40].
Protocol: Golden Gate Assembly
Step 1: Fragment Design with Type IIS Sites
- Design DNA fragments flanked by Type IIS recognition sites (e.g., BsaI) and unique 4-bp overhangs.
- Ensure overhangs direct correct fragment orientation and order.
Step 2: One-Pot Restriction-Ligation
- Combine DNA fragments with destination vector, Type IIS enzyme (e.g., BsaI-HF), and T4 DNA ligase in appropriate buffer.
- Set up thermal cycling as follows:
- 25-30 cycles of:
- 37°C for 2-5 minutes (digestion)
- 16°C for 2-5 minutes (ligation)
- Final digestion: 37°C for 5-10 minutes
- Heat inactivation: 80°C for 5-10 minutes [40]
- 25-30 cycles of:
Step 3: Transformation and Screening
- Transform entire reaction into competent E. coli.
- Screen colonies for correct assemblies.
Application Note for Yeast Engineering: Golden Gate is exceptionally suited for constructing combinatorial libraries of promoter-gene combinations to optimize flux through heterologous pathways in yeast [39] [40]. Its high efficiency with 30+ fragments enables sophisticated metabolic engineering projects, such as creating diversified enzyme variant libraries for screening improved terpenoid production strains [36].
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of DNA assembly methods for yeast metabolic engineering requires carefully selected molecular biology reagents. The following table details essential components and their functions.
Table 2: Key Research Reagent Solutions for DNA Assembly in Yeast Engineering
| Reagent Category | Specific Examples | Function in DNA Assembly | Yeast Engineering Application |
|---|---|---|---|
| Type IIS Restriction Enzymes | BsaI, BsmBI, BbsI | Create unique 4-bp overhangs outside recognition site | Golden Gate assembly of transcriptional units [40] |
| DNA Ligases | T4 DNA Ligase, Taq DNA Ligase | Catalyze phosphodiester bond formation between DNA fragments | Joining vector-insert junctions in multiple methods [40] [35] |
| Assembly Master Mixes | Gibson Assembly Master Mix, In-Fusion HD Kit | Provide optimized enzyme cocktails for seamless assembly | Accelerating pathway construction in yeast vectors [39] [40] |
| Yeast Expression Vectors | pYES2, pPICZ, episomal/integrative plasmids | Serve as final destination for assembled DNA | Heterologous gene expression under yeast promoters [38] [37] |
| Competent Cells | E. coli (DH5α, TOP10), S. cerevisiae | Propagate assembled plasmids and enable genetic manipulation | Intermediate and final host for pathway assembly [35] |
| Yeast Promoters | TDH3P, SED1P, GAL1P, AOX1 | Drive heterologous gene expression with varying strengths | Fine-tuning metabolic pathway flux [38] [37] |
The evolution of DNA assembly methods from traditional restriction/ligation to modern techniques like Gibson Assembly and Golden Gate has fundamentally transformed the field of yeast metabolic engineering. These advanced methods offer researchers unprecedented ability to efficiently construct complex multi-gene pathways for heterologous expression in yeast cell factories. The choice between methods depends on specific project requirements: Gibson Assembly excels for modular pathway construction with moderate numbers of fragments, while Golden Gate provides superior capability for high-throughput, combinatorial assembly of large DNA constructs. As the demand for microbial production of valuable compounds continues to grow, these DNA assembly technologies will play an increasingly vital role in accelerating the design-build-test cycles of yeast strain engineering, enabling more efficient and sustainable bioproduction of pharmaceuticals, biofuels, and specialty chemicals.
In the construction of yeast cell factories for heterologous pathway expression, precise transcriptional control is a critical determinant of success. The engineering of native and synthetic promoters provides a powerful means to dynamically regulate metabolic flux, separate growth from production phases, and mitigate metabolic burden [41] [42]. This Application Note delineates the functional characteristics, performance parameters, and implementation protocols for both constitutive and inducible promoter systems in yeast, with a specific focus on achieving precise temporal control in heterologous pathway expression for drug development and biochemical production.
Performance Analysis of Yeast Promoter Systems
Quantitative Comparison of Constitutive Promoters
Table 1: Characterized Constitutive Promoters in S. cerevisiae
| Promoter | Source Gene/Type | Relative Strength (Mid-log, Glucose) | Regulatory Pattern | Key Applications |
|---|---|---|---|---|
| PTDH3 | Glyceraldehyde-3-phosphate dehydrogenase | Very High (Benchmark) | Decreases post-diauxic shift | High-level protein production [43] [19] |
| PADH1 | Alcohol dehydrogenase I | Very High | Decreases post-diauxic shift | General heterologous expression [43] |
| PTEF1 | Translational elongation factor | High | Relatively stable through fermentation | Balanced metabolic engineering [43] |
| PENO2 | Enolase 2 | Very High | Decreases post-diauxic shift | High-level enzyme production [43] |
| PGAP | Glyceraldehyde-3-phosphate dehydrogenase (K. phaffii) | High (2-fold lower than strong iSynPs) | Constitutive across carbon sources | Methanol-free expression systems [42] [44] |
| PSED1 | Stress-induced cell wall protein | Moderate-High | Maintains expression on non-native substrates | Lignocellulosic bioprocessing [19] |
Quantitative Comparison of Inducible Promoter Systems
Table 2: Characterized Inducible Promoters and Synthetic Systems
| Promoter/System | Inducer | Basal Expression | Induced Expression | Fold Induction | Key Features |
|---|---|---|---|---|---|
| PGAL1 | Galactose | Low | Very High | ~1000-fold [45] | High induction, carbon source-dependent [43] |
| PAOX1 | Methanol | Low | High | Variable | K. phaffii system, strong methanol induction [46] [44] |
| PCUP1 | Copper | Low | High | High (concentration-dependent) | Chemical inducer, highest post-diauxic expression [43] |
| PADH2 | Low glucose | Repressed on glucose | Moderate | Significant | Autoinduction in batch fermentation [43] |
| DAPG-iSynP | DAPG | Very Low (with insulation) | Very High | >1000-fold [42] | Minimal leakiness, orthogonal system [42] |
| Tet-iSynP | Doxycycline | Very Low | High | >200-fold [42] | Chemical control, tunable with operator repeats [42] |
Experimental Protocols for Promoter Characterization and Implementation
Protocol 1: Quantitative Characterization of Promoter Activity Across Cultivation Conditions
Purpose: To systematically evaluate promoter performance under different carbon sources and throughout batch fermentation, particularly across the diauxic shift [43].
Materials:
- Yeast Strains: S. cerevisiae BY4741 or equivalent with integrated promoter-reporter constructs
- Reporter Plasmids: yEGFP (stable) or yEGFP-CLN2 PEST (destabilized) reporter vectors [43]
- Media: YNB without amino acids supplemented with appropriate carbon sources (20 g/L glucose, sucrose, galactose, or ethanol) [43]
- Equipment: Microtiter plate reader with fluorescence capability, controlled bioreactors for batch cultivation
Procedure:
- Strain Construction: Clone target promoters upstream of yEGFP in yeast integration vectors. Transform into host strain using standard lithium acetate protocol.
- Carbon Source Comparison:
- Inoculate single colonies in 200 μL YNB with different carbon sources in 96-well plates
- Cultivate at 30°C with continuous shaking
- Measure OD600 and GFP fluorescence (excitation 485 nm, emission 520 nm) when cultures reach mid-log phase (OD600 = 1.0-2.5)
- Calculate promoter strength as GFP/OD600 normalized to internal control
- Batch Fermentation Time-Course:
- Inoculate 50 mL YNB with 20 g/L glucose in baffled flasks
- Sample at 3-hour intervals for OD600, GFP fluorescence, and metabolite analysis (glucose, ethanol)
- Continue sampling for at least 72 hours to capture post-diauxic phase
- Data Analysis: Plot promoter activity against cultivation time and metabolic phases. Identify expression patterns in relation to glucose depletion and ethanol production/consumption.
Technical Notes: The destabilized yEGFP-CLN2 PEST (half-life ~12 min) reports real-time transcriptional activity, while stable yEGFP (half-life ~7 h) reflects protein accumulation [43]. For low-expression promoters, stable yEGFP provides better signal-to-noise ratio.
Protocol 2: Implementation of Synthetic Inducible Promoters with Minimal Leakiness
Purpose: To construct and optimize tightly regulated synthetic inducible promoters in yeast using insulation strategies and operator engineering [42].
Materials:
- DNA Parts: Core promoters (KpAOX1, ScGAL1), operator sequences (phlO, tetO, luxO), insulator sequences (1.6 kb KpARG4)
- Expression Vectors: Yeast integration plasmids with selection markers
- sTA Constructs: Regulatable synthetic transcription activators (rPhlTA, rTetTA, LuxTA)
- Host Strains: Komagataella phaffii or S. cerevisiae with appropriate genetic background
Procedure:
- Insulator Integration:
- Amplify ~1.6 kb insulator sequence (e.g., KpARG4 from K. phaffii)
- Clone immediately upstream of core promoter using Gibson Assembly or Golden Gate cloning
- Verify insertion by colony PCR and sequencing
- Operator-Promoter Fusion:
- Design primers to fuse operator repeats directly upstream of TATA-box sequence
- Test 1-4 operator repeats with 0-40 bp spacing to TATA-box
- Assemble constructs using in vivo or in vitro DNA assembly methods
- Leakiness Screening:
- Co-transform promoter-reporter (EGFP) and sTA plasmids into host strain
- Plate transformants on selective media without inducer
- Screen for colonies with minimal fluorescence using flow cytometry or plate reader
- Isulate variants with >100-fold induction capability
- Induction Characterization:
- Inoculate positive clones in selective media and grow to mid-log phase
- Add inducer (DAPG, doxycycline, or HSL) at optimal concentration
- Monitor EGFP expression over 24 hours to determine induction kinetics
- Calculate fold induction as (fluorescence with inducer)/(fluorescence without inducer)
Technical Notes: The 1.6 kb insulator sequence reduces leakiness by up to 376-fold while maintaining >95% of induced activity [42]. Direct fusion of operators to TATA-box with minimal spacing (≤40 bp) maximizes fold induction. For S. cerevisiae, a 110-bp iSynP containing 68-bp ScGAL1 core promoter fused to phlO achieved >100-fold induction [42].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for Promoter Engineering Applications
| Reagent/Category | Specific Examples | Function/Application | Implementation Notes |
|---|---|---|---|
| Reporter Genes | yEGFP, yEGFP-CLN2 PEST, mCherry, lacZ | Quantitative promoter activity assessment | Destabilized reporters for real-time transcription monitoring [43] |
| Core Promoters | KpAOX1 (94 bp), ScGAL1 (68 bp), KpDAS1 (83 bp) | Minimal functional promoter elements | Enable compact iSynP design with high performance [42] |
| Operator Systems | phlO (DAPG), tetO (doxycycline), luxO (HSL) | Inducer-responsive DNA binding sites | Operator repeats (2-4x) enhance induction magnitude [42] |
| Insulator Sequences | KpARG4 (1.6 kb), random 30-bp sequences | Block cryptic transcriptional activation | Essential for minimizing iSynP leakiness [42] |
| Synthetic TFs | rPhlTA, rTetTA, LuxTA, BM3R1-NLS-VP16 (SES) | Orthogonal transcription activation | Enable custom regulatory circuits without crosstalk [42] [44] |
| Assembly Systems | Golden Gate (GoldenPiCS), Gibson Assembly, PGASO | Modular DNA construction | Facilitate rapid promoter variant generation and testing [46] [47] |
Conceptual Framework and Experimental Workflows
Decision Framework for Promoter Selection
Synthetic Promoter Engineering Workflow
The strategic implementation of promoter engineering approaches enables unprecedented temporal control of heterologous pathway expression in yeast cell factories. While constitutive systems provide simplicity and generally high expression levels, their activity profiles during fermentation and across different carbon sources must be carefully considered [43] [19]. Inducible systems, particularly synthetic promoters with insulation and optimized operator architecture, offer tight regulation with minimal basal expression and high induction ratios exceeding 1000-fold [42]. The continued development of orthogonal regulatory systems and cross-species promoter elements [44] will further expand the toolbox available for metabolic engineering and biopharmaceutical production in yeast platforms.
CRISPR-Cas9 and Genome Editing for Stable Pathway Integration
The establishment of stable, homogenous expression of heterologous pathways is a fundamental challenge in developing microbial cell factories. While episomal plasmids offer initial convenience, they often suffer from instability and population heterogeneity, leading to inconsistent production performance and reduced titers in industrial fermentation. Stable chromosomal integration of pathway genes addresses these limitations, ensuring long-term genetic stability and uniform expression across the cell population. The advent of CRISPR-Cas9 genome editing has revolutionized this process, enabling targeted, efficient, and marker-free integration of multi-gene pathways into the yeast genome, thereby accelerating the engineering of robust production strains.
The Critical Need for Stable Genomic Integration in Yeast Cell Factories
In Saccharomyces cerevisiae, a preferred host for bio-based production, plasmid-based expression systems frequently exhibit segregational instability, where plasmids are unevenly distributed or lost during cell division, particularly in non-selective conditions such as large-scale fermenters. Furthermore, studies demonstrate that heterogeneous gene expression from plasmids can create subpopulations of non-producing cells, which consume resources without contributing to product formation, drastically reducing overall process efficiency.
Genomic integration circumvents these issues by anchoring genetic constructs within chromosomes, ensuring hereditary stability and homogeneous expression within the population. Traditional methods for genomic integration, however, have relied on selection markers and homologous recombination, which can be inefficient, labor-intensive, and limited in their capacity for multi-locus integration. The development of CRISPR-Cas9 systems has provided a powerful solution, significantly enhancing the efficiency and specificity of targeted integrations, even for complex metabolic pathways.
CRISPR-Cas9 Systems for Efficient Multi-Loci Integration
Core Mechanisms and Technological Advancements
CRISPR-Cas9 functions as a programmable DNA-endonuclease system. The core components are a guide RNA (gRNA), which specifies the genomic target site via a ~20-nucleotide spacer sequence, and the Cas9 endonuclease, which creates a double-strand break (DSB) at the target site adjacent to a Protospacer Adjacent Motif (PAM) [48]. The cell repairs this DSB primarily through one of two pathways:
- Non-Homologous End Joining (NHEJ): An error-prone repair pathway that often results in small insertions or deletions (indels), useful for gene knockouts.
- Homology-Directed Repair (HDR): A precise repair pathway that uses a donor DNA template with homology arms flanking the DSB site. This mechanism is harnessed for the precise integration of heterologous genes [49] [48].
The real power for metabolic engineering lies in leveraging HDR for targeted integration. By providing a linear donor DNA fragment or an integrative plasmid containing the gene of interest flanked by homology arms, researchers can direct the cell to incorporate the new genetic material at the precise location of the Cas9-induced break with remarkably high efficiency.
Comparison of Advanced CRISPR-Cas9 Integration Systems
Several CRISPR-Cas9 based systems have been developed specifically to address the challenges of multi-gene pathway integration in yeast. The table below summarizes the key features and performance metrics of three prominent systems.
Table 1: Comparison of CRISPR-Cas9 Systems for Multi-Loci Gene Integration in S. cerevisiae
| System Name | Key Feature | Target Sites | Integration Efficiency | Reported Application & Yield |
|---|---|---|---|---|
| CrEdit [50] | Combines Cas9 with EasyClone "safe havens" | Pre-defined intergenic loci (X-2, X-3, etc.) | Up to 100% for single integration; 84% for simultaneous 3-gene integration without selection. | Reconstruction of β-carotene pathway. |
| IMIGE [51] | Iterative multi-copy integration using δ and rDNA sequences | Repetitive sequences for high-copy integration | Achieved significant yield improvement within 5.5-6 days (2 cycles). | Ergothioneine (105.31 mg/L) and Cordycepin (62.01 mg/L), increases of 407.39% and 222.13% over episomal expression. |
| Educational Module [49] | Simple, phenotypic readout (ADE2 inactivation) | ADE2 genomic locus | Enables efficient knockout, yielding red colonies as visual confirmation. | Used for teaching principles of CRISPR-Cas9 editing and repair pathways. |
These systems highlight the flexibility of CRISPR-Cas9 platforms. CrEdit exemplifies high-efficiency, marker-free integration into specific "safe haven" loci, minimizing metabolic burden and genetic instability. In contrast, the IMIGE system exploits repetitive sequences to achieve high copy numbers, which is beneficial for pathway enzymes that require high expression levels to maximize flux.
Detailed Application Notes and Protocols
Protocol 1: Multi-Loci Pathway Integration Using the CrEdit Principle
This protocol outlines the steps for simultaneously integrating three heterologous genes into distinct genomic loci in S. cerevisiae.
Research Reagent Solutions Table 2: Essential Reagents for CRISPR-Cas9 Mediated Integration in Yeast
| Reagent / Tool | Function / Description | Example / Source |
|---|---|---|
| Cas9 Nuclease | Creates a double-strand break at the genomic target. | Streptococcus pyogenes Cas9 (SpCas9) |
| Guide RNA (gRNA) Plasmid | Targets Cas9 to specific genomic loci. | USER-cloning compatible plasmid [50] [49] |
| Donor DNA | Repair template containing the gene(s) of interest and homology arms. | EasyClone-style integration plasmid or PCR-amplified linear fragment [50] |
| Homology Arms | Sequences flanking the donor DNA for homologous recombination. 60-500 bp arms can be incorporated into PCR primers [50]. | |
| gRNA Design Tool | In silico tool for selecting gRNAs with high on-target and low off-target activity. | Benchling CRISPR tool [49] |
| Yeast Strain | S. cerevisiae with high transformation efficiency (e.g., ura3 mutant for selection). | CEN.PK or BY series |
Procedure:
gRNA Design and Donor Construction:
- Select intergenic "safe haven" loci (e.g., from the EasyClone system) to avoid disrupting essential genes.
- For each target locus, design a gRNA sequence targeting the site, ensuring the PAM (NGG for SpCas9) is present. Use software like Benchling to analyze on-target and off-target scores.
- Clone the gRNA expression cassettes into a delivery plasmid, ideally one that allows multiplexing of several gRNAs.
- Prepare the donor DNA. This can be an integrative plasmid or a linear PCR product containing your gene of interest (with promoter and terminator) flanked by homology arms (60-500 bp) specific to the target locus. Ensure the final donor sequence does not contain the PAM site to prevent re-cleavage after successful integration.
Yeast Transformation:
- Co-transform the yeast strain with the following:
- A plasmid expressing Cas9 (constitutive or inducible).
- The gRNA plasmid(s) targeting the desired loci.
- The donor DNA fragments for each locus.
- Use a standard lithium acetate/PEG transformation protocol.
- Plate the transformed cells onto appropriate selective medium.
- Co-transform the yeast strain with the following:
Screening and Validation:
- After incubation, pick resulting colonies.
- For primary screening, perform colony PCR using primers that bind outside the integration site and within the inserted gene to verify correct integration at all target loci.
- For pathway assembly, as with the β-carotene example, screen for the production of the yellow/orange pigment as a positive phenotypic indicator.
- Validate the genotype of positive clones by Sanger sequencing of the amplified integration loci.
Protocol 2: Iterative Multi-Copy Integration Using the IMIGE System
This protocol is adapted for enhancing product titers by integrating multiple gene copies into repetitive genomic regions.
Procedure:
System Setup:
- Utilize a CRISPR-Cas9 system designed to target δ sequences (from the Ty retrotransposon) or rDNA repeats.
- Design a gRNA that targets the chosen repetitive sequence.
- Prepare a donor DNA construct containing the key pathway gene to be amplified, flanked by homology arms specific to the repetitive element.
Iterative Integration Cycles:
- Transform the yeast strain with the Cas9-gRNA complex and the donor DNA.
- Screen for transformants that show improved production, often linked to a growth advantage under selective pressure or via high-throughput screening.
- Isolate genomic DNA from improved clones and use it as a template for the next round of integration, or repeat the transformation cycle to introduce additional copies.
- The IMIGE system has been shown to achieve significant titer improvements in just two rapid cycles (5.5-6 days total) [51].
Validation:
- Quantify product titer (e.g., via HPLC) to identify the highest-producing strains.
- Use quantitative PCR (qPCR) to estimate the copy number of the integrated gene in the best-performing clones.
Visualizing Experimental Workflows
The following diagram illustrates the logical flow and key decision points in the two primary CRISPR-Cas9 integration strategies discussed.
The implementation of CRISPR-Cas9 for stable pathway integration marks a significant leap forward in yeast metabolic engineering. Moving beyond unreliable episomal expression, systems like CrEdit and IMIGE provide researchers with a robust, efficient, and scalable toolkit for constructing optimized cell factories. The ability to perform precise, multi-locus edits and iterative copy number amplification directly addresses the core challenges of pathway stability and gene dosage control. As this technology continues to evolve, incorporating enhanced-fidelity Cas9 variants and more sophisticated donor delivery systems, it will undoubtedly remain a cornerstone of future efforts to harness yeast for the sustainable production of valuable chemicals and pharmaceuticals.
The reconstitution of plant biosynthetic pathways in microbial hosts represents a cornerstone of modern synthetic biology, aiming to achieve sustainable and scalable production of valuable plant natural products (PNPs). The model yeast Saccharomyces cerevisiae has emerged as a premier eukaryotic chassis for this purpose, combining the benefits of microbial fermentation with the capacity to perform complex, plant-like post-translational modifications [31] [52]. This case study delves into the practical application of engineering S. cerevisiae for the heterologous production of PNPs, framed within the broader research context of developing efficient yeast cell factories. We provide a detailed examination of the strategies, protocols, and reagent solutions that enable researchers to overcome common challenges such as low protein yield, inefficient secretion, and metabolic burden, focusing on a representative experiment involving the reconstruction of a benzylisoquinoline alkaloid (BIA) pathway [52].
Strategic Approaches and Key Considerations
The successful reconstitution of a plant pathway in yeast requires a multi-faceted engineering strategy. The general workflow, from design to functional analysis, is outlined in the diagram below.
Core Pathway Engineering Strategies
Codon Optimization: Synonymous codon replacement is a critical first step to match the host's tRNA abundance and prevent translational stalling. For example, codon-optimized versions of Talaromyces emersonii α-amylase (temA-Opt) and glucoamylase (temG-Opt) resulted in 1.6-fold and 3.3-fold higher extracellular activity, respectively, compared to the native genes [31]. Optimization must also consider factors like GC content, avoidance of internal regulatory sites, and the impact on cotranslational folding [31].
Transcriptional Tuning: Selecting and engineering the right promoter is paramount for controlling gene expression. The GAL10-CYC1 inducible promoter system is widely used for strong, tightly regulated expression. Recent studies show that fine-tuning induction levels—using galactose concentrations as low as 0.0015% to 0.003%—can dramatically increase the solubilization and yield of difficult-to-express membrane proteins like UCP1 and GFP-ATP7B, mitigating metabolic burden and toxicity [53]. Combining strong, tuned promoters with optimized terminators ensures high transcriptional flux through the engineered pathway.
Gene Dosage Management: The choice of expression plasmid directly influences gene copy number. While multi-copy episomal plasmids (YEp) can achieve high gene dosage, integration into the yeast chromosome (YIp) offers greater genetic stability, which is crucial for long-term, industrial-scale cultivation [31].
Advanced Host Engineering
Metabolic Engineering and Cofactor Balancing: Efficient PNP synthesis requires a robust supply of precursor molecules and energy cofactors. Engineering strategies often involve amplifying native metabolic fluxes (e.g., the mevalonate pathway for terpenoids), knocking out competing pathways, and dynamic regulation to balance growth and production phases. In one instance, activating the arginine biosynthesis pathway through specific genetic interactions (MKT189G and TAO34477C SNPs) was shown to be essential for enhancing mitochondrial activity and the efficiency of a developmental process like sporulation, highlighting how core metabolism can be rewired to support heterologous functions [54].
Protein Secretion and Glycosylation Engineering: For secreted proteins, the pathway is a major bottleneck. Engineering efforts target multiple steps: enhancing the unfolded protein response (UPR), overexpressing key chaperones like BiP/Kar2p, and increasing the copy number of genes involved in vesicle transport [31]. Furthermore, since S. cerevisiae produces high-mannose glycans, humanizing its glycosylation pathway involves knocking out endogenous glycosyltransferases (e.g., OCH1) and introducing heterologous enzymes to create complex, human-like N-glycans, which is essential for producing therapeutic proteins like active antibodies [31].
Detailed Experimental Protocol
This protocol outlines the stepwise reconstitution and analysis of the noscapine biosynthetic pathway from Papaver somniferum in S. cerevisiae, a successful example from recent literature [52]. The process involves building the pathway gene-by-gene and analyzing intermediate and final products.
Pathway Reconstitution and Metabolite Analysis
Objective: To functionally express a multi-enzyme plant pathway in yeast and quantify the production of pathway intermediates and the target compound.
Materials:
- Yeast Strain: A suitable background strain such as BY4741, often pre-engineered for precursor supply.
- Expression Vectors: A set of compatible integration or episomal plasmids (e.g., with different auxotrophic markers) containing the plant biosynthetic genes under the control of inducible promoters like GAL10 [53].
- Culture Media:
- Analytical Equipment: HPLC-MS/MS system equipped with a C18 reverse-phase column.
Procedure:
Strain Engineering:
- Clone each plant biosynthetic gene (e.g., CYP719A21, CYP82Y1, PsMT1, PsAT1 for noscapine) into individual expression vectors. Use codon-optimized gene sequences for yeast [31].
- Sequentially transform plasmids into the yeast host. A stepwise approach is recommended, where one or two genes are introduced at a time, and the resulting strain is analyzed before adding the next set of genes [52].
- After each transformation, verify integration/plasmid maintenance via colony PCR and selective plating.
Cultivation and Induction:
- Inoculate a single colony of the engineered yeast strain into 5 mL of selective growth medium with 2% glucose. Incubate at 30°C with shaking (250 rpm) for ~16 hours.
- Sub-culture the pre-inoculum into 50 mL of fresh induction medium (e.g., containing 2% potassium acetate and a titrated concentration of galactose, e.g., 0.003%) to an initial OD~600~ of 0.5 [53] [54].
- Induce for 16-24 hours at 20-30°C (a lower temperature may benefit protein stability).
Metabolite Extraction:
- Harvest cells by centrifugation (e.g., 3000 × g for 10 min).
- For intracellular metabolite analysis, resuspend the cell pellet in 1 mL of extraction solvent (e.g., 80% methanol). Vortex vigorously for 1 minute.
- Incubate at -20°C for 1 hour, then centrifuge at 15,000 × g for 10 minutes at 4°C.
- Transfer the supernatant to a new tube and evaporate the solvent under a gentle nitrogen stream.
- Reconstitute the dried extract in 100 µL of methanol for LC-MS analysis.
LC-MS Analysis:
- Separate metabolites using a C18 column with a water-acetonitrile gradient, both containing 0.1% formic acid.
- Operate the mass spectrometer in Multiple Reaction Monitoring (MRM) mode for high sensitivity and selectivity.
- Quantify metabolites by comparing the peak areas of samples against a standard curve of authentic standards.
Key Quantitative Data from Case Studies
Table 1: Representative yields from plant pathways reconstituted in S. cerevisiae.
| Metabolite (Class) | Plant Source | Key Pathway Genes Expressed | Reported Yield / Effect | Citation |
|---|---|---|---|---|
| Noscapine (Alkaloid) | Papaver somniferum | CYP719A21, CYP82Y1, PsMT1, PsAT1 (10 genes total) | Successful de novo production in yeast via stepwise pathway expansion | [52] |
| UCP1 (Membrane Protein) | Rattus norvegicus | UCP1 (model protein) | 0.2 mg/L purified, active protein; 70% solubilization efficiency with low galactose induction | [53] |
| GFP-ATP7B (Membrane Protein) | Homo sapiens | GFP-ATP7B (model protein) | Optimal production at 0.0015% galactose, enhancing solubilization | [53] |
| 5,10-Diketo-casbene (Diterpene) | Oryza sativa | OsTPS28, OsCYP71Z2, OsCYP71Z21 | Functional characterization achieved via in vitro assays with yeast microsomes | [52] |
The Scientist's Toolkit: Research Reagent Solutions
A successful pathway reconstitution project relies on a suite of reliable reagents and tools. The following table details essential components and their functions.
Table 2: Key research reagents and materials for pathway reconstitution in yeast.
| Reagent / Material | Function / Description | Example Use Case |
|---|---|---|
| pYeDP60 Vector | An episomal plasmid with a strong, inducible GAL10-CYC1 promoter and URA3 selection marker. | Used for controlled, high-level expression of heterologous genes, such as UCP1 [53]. |
| W303.1b/Gal4 Strain | An engineered S. cerevisiae strain with a GAL4 transactivator integrated at the GAL10 locus for enhanced regulation of GAL promoters. | Provides tight control over induction for toxic or hard-to-express proteins [53]. |
| S-Lactate Medium | A defined culture medium used for metabolic studies and protein production. | Used in mitochondrial protein production protocols; can be supplemented with 0.1% casamino acids to relieve metabolic burden and boost yield [53]. |
| DDM Detergent | n-Dodecyl-β-D-maltopyranoside, a mild, non-ionic detergent. | Effective for solubilizing functional membrane proteins from yeast membranes at a 10:1 (w:w) detergent-to-protein ratio [53]. |
| Codon Optimization Tools | In silico services (e.g., from IDT or Thermo Fisher) that redesign gene sequences for optimal expression in S. cerevisiae. | Increased extracellular activity of T. emersonii α-amylase by 1.6-fold [31]. |
Pathway Visualization and Functional Analysis
The functional analysis of a reconstituted pathway often reveals interconnected molecular events. The diagram below illustrates a discovered genetic interaction where two SNPs activated a latent metabolic pathway, leading to a enhanced phenotype.
Diagram Interpretation: This diagram is based on a study investigating the interaction between two causal SNPs, MKT189G and TAO34477C [54]. Individually, each SNP activates distinct, independent pathways (e.g., mitochondrial signaling or the TCA cycle), leading to a moderate increase in sporulation efficiency. However, only when combined in the MMTT strain do these SNPs interact to activate a unique, latent pathway—arginine biosynthesis. This new metabolic activity, not present in the single-SNP strains, provides a unique metabolic advantage that dramatically enhances the final phenotype, demonstrating how genetic interactions can rewire core metabolism in a yeast host [54].
The burgeoning demand for recombinant proteins in the biopharmaceutical, industrial enzyme, and sustainable food sectors has positioned microbial cell factories, particularly the yeast Saccharomyces cerevisiae, at the forefront of bioproduction technology [55]. As a generally recognized as safe (GRAS) organism with a well-characterized genetics, eukaryotic protein folding machinery, and capacity for post-translational modifications, S. cerevisiae is an indispensable host for complex heterologous proteins [55] [11]. However, a significant bottleneck constraining the industrial scalability of yeast-based production is the inherent inefficiency of its secretory pathway, often resulting in poor yields of extracellular protein [56]. The secretory pathway is a complex sequence of intracellular processes—encompassing transcription, translation, endoplasmic reticulum (ER) folding, Golgi processing, and vesicular trafficking—that must operate in concert to ensure efficient protein export [55] [11]. Breaks in this chain, such as ER stress-induced misfolding, inefficient vesicle transport, or proteolytic degradation, can lead to intracellular accumulation and diminished extracellular titers [56] [17]. This Application Note details targeted genetic engineering and automated screening protocols designed to alleviate these bottlenecks, providing researchers with a methodological framework for enhancing recombinant protein secretion in yeast cell factories. The strategies outlined herein are framed within the broader thesis that rational host strain engineering, coupled with systems-level metabolic optimization, is pivotal for unlocking the full potential of heterologous pathway expression.
Key Engineering Targets and Quantitative Outcomes
Engineering the secretory pathway requires a multi-faceted approach. The following table summarizes key genetic targets, their physiological roles, and the documented impact on protein secretion.
Table 1: Key Genetic Engineering Targets for Enhanced Secretion in S. cerevisiae
| Target Gene | Modification | Biological Function | Effect on Secretion | Experimental Context |
|---|---|---|---|---|
| PAH1 | Knockout | Phosphatidate phosphatase; negative regulator of phospholipid biosynthesis [56] | ↑ 74% Ovalbumin secretion (5.68 mg/L) [56] | Recombinant ovalbumin production [56] |
| GOS1 | Knockout | Golgi-to-ER SNARE protein; mediates retrograde trafficking [56] | Enhanced secretion; reduced ER retention [56] | Recombinant ovalbumin production [56] |
| VPS5 | Knockout | Component of retromer complex; endosome-to-Golgi retrograde transport [56] | Complete secretion failure; intracellular OVA accumulation [56] | Recombinant ovalbumin production [56] |
| ERG26 | Overexpression | Sterol biosynthesis enzyme [57] | ↑ 2- to 5-fold Verazine production [57] | Verazine biosynthetic pathway screening [57] |
| DGA1 | Overexpression | Diacylglycerol acyltransferase; lipid droplet formation [57] | ↑ 2- to 5-fold Verazine production [57] | Verazine biosynthetic pathway screening [57] |
| INO2 | Overexpression | Transcription factor activator of phospholipid biosynthesis [56] | Promotes ER membrane biogenesis [56] | Recombinant ovalbumin production [56] |
| ERV29 | Overexpression | COPII vesicle cargo receptor [56] | Facilitates ER export of soluble proteins [56] | Recombinant ovalbumin production [56] |
Visualizing the Engineered Secretory Pathway
The following diagram illustrates the eukaryotic secretory pathway in S. cerevisiae, highlighting the key genetic engineering targets and their points of intervention to enhance heterologous protein export.
Diagram Title: Engineered Secretory Pathway in S. cerevisiae
Detailed Experimental Protocols
Protocol 1: CRISPR/Cas9-Mediated Gene Knockout for Secretory Pathway Engineering
This protocol details the steps for targeted gene knockout (e.g., PAH1, GOS1) in S. cerevisiae using a CRISPR/Cas9 system to alleviate secretory constraints [56].
Key Research Reagent Solutions:
- Plasmid pCas9_AUR: Harbors the CRISPR/Cas9 system for targeted DNA cleavage [56].
- Guide RNA (gRNA) Plasmids: pgRNA-TRP1-HYB plasmid backbone for expressing target-specific gRNAs [56].
- Repair DNA Fragments: PCR-amplified DNA fragments with 50 bp homology arms flanking the target deletion region (56-200 bp within the ORF) [56].
- Yeast Strain: S. cerevisiae D452-2 or other suitable expression host [56].
- Transformation Kit: Yeast EZ-transformation kit [56].
Methodology:
- gRNA Plasmid Construction: Design primers with 20 bp target sequences for the gene of interest. Amplify the pgRNA-TRP1-HYB plasmid and assemble using a NEBuilder HiFi DNA Assembly Master Mix to create the final gRNA plasmid [56].
- Repair Template Preparation: Generate repair DNA fragments via PCR to precisely delete the target genomic region [56].
- Yeast Transformation: Co-transform the competent S. cerevisiae strain (harboring pCas9_AUR) with the constructed gRNA plasmid and the corresponding repair DNA fragment using the yeast EZ-transformation kit [56].
- Selection and Validation: Plate transformants on solid yeast synthetic complete (YSC) medium lacking tryptophan to select for gRNA plasmid integration. Confirm successful gene knockout via colony PCR and DNA sequencing [56].
Protocol 2: Automated High-Throughput Strain Construction and Screening
This protocol describes an automated pipeline for building and screening yeast strain libraries, enabling rapid identification of genes that enhance the production of target compounds [57].
Key Research Reagent Solutions:
- Robotic Platform: Hamilton Microlab VANTAGE with iSWAP robotic arm and VENUS software [57].
- Off-deck Hardware: Integrated 96-well thermocycler (e.g., Inheco ODTC), plate sealer (e.g., 4titude_a4S), and plate peeler (e.g., HSLBrooksAutomationXPeel) [57].
- Transformation Reagents: Optimized lithium acetate/ssDNA/PEG mixture in a 96-well format [57].
- Competent Yeast & Plasmid Library: Chemically competent yeast and a plasmid library (e.g., pESC-URA with genes under pGAL1 promoter) [57].
Methodology:
- Workflow Setup: Program the robotic platform with a modular protocol: "Transformation set up and heat shock," "Washing," and "Plating." Key parameters (DNA volume, reagent ratios, heat shock time) are customizable via a user interface [57].
- Deck Preparation: Load the robotic deck with labware containing competent yeast cells, plasmid DNA library, and transformation reagents as per predefined positions [57].
- Automated Execution: Initiate the run. The system automatically pipettes reagents, performs heat shock by shuttling plates to the off-deck thermocycler, and seals/peels plate seals as needed. The entire process for a 96-well plate takes approximately 2 hours [57].
- Downstream Processing: The output is plated and compatible with automated colony picking (e.g., QPix 460). Picked colonies are inoculated in 96-deep-well plates for high-throughput culturing and analysis (e.g., LC-MS for metabolite quantification) [57].
The workflow for this automated pipeline is visualized below.
Diagram Title: Automated DBTL Cycle for Strain Engineering
The Scientist's Toolkit
Table 2: Essential Research Reagents and Hardware for Secretion Engineering
| Category | Item | Function/Application |
|---|---|---|
| Molecular Biology Tools | CRISPR/Cas9 System (pCas9_AUR, gRNA plasmids) [56] | Targeted genome editing for gene knockout/overexpression. |
| NEBuilder HiFi DNA Assembly Master Mix [56] | Cloning and assembly of DNA fragments with high efficiency. | |
| pESC-URA Plasmid Series [57] | Galactose-inducible expression vectors for pathway engineering. | |
| Strains and Culture | S. cerevisiae D452-2 [56] | A common expression host for recombinant protein production. |
| Yeast EZ-transformation Kit [56] | Facilitates efficient plasmid introduction into yeast cells. | |
| YSC, YPDA Media [56] [57] | Defined and rich media for yeast cultivation and selection. | |
| Analytical Equipment | Liquid Chromatography-Mass Spectrometry (LC-MS) [57] | Quantification of target metabolite titers from culture extracts. |
| Pulsed-Field Gel Electrophoresis (PFGE) [58] | Validation of large-scale chromosomal rearrangements. | |
| Automation & Hardware | Hamilton Microlab VANTAGE [57] | Robotic liquid handling platform for high-throughput workflows. |
| QPix 460 Automated Colony Picker [57] | Enables rapid screening and picking of thousands of colonies. | |
| 96-well Thermal Cycler, Plate Sealer/Peeler [57] | Off-deck hardware integrated for automated heat shock steps. |
Maximizing Titers and Overcoming Production Bottlenecks
Carbon Source Selection and Management Across the Diauxic Shift
The diauxic shift in Saccharomyces cerevisiae represents a critical metabolic transition from fermentative to respiratory growth, occurring upon depletion of preferred sugars like glucose [59]. For researchers engineering yeast cell factories, this shift presents both a challenge and an opportunity. The metabolic rewiring involves massive transcriptional reprogramming and changes in protein-metabolite interactions, significantly impacting heterologous pathway performance [59] [60]. Proper management of carbon sources before, during, and after this transition is therefore essential for optimizing yields, rates, and titers in industrial bioprocesses.
This Application Note provides detailed protocols and data frameworks for controlling heterologous gene expression by strategically leveraging carbon source selection and understanding the underlying regulatory hierarchies. We present quantitative promoter performance data across different metabolic phases and experimental approaches for analyzing and manipulating the diauxic shift in engineered strains.
Established Sugar Hierarchies Beyond Glucose
While glucose repression is a well-documented phenomenon, recent research reveals that yeast actively prioritize other carbon sources through specific regulatory mechanisms, not merely based on which sugars support the fastest growth rates [61].
Table 1: Sugar Preferences and Growth Characteristics in S. cerevisiae
| Sugar | Transport System | Relative Growth Rate | Preference over Palatinose | Key Regulatory Elements |
|---|---|---|---|---|
| Glucose | Hexose transporters | High [61] | Strong preference [61] | Multiple repression mechanisms |
| Galactose | Hexose transporters | Intermediate [61] | Strong preference [61] | GAL regulon (Gal4p) [61] |
| Fructose | Hexose transporters | High [61] | Weak or no preference [61] | Limited repression capability |
| Sucrose | Primarily extracellular hydrolysis | High [61] | Weak or no preference [61] | Limited repression capability |
| Palatinose | MAL regulon (proton symport) | Lower [61] | Reference | MAL regulon (Mal13p, Znf1p) [61] |
The preference for galactose over palatinose is actively enforced through repression of MAL genes by Gal4p, the master regulator of the GAL regulon [61]. This repression occurs through two primary mechanisms: downregulation of the MAL11 palatinose transporter gene, and expression of isomaltases IMA1 and IMA5 that catabolize palatinose [61]. This hierarchy is not determined solely by the growth rate each sugar supports, as fructose and sucrose enable faster growth than palatinose yet are not strongly preferred over it [61].
Regulatory Network Governing Carbon Source Decisions
The following diagram illustrates the key regulatory interactions that determine carbon source prioritization during the diauxic shift:
Diagram 1: Regulatory network of carbon source utilization. This map illustrates the key interactions that determine sugar preferences, highlighting how Gal4p mediates cross-regulation between galactose and palatinose utilization genes [61].
Promoter Selection Strategies for Different Metabolic Phases
Selection of appropriate promoters is crucial for maintaining predictable heterologous expression throughout fermentation. Performance varies significantly across carbon sources and metabolic phases.
Table 2: Promoter Activity Across Carbon Sources and Growth Phases [27] [43]
| Promoter | Type | Glucose | Sucrose | Galactose | Ethanol | Diauxic Shift Response |
|---|---|---|---|---|---|---|
| P_TDH3 | Constitutive (Glycolytic) | High | High | High | Low | Sharp decrease post-shift |
| P_ENO2 | Constitutive (Glycolytic) | High | High | High | Low | Sharp decrease post-shift |
| P_ADH1 | Constitutive (Glycolytic) | High | High | High | Low | Sharp decrease post-shift |
| P_PGK1 | Constitutive (Glycolytic) | Medium-High | Medium-High | Medium-High | Low | Sharp decrease post-shift |
| P_TEF1 | Constitutive (Translation) | Medium | Medium | Medium | Low | Decrease post-shift |
| P_TEF2 | Constitutive (Translation) | Medium | Medium | Medium | Low | Decrease post-shift |
| P_GAL1 | Inducible (Galactose) | Low | Low | Very High | Low | Strong induction on galactose |
| P_SSA1 | Low-glucose inducible | Low | Medium | Medium | High | Increases during/post-shift |
| P_HXT7 | Low-glucose inducible | Low | Medium | Medium | Medium | Increases during/post-shift |
| P_ADH2 | Low-glucose inducible | Low | Medium | Medium | High | Increases during/post-shift |
| P_CUP1 | Chemically inducible | Low | Low | Low | Medium | Inducible post-shift with Cu²⁺ |
Strategic Promoter Deployment Framework
For predictable heterologous pathway expression:
- Rapid biomass accumulation phase: Utilize strong constitutive promoters (PTDH3, PENO2, P_ADH1) when the goal is maximum protein production during exponential growth on fermentable sugars [27] [43].
- Transition phase: Employ low-glucose inducible promoters (PSSA1, PHXT7, P_ADH2) to maintain expression as glucose depletes and cells prepare for metabolic restructuring [27] [43].
- Respiratory phase: For production during ethanol utilization, PSSA1 provides higher expression than traditional constitutive promoters, while PCUP1 offers chemically inducible control [27] [43].
- Galactose-based processes: P_GAL1 provides extremely high, specific expression on galactose, making it ideal for pathways where tight control is needed [27] [43].
Experimental Protocols
Protocol 1: Characterizing Diauxic Growth in Sugar Mixtures
Purpose: To quantitatively analyze yeast growth and sugar preference in mixed carbon source environments.
Materials:
- Yeast strains (e.g., BY4741 or engineered derivatives)
- SC medium with appropriate auxotrophic supplements
- Sugar stocks: glucose, galactose, fructose, sucrose, palatinose
- Plate reader with temperature control and shaking
- HPLC system or metabolomics platform for extracellular metabolite quantification
Procedure:
- Prepare pre-cultures in SC medium with 2% glucose and grow overnight at 30°C with shaking at 220 rpm.
- Dilute cultures to OD600 = 0.05 in fresh SC medium containing mixtures of sugars (e.g., 0.5% galactose + 1% palatinose).
- Transfer 200 µL aliquots to 96-well plates and incubate in plate readers with continuous shaking at 30°C.
- Measure OD600 every 15 minutes for 48-72 hours.
- For GFP-tagged strains (e.g., IMA1-GFP, IMA5-GFP), include fluorescence measurements to monitor gene expression dynamics.
- Use software such as
omniplatefor automated growth curve analysis, correcting for nonlinear OD-cell density relationships [61]. - Extract growth rates using Gaussian processes to identify local maxima corresponding to consumption of different sugars [61].
- For metabolite tracking, perform parallel flask cultures and sample periodically for extracellular sugar quantification via HPLC or metabolomics [61].
Data Analysis:
- Identify ODswitch at the local minimum of growth rate between two growth phases [61].
- Calculate growth yields for each phase: (ODswitch - initial OD) correlates with first sugar concentration, (final OD - ODswitch) correlates with second sugar concentration [61].
- Determine sugar consumption profiles from metabolite data to confirm sequential utilization.
Protocol 2: Transcriptomic Analysis During Metabolic Transitions
Purpose: To monitor global gene expression changes and heterologous pathway performance across the diauxic shift.
Materials:
- Yeast strains (wild-type and engineered)
- SC medium with 2% glucose
- RNA extraction kit (e.g., RNeasy from QIAGEN)
- RNA quality assessment tools (NanoDrop, BioAnalyzer)
- RNAseq library preparation kit (e.g., Illumina TruSeq Stranded mRNA)
- Sequencing platform
Procedure:
- Grow pre-cultures overnight in SC medium with 2% glucose at 30°C, 220 rpm.
- Dilute to OD600 = 0.05 in fresh medium and incubate in baffled shake flasks at 30°C, 220 rpm.
- Sample cultures at key time points:
- Exponential phase (OD600 ~1-2, glucose-replete)
- Diauxic shift (OD600 at growth rate minimum)
- Post-diauxic phase (ethanol utilization, OD600 stable or increasing slowly) [62]
- Harvest 1.0-1.5 mL cell broth equivalent to OD600 = 10 by centrifugation (5,000 rcf, 5 min).
- Extract RNA immediately using RNeasy kit, elute in 30 µL RNase-free water, and store at -80°C.
- Verify RNA quality (RIN > 8.0) and quantity before library preparation.
- Prepare libraries using TruSeq Stranded mRNA kit and sequence with minimum 30 million reads per sample on Illumina platform [62].
- For targeted analysis, use qRT-PCR to monitor specific heterologous pathway genes.
Data Analysis:
- Perform differential expression analysis comparing different time points or engineered vs. reference strains.
- Conduct gene set enrichment analysis to identify affected pathways (e.g., mitochondrial translation, glycolytic processes) [62].
- Use transcription factor enrichment analysis to identify regulatory mechanisms (e.g., HAP-complex, Mig1p targets) [62].
Metabolic Engineering Applications
Engineering Crabtree-Negative Phenotypes for Improved Product Yields
Manipulating sugar phosphorylation presents a powerful strategy for altering yeast central metabolism and redirecting carbon flux from ethanol to desired products:
Diagram 2: Metabolic engineering to attenuate the Crabtree effect. Strategic interventions at sugar uptake and phosphorylation enable redirection of carbon flux from ethanol to valuable products [63].
Key engineering approaches include:
- Hexose transporter engineering: Replace multiple HXT genes with a single chimeric transporter to limit glucose uptake and prevent carbon overflow to ethanol [63].
- MTH1 manipulation: Utilize MTH1-ΔT or MTH1A81D mutants to decrease hexose transporter expression and attenuate the Crabtree effect [63].
- Pyruvate decarboxylase elimination: Create Pdc− strains to abolish ethanol formation, then introduce alternative acetyl-CoA bypass pathways (pyruvate oxidase + phosphotransacetylase or ATP-citrate lyase) [63].
- Dynamic regulation of sugar phosphorylation: Implement sensor-driven autoregulation of sugar phosphorylation for more resilient control across different bioprocess conditions [63].
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Diauxic Shift Studies
| Reagent/Strain | Function/Application | Key Features | Example Sources |
|---|---|---|---|
| BY4741 strain | Reference strain for physiological studies | Prototrophic, well-characterized | EUROSCARF [62] |
| Yeast knockout collection | Gene function analysis | Systematic single-gene deletions | EUROSCARF [62] |
| P_GAL1 vectors | Tightly regulated heterologous expression | Strong, galactose-specific induction | Commercial suppliers [27] [43] |
| PTDH3/PENO2 vectors | Constitutive high-level expression | Strong activity during exponential growth | Commercial suppliers [27] [43] |
| P_SSA1 vectors | Post-diauxic expression | Induced during ethanol utilization phase | Commercial suppliers [27] [43] |
| yEGFP reporters | Promoter activity quantification | Fluorescent reporter for expression levels | Commercial suppliers [27] [43] |
| HPLC systems | Extracellular metabolite quantification | Sugar and ethanol measurement | Various manufacturers [61] |
| RNAseq kits | Transcriptomic analysis | Global gene expression profiling | Illumina, QIAGEN [62] |
Strategic carbon source management across the diauxic shift requires understanding both the native regulatory hierarchies and the tools available for engineering controllable expression systems. The data and protocols presented here enable researchers to make informed decisions about promoter selection, strain engineering, and process optimization for heterologous pathway expression in yeast. By aligning expression timing with metabolic capacity through careful carbon source selection and engineered control systems, significant improvements in product titers, yields, and productivity can be achieved in yeast-based biomanufacturing platforms.
The engineering of microbial cell factories, particularly the yeast Saccharomyces cerevisiae, for heterologous pathway expression is a cornerstone of modern industrial biotechnology, enabling the production of pharmaceuticals, biofuels, and specialty chemicals [36] [21]. A critical challenge in this field is balancing the metabolic burden between robust host cell growth and high-yield production of the target compound. Dynamic metabolic control strategies, which decouple the growth phase from the production phase, offer a powerful solution to this challenge [38]. Central to these strategies are inducible promoters, which allow for precise, external regulation of gene expression.
Inducible promoters initiate transcription in response to specific environmental stimuli or chemical inducers [38]. Unlike constitutive promoters, which provide a constant level of expression, inducible systems provide temporal control, enabling researchers to first build up high cell density before activating resource-intensive heterologous pathways. This control is essential for expressing genes that might be toxic to the host or for optimizing the flux through engineered pathways. The low-glucose promoter is one such system, naturally activated as the readily available carbon source is depleted, triggering a metabolic shift that can be harnessed to drive product synthesis. This protocol details the application of low-glucose and other inducible promoters for dynamic control of heterologous pathways in yeast, providing a framework for enhancing the performance of cell factories in the production of high-value molecules such as terpenoids [36] and other small molecules [21].
Promoter Characteristics and Selection Criteria
Selecting the appropriate promoter is a critical first step in designing a yeast cell factory, as promoter performance is highly dependent on the specific experimental conditions and the protein being expressed [38]. The table below summarizes the key characteristics of common promoters used in yeast metabolic engineering.
Table 1: Key Promoters for Heterologous Expression in Saccharomyces cerevisiae
| Promoter Name | Type | Inducer/Regulation | Typical Application Context | Strengths | Considerations |
|---|---|---|---|---|---|
| ADH2P | Inducible | Low Glucose/Glucose Depletion | Dynamic control of pathways post-growth phase [38] | Strong, native yeast system; avoids catabolite repression | Requires glucose depletion for full induction |
| GAL1/GAL7/GAL10P | Inducible | Galactose | High-level protein production [38] | Very strong, tight regulation | Cost of inducer (galactose); metabolic shift required |
| CUP1P | Inducible | Cu²⁺ Ions | Expression of potentially toxic genes [38] | Tight regulation with simple, cheap inducer | Potential cellular toxicity of copper ions |
| TDH3P | Constitutive | N/A (Strong constitutive) | High-level enzyme expression under various conditions [38] | One of the strongest native yeast promoters | No temporal control; constant metabolic burden |
| TEF1P | Constitutive | N/A (Strong constitutive) | General-purpose protein and pathway expression [21] | Strong, reliable expression levels | No temporal control |
| SED1P | Constitutive | N/A | Expression under various cultivation conditions [38] | Effective performance on non-native substrates like xylan [38] | Constitutive nature may not be ideal for all pathways |
The choice between an inducible and a constitutive promoter depends on the specific goals of the experiment. Constitutive promoters like TDH3P and TEF1P are valuable for their stable expression levels and are often used for genes encoding central metabolic enzymes or when constant, moderate expression is desirable [38] [21]. However, for dynamic metabolic control, inducible promoters are superior. The low-glucose promoter ADH2P is particularly useful because its induction is linked to the depletion of the primary carbon source, creating a natural transition from growth to production without the need for expensive inducers. It is crucial to test promoter performance under the intended fermentation conditions, as strength and regulation can be unpredictable and are highly context-dependent [38].
Experimental Protocol: Characterization and Application of a Low-Glucose Inducible System
This protocol describes a method for characterizing a low-glucose inducible promoter and applying it to control a heterologous pathway in S. cerevisiae. The example involves controlling the expression of a terpenoid biosynthetic pathway [36], but the principles can be adapted for other products.
Materials and Reagents
Table 2: Research Reagent Solutions and Essential Materials
| Item | Function/Description | Example or Specification |
|---|---|---|
| S. cerevisiae Strain | Host organism for heterologous pathway expression. | e.g., CEN.PK113-7D or other lab strain with relevant auxotrophic markers [21]. |
| Expression Plasmid | Vector containing the gene of interest under control of the inducible promoter. | Contains ADH2P upstream of your heterologous gene and a selectable marker (e.g., URA3). |
| CRISPR/Cas9 System | For genomic integration of expression cassettes [38]. | Cas9, gRNA, and donor DNA template for targeted integration. |
| Synthetic Complete (SC) Medium | Defined medium for selective growth of transformed yeast. | Lacks specific amino acids/nucleotides to maintain plasmid or genomic integration selection. |
| High-Glucose Feed | Initial growth phase carbon source. | 20 g/L glucose in batch culture. |
| Low-Glucose Feed/Stat | Induction phase carbon source. | < 2 g/L glucose, often maintained via fed-batch system or a single batch transition. |
| qPCR Reagents | For quantifying mRNA expression levels of the target gene. | Primers for target gene and housekeeping genes (e.g., ACT1). |
| Enzymatic Assay Kits | For quantifying extracellular glucose concentration. | Glucose oxidase-based assay kit. |
| Analytical Chemistry Equipment | For quantifying the final product titer. | HPLC or GC-MS for terpenoid analysis [36]. |
Step-by-Step Methodology
Step 1: Strain Construction and Pathway Integration
- Design Expression Cassette: Clone your heterologous gene(s) of interest (e.g., a terpene synthase) downstream of the ADH2 promoter (ADH2P) in an expression plasmid or a donor DNA template for genomic integration. Use a strong terminator such as
CYC1orADH1[21]. - Transform Yeast: Introduce the expression cassette into your chosen S. cerevisiae host strain. For genomic integration, the CRISPR/Cas9 system is recommended for its precision and efficiency [38]. For a multi-gene pathway, consider integrating genes into well-characterized genomic loci (e.g.,
X-2,X-4, orXII-5) known to support high and stable expression [21]. - Validate Integrants: Confirm correct genomic integration via colony PCR and verify the absence of unwanted mutations by sequencing the integration site.
Step 2: Cultivation and Induction Regime
- Pre-culture: Inoculate a single colony of the engineered yeast strain into 5 mL of SC medium with high glucose (20 g/L) and the appropriate selective pressure. Incubate overnight at 30°C with shaking at 200-250 rpm.
- Main Culture: Dilute the pre-culture into fresh SC medium with high glucose to an initial OD600 of ~0.1 in a baffled flask. Continue incubation under the same conditions.
- Monitor Growth and Glucose: Periodically sample the culture to measure OD600 and extracellular glucose concentration using an enzymatic assay.
- Induction Phase: When the glucose concentration falls below 2 g/L (typically in mid-to-late exponential phase), the ADH2P will begin to induce. To maintain induction, you can either:
- Allow the culture to enter a stationary phase maintained by the residual, low level of glucose.
- Initiate a fed-batch process with a controlled feed of low-glucose medium to prolong the production phase.
Step 3: Sampling and Analytical Procedures
- Sample Collection: Collect samples at regular intervals before, during, and after induction (e.g., every 2-3 hours). Centrifuge samples to separate cells (pellet) and supernatant.
- mRNA Quantification (qPCR): Extract total RNA from cell pellets. Synthesize cDNA and perform qPCR with primers specific to your heterologous gene and a housekeeping gene for normalization. This data will generate a profile of gene expression dynamics relative to glucose concentration.
- Product Titer Analysis: Extract the product (e.g., terpenoid) from the cell pellet or supernatant using an appropriate organic solvent (e.g., ethyl acetate or hexane for hydrophobic compounds). Analyze the extract using HPLC or GC-MS to quantify the titer and calculate the yield [36].
Data Analysis and Interpretation
Plot the glucose concentration, OD600 (biomass), mRNA level of the target gene, and product titer over time. A successful dynamic control experiment will show a clear correlation: high biomass accumulation during the high-glucose phase, followed by a sharp increase in target gene mRNA and subsequent product accumulation as glucose depletes and ADH2P is induced. Compare the final titer and yield to a control strain using a strong constitutive promoter (e.g., TDH3P) to quantify the improvement afforded by dynamic control.
Pathway Visualization and Experimental Workflow
The following diagrams, generated using Graphviz DOT language, illustrate the logical flow of the low-glucose induction mechanism and the experimental protocol.
Low-Glucose Promoter Induction Logic
Experimental Workflow for Promoter Characterization
Advanced Applications and Synergistic Strategies
The utility of dynamic promoters can be significantly enhanced by combining them with other metabolic engineering strategies.
- Multi-Promoter Tuning for Pathway Balancing: In a multi-gene heterologous pathway, different inducible and constitutive promoters can be used in combination to balance the flux and avoid the accumulation of toxic intermediates [36]. For instance, a rate-limiting enzyme could be placed under a strong, glucose-sensitive promoter like ADH2P, while other enzymes are expressed constitutively.
- Chaperone Co-expression for Enzyme Folding: The overexpression of endogenous cytosolic chaperones, such as Ydj1 and Ssa1, has been shown to boost the production of heterologous small molecules in yeast by aiding the proper folding of complex, multi-domain enzymes like non-ribosomal peptide synthetases (NRPS) and polyketide synthases (PKS) [21]. Combining a dynamically induced pathway with the constitutive overexpression of such chaperones can lead to significant yield improvements, as demonstrated by an 84% increase in aspulvinone E production [21].
The implementation of dynamic metabolic control using low-glucose and other inducible promoters represents a sophisticated and highly effective strategy for optimizing heterologous pathway expression in yeast cell factories. By temporally separating cell growth from product synthesis, this approach mitigates metabolic burden and can lead to substantially improved titers of valuable compounds. The protocols outlined here for promoter characterization and application, combined with synergistic strategies like chaperone engineering, provide a robust framework for researchers and industrial scientists to advance the development of efficient microbial production platforms for pharmaceuticals and other bioproducts.
Within the field of yeast metabolic engineering, a paradigm shift is underway, moving from strategies that maximize short-term productivity to those that enhance long-term cellular robustness. Chronological lifespan (CLS), which measures the time a non-dividing cell remains viable and metabolically active, is emerging as a critical determinant for the performance of microbial cell factories in industrial fed-batch fermentations [64]. Engineering a longer CLS automatically remodels cellular metabolism toward a more robust state, leading to significantly improved synthesis of valuable compounds like the diterpenoid sclareol [64]. This application note details the principles and protocols for leveraging lifespan engineering to enhance the biosynthetic capacity of Saccharomyces cerevisiae.
Key Concepts and Evidence
The Link Between Lifespan and Production: Metabolic rewiring for heterologous pathway expression often imposes substantial stress on host cells, leading to premature metabolic decline during long-term cultivation. Research demonstrates that systematically extending CLS directly addresses this issue. In one study, engineering CLS in yeast resulted in a 70.3% improvement in sclareol production, achieving a titer of 20.1 g/L. Further enhancement of central metabolism boosted production to 25.9 g/L, the highest reported titer in microbes at the time [64]. Multi-omics data confirmed that extending CLS via the upregulation of lifespan-related genes led to a comprehensive metabolic remodeling that improved overall cellular robustness [64].
Core Longevity Pathways: The primary metabolic targets for CLS extension are highly conserved nutrient-sensing and stress-response pathways. Key interventions include:
- Weakening nutrient-sensing pathways such as Target of Rapamycin (TOR) and Protein Kinase A (PKA) signaling.
- Enhancing mitophagy, the selective autophagy of damaged mitochondria.
- Activating stress response systems that maintain proteostasis and reduce oxidative damage [65] [64].
Quantitative Data on Lifespan-Extending Interventions
Table 1: Metabolic Pathway Manipulations that Extend Lifespan in Model Organisms
| Target / Intervention | Metabolic Pathway | Effect on Lifespan | Organism |
|---|---|---|---|
| 2-Deoxyglucose (2-DG) | Glycolysis inhibitor | 17% increase [65] | C. elegans |
| fgt-1 (Glucose transporter) | Glucose uptake | 25% increase [65] | C. elegans |
| gfat-1 | Hexosamine pathway | 42% increase [65] | C. elegans |
| Fructose | Carbohydrate metabolism | 45% increase [65] | C. elegans |
| Weakened TOR/Sch9 Signaling | Nutrient sensing / Ribosome biogenesis | Increased [66] | S. cerevisiae |
| Enhanced Mitophagy | Mitochondrial quality control | Synergistic improvement [64] | S. cerevisiae |
Table 2: Production Outcomes from Chronological Lifespan Engineering in Yeast
| Engineering Strategy | Product | Titer (g/L) | Yield (g/g glucose) | Improvement |
|---|---|---|---|---|
| Reference Strain | Sclareol | ~11.8 | ~0.027 | Baseline [64] |
| CLS Engineering (Weakened nutrient-sensing + Enhanced mitophagy) | Sclareol | 20.1 | 0.046 | +70.3% [64] |
| CLS Engineering + Central Metabolism Enhancement | Sclareol | 25.9 | 0.051 | +~119% [64] |
| CLS Engineering Strategy | β-elemene & Phenolic acids | Significantly improved | N/R | Validated [64] |
Signaling Pathways in Lifespan Engineering
The following diagram illustrates the core signaling pathways and their interactions that can be engineered to extend chronological lifespan in yeast.
Experimental Protocols
Protocol: Engineering Yeast for Extended Chronological Lifespan
Objective: Genetically modify S. cerevisiae to weaken nutrient-sensing pathways and enhance mitophagy for improved CLS and biosynthetic capacity.
Materials:
- Yeast strain with heterologous biosynthetic pathway (e.g., for sclareol production)
- CRISPR/Cas9 system for yeast [31]
- Knockout cassettes for TOR1 and SCH9
- Overexpression cassette for mitophagy gene ATG32
- Rich medium (YPD) and synthetic complete (SC) medium
- Microfluidic culturing device or shake flasks [67]
Procedure:
- Strain Development:
- Weaken Nutrient Sensing: Co-transform yeast with CRISPR/Cas9 plasmids and repair templates to delete or downregulate key genes in the TOR and Sch9 pathways [64].
- Enhance Mitophagy: Integrate a strong, constitutive promoter (e.g., pTDH3) upstream of the native ATG32 gene or introduce an extrachromosomal expression cassette [64].
- Verify all genetic modifications by colony PCR and sequencing.
Chronological Lifespan Assay:
- Inoculate single colonies into 5 mL of SC medium with 2% glucose and grow overnight at 30°C.
- Dilute the culture to an OD600 of 0.2 in fresh medium and allow cells to proliferate until they reach the post-diauxic phase (OD600 ~5.0-8.0), marking day 0 of the CLS assay.
- Maintain cultures at 30°C with constant shaking. At regular intervals (e.g., every 2-3 days), vortex cultures and remove aliquots.
- Perform serial dilutions and spot cells onto YPD agar plates to determine the number of colony-forming units (CFUs). Alternatively, use a microfluidic device to track viability and morphological changes in real-time [67].
- Calculate percent viability relative to day 0.
Production Analysis:
- For production strains, cultivate engineered and control strains in production medium.
- At peak production time (determined empirically), extract metabolites from the culture medium using an organic solvent (e.g., ethyl acetate).
- Analyze product titer and yield using HPLC or GC-MS [64].
Protocol: Quantitative Proteomic Analysis of Aging Yeast
Objective: Compare proteomes of young and aged yeast cells under normal and calorie-restricted conditions using SILAC.
Materials:
- BY4741 yeast strain [66]
- SILAC media: SC media with 2% (NR) or 0.05% (CR) glucose, containing either regular Lysine or 13C-labeled Lysine [66]
- Biotinylation reagent
- Magnetic streptavidin beads
- LysC protease
- LTQ-Orbitrap mass spectrometer [66]
- MaxQuant software [66]
Procedure:
- SILAC Labeling:
- Grow the common reference strain in heavy medium (13C-Lys) for >40 doublings to ensure full label incorporation [66].
- Grow experimental cells (young and old, under NR and CR) in light medium (regular Lys).
Old Cell Isolation:
- Label cells from light cultures with biotin.
- Bind biotinylated old cells to magnetic streptavidin beads and separate them from unbiotinylated young cells magnetically.
- Confirm old cell purity by bud scar staining [66].
Sample Preparation and MS Analysis:
- Mix whole-cell extracts from light experimental cells with heavy reference cells at a 1:1 protein ratio.
- Separate proteins on a 4-12% gradient NuPAGE gel and cut the lane into 18 slices.
- Perform in-gel digestion with LysC protease.
- Desalt peptides and analyze by LC-MS/MS on an LTQ-Orbitrap instrument [66].
Data Processing:
- Process raw data with MaxQuant software using the yeast UP000002311 database.
- Identify proteins and quantify heavy/light ratios to determine relative abundance changes in the aging proteome [66].
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Lifespan Engineering
| Reagent / Tool | Function / Application | Key Features / Examples |
|---|---|---|
| CRISPR/Cas9 System | Genome editing for precise gene knockout (e.g., TOR1, SCH9) or gene insertion. | Enables efficient and multiplexed genetic modifications in yeast [31]. |
| Microfluidic Devices | Automated, high-throughput replicative and chronological lifespan analysis. | Allows real-time, single-cell monitoring of aging and physiology [67]. |
| SILAC (Stable Isotope Labeling by Amino acids in Cell culture) | Quantitative proteomic analysis of aging cells. | Uses 13C-labeled lysine to compare protein abundance between young and old cells [66]. |
| Metabolomic Aging Clocks | Profiling metabolite levels to assess biological age and metabolic health. | NMR-based platforms can identify aging-induced shifts like NAD+ depletion [68]. |
| Hyperexpression Systems | Maximizing expression of heterologous pathway enzymes and longevity factors. | Combines codon optimization, strong promoters, and increased gene copy number [31]. |
| Compartmentalization Tools | Targeting pathways to organelles like peroxisomes. | Enhances local substrate concentration and shields toxic intermediates [69]. |
The efficient expression of heterologous pathways in yeast cell factories is a cornerstone of modern industrial biotechnology, enabling the sustainable production of therapeutics, nutraceuticals, and biofuels. A significant barrier to achieving high-yield production lies in sequence-based incompatibilities between the introduced genes and the host's translational machinery and genomic architecture. Two critical factors demanding meticulous attention are codon optimization—the strategic matching of synonymous codons to the host's preference—and the management of AT-rich regions, which can destabilize DNA and form inhibitory secondary structures in mRNA. This Application Note provides a structured framework and detailed protocols to address these factors in Saccharomyces cerevisiae, a premier microbial chassis, thereby enhancing protein expression, maximizing metabolic flux, and improving the overall performance and predictability of engineered yeast strains.
Core Principles and Quantitative Analysis
Codon Optimization Strategies
Codon optimization moves beyond simple synonymity, employing various algorithms to refine gene sequences for a heterologous host. The following strategies are foundational [70]:
- Codon Usage Tables: Replacing rare codons with those most frequently used in the host's highly expressed genes.
- Codon Adaptation Index (CAI): A quantitative measure (0 to 1) of how well a gene's codon usage matches the host preference. A higher CAI correlates with a higher potential for expression.
- Codon Pair Bias (CPB) Optimization: Refining the non-random pairing of adjacent codons to enhance translational efficiency and accuracy.
- GC Content and mRNA Secondary Structure Control: Adjusting the guanine-cytosine content and avoiding sequences that form stable secondary structures (e.g., hairpins) in the 5' end, which can impede translation initiation [71].
Comparative Analysis of Codon Optimization Tools
The selection of an optimization tool significantly impacts the final sequence and expression outcome. An analysis of prominent tools reveals distinct strategic emphases. The following table summarizes key parameters and outputs for the optimization of a theoretical gene across three industrially relevant hosts [71].
Table 1: Performance of Codon Optimization Tools Across Different Host Organisms
| Tool / Parameter | E. coli | S. cerevisiae | CHO Cells | |||
|---|---|---|---|---|---|---|
| CAI | GC% | CAI | GC% | CAI | GC% | |
| JCat | 0.89 | 54.2 | 0.91 | 40.1 | 0.87 | 52.5 |
| OPTIMIZER | 0.87 | 53.8 | 0.90 | 39.8 | 0.86 | 51.9 |
| ATGme | 0.88 | 54.5 | 0.92 | 40.3 | 0.88 | 52.8 |
| TISIGNER | 0.85 | 52.1 | 0.88 | 38.5 | 0.84 | 50.2 |
| IDT | 0.86 | 53.0 | 0.89 | 39.2 | 0.85 | 51.0 |
| GeneOptimizer | 0.90 | 54.0 | 0.93 | 40.5 | 0.89 | 52.0 |
The Challenge of AT-Rich Regions
AT-rich sequences, while common in native genes from certain organisms, are problematic in yeast. They are associated with [72]:
- Genomic Instability: Acting as hotspots for DNA deletion and recombination events.
- Transcriptional Issues: Potential unintended transcription termination.
- mRNA Destabilization: Reduced transcript half-life. Furthermore, AT-richness in the coding sequence often correlates with low GC content, which can exacerbate the formation of stable mRNA secondary structures that occlude ribosome binding and impede translation elongation [71].
Experimental Protocols
Protocol: A Multi-Step Codon Optimization and Validation Pipeline
This protocol outlines a comprehensive workflow for designing, synthesizing, and validating codon-optimized genes for expression in S. cerevisiae.
I. Gene Optimization and In Silico Analysis
- Input Sequence: Provide the amino acid sequence or native nucleotide sequence of the heterologous gene.
- Tool Selection: Utilize multiple algorithms from Table 1 (e.g., JCat, OPTIMIZER, ATGme) to generate several candidate optimized sequences.
- Parameter Analysis: For each candidate sequence, calculate and compare the CAI, GC content, and Codon Pair Bias (CPB) using resources like the CAI Calculator [71] and codon usage tables for S. cerevisiae (e.g., from the GEO dataset GSE208095).
- mRNA Structure Prediction: Analyze the 5' end of the coding sequence (approximately ~50 codons) for stable secondary structures using RNAfold [73] or similar tools. Select candidates with lower predicted stability (higher ΔG) to ensure efficient translation initiation.
- Sequence Finalization: Choose the top 2-3 candidate sequences that exhibit a balance of high CAI (>0.9), host-appropriate GC content (~40%), and minimal 5' secondary structure.
II. Gene Synthesis and Cloning
- Synthesis: Order the full-length, optimized gene sequences as clonal DNA fragments from a commercial supplier.
- Vector Assembly: Clone each synthesized gene into a yeast expression vector (e.g., a CEN/ARS plasmid with a strong constitutive promoter like PTEF1 and a selectable marker) using standard assembly techniques (e.g., Gibson Assembly).
- Transformation: Transform the assembled plasmids into a standard S. cerevisiae laboratory strain (e.g., CEN.PK) and select on appropriate solid medium.
III. In Vivo Validation
- Culturing: Inoculate single colonies of transformed yeast into liquid selection medium and culture to mid-log phase.
- mRNA Quantification: Harvest cells and extract total RNA. Perform reverse transcription followed by quantitative PCR (RT-qPCR) using primers specific to the heterologous gene and a reference housekeeping gene (e.g., ACT1). This assesses transcript abundance and stability.
- Protein Analysis:
- Western Blot: Use protein-specific antibodies to detect and semi-quantify the target protein levels.
- Fluorescence Assay: If the target protein is a fluorescent fusion (e.g., MelA-mRFP [21]), measure fluorescence intensity directly from cell cultures using a plate reader.
- Functional Assay: Perform a growth-based or enzymatic assay specific to the protein's function to confirm biological activity.
Protocol: Identification and Engineering of AT-Rich Regions
This protocol describes a method to identify problematic AT-rich segments and engineer them for stable expression.
I. Sequence Identification and Analysis
- Sequence Scan: Input the heterologous gene sequence into a bioinformatics tool (e.g., using a custom Python script with Biopython) to scan for regions where the AT content exceeds 70% over a sliding window of 50 base pairs.
- MFE Calculation: For each identified AT-rich window, use an RNA folding program (e.g., RNAfold) to predict the Minimum Free Energy (MFE) of the corresponding mRNA sequence. Regions with highly negative MFE are likely to form stable secondary structures.
II. In Silico Engineering and Synthesis
- Synonymous Codon Replacement: For each problematic AT-rich region, use a codon optimization tool that allows for user-defined constraints to replace AT-rich codons with GC-rich synonymous codons that are still common in S. cerevisiae. The goal is to reduce the local AT content to below 60% without altering the amino acid sequence.
- Structure Re-check: Re-run the MFE prediction on the engineered sequence to verify the reduction in mRNA secondary structure stability.
- Full Gene Synthesis: Synthesize the entire engineered gene, which incorporates the optimized codons for both general usage and AT-rich remediation.
III. Experimental Validation
- Plasmid Stability Assay: Co-transform yeast with the plasmid containing the wild-type (AT-rich) gene and the engineered version. Passage the cultures serially for ~50-60 generations without selection.
- Plasmid Loss Rate: At intervals, plate cells on non-selective and selective media to count the number of plasmid-retaining colonies. A lower plasmid loss rate for the engineered construct indicates improved genomic stability.
- Expression Comparison: Compare the protein expression yield of the engineered gene versus the wild-type version using the methods described in Protocol 3.1, Part III.
Workflow Visualization
The following diagram illustrates the logical and experimental workflow integrating the protocols for codon optimization and AT-rich region management.
The Scientist's Toolkit: Research Reagent Solutions
Successful implementation of these protocols relies on key reagents and tools. The following table details essential solutions for codon optimization and heterologous expression in yeast.
Table 2: Key Research Reagents and Materials
| Item | Function / Application | Example / Specification |
|---|---|---|
| Codon Optimization Tools | Generate host-specific optimized nucleotide sequences. | JCat, OPTIMIZER, ATGme, GeneOptimizer [71]. |
| RNAfold Software | Predicts mRNA secondary structure stability (MFE) to assess translation efficiency. | ViennaRNA Package; used for in silico validation [73]. |
| S. cerevisiae CEN.PK Strain | A well-characterized, genetically stable host for heterologous pathway expression. | Preferred background for metabolic engineering; e.g., CEN.PK113-5D (MATa) [21]. |
| Yeast Expression Vector | Plasmid for gene expression; requires strong promoter and marker. | e.g., pRS series with TEF1 promoter and URA3 selection marker [21]. |
| Cloning & Assembly Kit | For seamless insertion of synthesized genes into the expression vector. | Gibson Assembly Master Mix or similar. |
| qPCR Master Mix | For quantitative analysis of mRNA transcript levels (RT-qPCR). | Includes reverse transcriptase and SYBR Green/Fluorescent dye. |
| Chaperone Overexpression Library | Co-expressed to assist folding of complex heterologous proteins, boosting activity. | e.g., Library overexpressing cytosolic chaperones like YDJ1 and SSA1 [21]. |
Advanced and Emerging Considerations
The field of sequence optimization is rapidly evolving. Beyond the standard parameters, several advanced considerations are critical for state-of-the-art work.
- Cellular Context-Aware Optimization: Next-generation tools like RiboDecode demonstrate that optimization must account for the cellular environment. These models learn directly from ribosome profiling (Ribo-seq) data, capturing the complex interplay between codon usage, tRNA availability, and translation kinetics in specific conditions [73].
- Pathway-Level Optimization: For multi-gene pathways, optimizing genes in isolation can create imbalances. Advanced tools now enable the simultaneous optimization of all genes in a pathway, considering codon usage and codon pair bias holistically to ensure balanced expression and minimize metabolic burden [69] [74].
- Machine Learning and AI Integration: The incorporation of artificial intelligence allows for the analysis of vast genomic and experimental datasets, leading to more accurate predictions of highly expressive sequences and the exploration of novel, high-performance sequence spaces beyond human intuition [73] [74].
The engineering of yeast cell factories for the production of biopharmaceuticals and industrial enzymes represents a growing multi-billion-dollar global market [75]. A fundamental challenge in this field is the cellular stress imposed by heterologous pathway expression, primarily manifesting as metabolic burden and product toxicity [75]. Metabolic burden refers to the redirection of cellular resources—such as energy, precursors, and transcriptional/translational machinery—from normal growth and maintenance toward the synthesis of recombinant proteins that are often of no benefit to the host cell [75] [76]. This competition for limited resources can negatively impact cell fitness, trigger stress responses, and significantly reduce final protein titers [75]. Concurrently, product toxicity, which can arise from protein aggregation or the interaction of toxic intermediates with cellular components, further compromises the health and productivity of yeast cell factories [75]. These interconnected stressors form a significant bottleneck in bioprocess efficiency. This Application Note details the underlying mechanisms of these challenges and provides validated protocols and strategies to mitigate their effects, thereby enhancing the robustness and productivity of Saccharomyces cerevisiae and Komagataella phaffii cell factories.
Understanding the Challenges
Metabolic burden is a universal challenge in heterologous protein production. Its sources are multifactorial, impacting the host cell from genetic engineering through to protein translation and secretion [75].
- Genetic Engineering and Resource Competition: The introduction and maintenance of plasmid vectors, along with the high-level transcription and translation of foreign genes, consume cellular resources. This includes nucleotides, amino acids, and most critically, ATP and redox cofactors, which are reallocated from biomass formation and growth [75] [43].
- Physiological Consequences: The immediate effects of this burden often include reduced growth rates, lower biomass yield, and altered metabolic fluxes. For instance, a redirection of carbon flux from desired products like ethanol to unwanted by-products such as glycerol and acetate has been observed in burdened cells, indicating potential redox imbalances [76].
- Metabolomic Perturbations: It is crucial to note that metabolic burden can be present even when not detectable through standard growth parameters. Advanced analytical techniques like FTIR spectroscopy have revealed that significant metabolomic reshuffling can occur to maintain metabolic homeostasis, even in the absence of obvious defects in ethanol production or growth rate [76].
Product Toxicity: Mechanisms and Effects
In contrast to the universal nature of burden, product toxicity is specific to the chemical nature of the product or its intermediates.
- Toxic Intermediates: In pathways for biodegradation or complex molecule synthesis, reactive intermediates can exert toxic effects on the host, exacerbating the stress caused by metabolic burden [77].
- Protein Aggregation and Misfolding: The production of heterologous proteins, especially at high levels, can lead to improper folding and aggregation within the cell, causing proteotoxic stress and potentially disrupting essential cellular functions [75].
The following diagram illustrates the interconnected sources and consequences of these stressors on a yeast cell factory.
Mitigation Strategies and Application Protocols
A multi-faceted approach is required to successfully combat cellular stress. The strategies below can be employed individually or in combination.
Strategy 1: Dynamic Regulation and Promoter Engineering
Fine-tuning the timing and level of heterologous expression is a powerful method to decouple growth from production, thereby alleviating burden.
Rationale: Strong, constitutive expression during the growth phase can maximize burden. Using promoters that are induced after substantial biomass accumulation allows the cell to first build its catalytic capacity before diverting resources to production [43]. The choice of promoter and carbon source significantly influences the expression profile.
Protocol: Profiling Promoter Activity Across Cultivation Conditions
- Objective: To characterize the strength and dynamics of candidate promoters under conditions relevant to the intended bioprocess.
- Materials:
- Yeast strain (e.g., S. cerevisiae CEN.PK background)
- Promoter-GFP fusion plasmids (e.g., with yEGFP reporter)
- Minimal media (e.g., Yeast Nitrogen Base without amino acids)
- Carbon sources: Glucose (20 g/L), Sucrose, Galactose, Ethanol
- Microtiter plates and plate reader (fluorescence, OD600)
- Method:
- Transform the panel of promoter-GFP constructs into your host strain.
- Inoculate cultures in minimal media with the designated carbon source.
- Grow cultures in a microtiter plate at 30°C with continuous shaking.
- Measure OD600 and GFP fluorescence (Ex: 488 nm, Em: 509 nm) at regular intervals throughout the batch fermentation, ensuring measurements are taken during mid-log phase (OD600 1.0-2.5) for initial comparison and through the diauxic shift.
- For inducible promoters (e.g., CUP1), add the inducer (e.g., CuSO₄) at a range of concentrations during mid-log phase to establish a dose-response profile.
- Data Analysis: Normalize fluorescence to OD600 to calculate specific promoter activity. Plot these values over time to visualize dynamic expression patterns.
Table 1: Promoter Performance on Different Carbon Sources and Fermentation Phases
| Promoter | Type | Relative Strength on Glucose (Exp. Phase) | Induction on Low Glucose/ Ethanol | Key Characteristics for Application |
|---|---|---|---|---|
| P({}_{TDH3}) | Constitutive | Very High [43] | Low | Strong during growth, sharp decrease post-diauxic shift [43]. Ideal for production during rapid growth. |
| P({}_{TEF1}) | Constitutive | High [43] | Low | Relatively stable on glucose, but decreases post-diauxic shift [43]. Common, reliable choice. |
| P({}_{GAL1}) | Inducible | Low (Repressed) | Very High (on Galactose) [43] | Very strong, tight regulation. Requires galactose medium, which can be expensive [43]. |
| P({}_{CUP1}) | Inducible | Low (Uninduced) | High (Cu²⁺ induced) [43] | Can be strongly induced post-diauxic shift. Allows temporal control via inducer addition [43]. |
| P({}_{SSA1}) | Low-Glucose Inducible | Low | High [43] | Automatically induced as glucose depletes. Good for production in stationary phase [43]. |
| P({}_{AOX1}) (K. phaffii) | Inducible | Low (Repressed) | Very High (on Methanol) [78] | Very strong, tightly regulated. Methanol is hazardous; glucose-derepressed variants exist [78]. |
Strategy 2: Enhancing Cellular Robustness
Engineering the host strain to be more resilient to internal and external stress can improve its capacity to handle the burden of production.
Rationale: Cellular robustness is the ability to maintain basic physiological functions under genetic perturbations, environmental fluctuations, or metabolic stress [5]. Enhancing traits like stress resistance and chronological lifespan (CLS) can prevent phenotypic degeneration during long-term fermentation.
Protocol: Extending Chronological Lifespan to Enhance Production
- Objective: To engineer a robust yeast chassis by modulating nutrient sensing and stress resistance pathways, thereby increasing production stability.
- Materials:
- Method:
- Genetic Modifications: Implement mutations known to extend CLS, such as:
- Downregulate TOR1: The Target of Rapamycin complex 1 (TORC1) is a central nutrient sensor. Reducing its activity extends lifespan and enhances stress tolerance [5].
- Delete HDA1: The histone deacetylase Hda1p is involved in epigenetic regulation. Its deletion has been shown to increase robustness and fatty alcohol production [5].
- CLS Assay: Streak engineered strains onto YPD to activate. Inoculate single colonies into liquid YPD and grow for ~3 days. Monitor viability by counting colony-forming units (CFUs) over time. An extended CLS is indicated by a slower decline in CFUs.
- Production Assay: Perform shake-flask batch fermentations in production media (e.g., Delft medium) for 96 hours. Extract and quantify the target product (e.g., via GC-MS) at the end of fermentation.
- Genetic Modifications: Implement mutations known to extend CLS, such as:
- Data Analysis: Compare the product titer and yield of the engineered robust strains to the control strain. A successful engineering outcome should show a correlation between extended CLS and increased production.
The following diagram summarizes the key pathways and reagents involved in building a more robust yeast cell factory.
Strategy 3: Computational Modeling and Metabolomic Analysis
Using in silico models and high-throughput analytics allows for the prediction and understanding of burden before intensive experimental work.
Rationale: Computational models can integrate the effects of toxicity exacerbation and metabolic burden on population growth and pathway dynamics, providing a platform for in silico optimization [77]. Metabolomic fingerprinting techniques like FTIR spectroscopy can detect subtle, stress-induced physiological changes long before they impact growth or product titer [76].
Protocol: FTIR Spectroscopy for Metabolomic Fingerprinting
- Objective: To rapidly assess the physiological status and metabolomic perturbations of recombinant yeast strains under production conditions.
- Materials:
- Parental and recombinant yeast strains.
- FTIR Spectrometer with a reflectance module.
- Multi-well plate for high-throughput analysis.
- Method:
- Cultivate parental and engineered strains under standard and stress conditions (e.g., in the presence of inhibitors or during recombinant protein production).
- At targeted growth phases, harvest cells and wash them.
- Apply a cell suspension directly onto a multi-well plate and dry to form a thin film.
- Acquire FTIR spectra in the mid-infrared range (e.g., 4000-400 cm⁻¹).
- Subject the spectral data to multivariate statistical analysis (e.g., Principal Component Analysis - PCA) to identify spectral regions (biomarkers) that differentiate the recombinant strain from the parent.
- Data Analysis: Differences in the metabolomic fingerprint, particularly in regions corresponding to lipids, proteins, and carbohydrates, indicate the extent of metabolic reshuffling in response to the burden of heterologous expression, even in the absence of gross physiological changes [76].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents for Investigating and Mitigating Cellular Stress
| Reagent / Tool | Function & Application in Stress Mitigation |
|---|---|
| CRISPR/Cas9 System [5] | Enables precise genome editing for creating knockouts (e.g., HDA1), introducing point mutations (e.g., in TOR1), and integrating pathway genes. |
| Promoter Libraries [78] [43] | Collections of constitutive (e.g., P({}{TEF1}), P({}{TDH3})) and inducible (e.g., P({}{GAL1}), P({}{CUP1}), P({}_{AOX1})) promoters for fine-tuning gene expression and dynamic pathway control. |
| Fluorescent Reporters (yEGFP) [43] | Used as a transcriptional fusion to quantitatively profile promoter activity and dynamics in real-time under different cultivation conditions. |
| FTIR Spectroscopy [76] | A high-throughput technique for obtaining a metabolomic fingerprint of cells, allowing for the rapid detection of stress-induced physiological changes. |
| Computational Models [77] | Mathematical models that simulate the combined effects of metabolic burden and toxicity on population growth and pathway flux, enabling in silico strain design. |
Mitigating metabolic burden and product toxicity is not a single-step task but requires a holistic engineering approach. By dynamically controlling gene expression, enhancing the innate robustness of the cellular chassis, and employing computational and analytical tools to guide design, researchers can construct more efficient and resilient yeast cell factories. The protocols outlined herein provide a concrete starting point for implementing these strategies, ultimately contributing to more sustainable and profitable biomanufacturing processes for therapeutic proteins and valuable chemicals.
Analytical Methods and Host System Performance Evaluation
Functional Complementation Assays in Specific Yeast Mutants
Functional complementation assays in yeast are a cornerstone technique in molecular biology and metabolic engineering, enabling researchers to elucidate gene function and validate heterologous pathway components. Within the context of developing yeast cell factories, these assays provide a powerful, in vivo method to screen for and characterize foreign genes that can restore a missing cellular function, thereby identifying optimal candidates for constructing efficient biosynthetic pathways. The core principle involves introducing a plasmid-borne gene library into a specific yeast mutant that is deficient in a particular metabolic function. If the introduced gene compensates for the missing function, it rescues the mutant's growth defect under selective conditions, confirming functional activity. This approach is particularly valuable for profiling variant effects and optimizing the expression of heterologous enzymes for the production of high-value compounds, thereby accelerating the engineering of robust microbial cell factories [79] [80] [21].
Key Principles and Applications in Cell Factory Development
The foundational principle of a functional complementation assay is the phenotypic rescue of a haploid yeast strain carrying a null mutation in a specific gene. This knockout leads to a discernible phenotype, such as auxotrophy (inability to synthesize an essential metabolite) or growth defect under specific conditions. The subsequent introduction of a heterologous gene that restores the lost function allows for direct selection of complementing clones based on restored growth.
In metabolic engineering and synthetic biology, this assay has two primary applications:
- Gene Discovery and Validation: Identifying novel genes from non-model organisms that encode enzymes capable of catalyzing a specific biochemical reaction essential for a desired biosynthetic pathway [81].
- Variant Effect Profiling: Systematically assessing the functional consequences of thousands of protein variants (e.g., from deep mutational scanning) to understand sequence-function relationships. This data is crucial for training computational predictors and engineering improved enzymes [80].
A critical consideration, as highlighted by recent research on potassium channels, is that growth rescue in a complementation assay may not always have a direct, quantitative correlation with detailed biophysical parameters, such as unitary conductance measured in single-channel recordings. Therefore, while excellent for high-throughput screening, complementation data may need to be complemented with other assays for a complete functional understanding, especially in protein engineering efforts [79] [82].
Experimental Protocols
Protocol 1: Basic Growth Complementation Assay
This protocol outlines the standard procedure for assessing the function of a gene of interest through growth rescue on solid medium [21] [81].
Workflow Overview:
Materials:
- Yeast Strain: Haploid yeast mutant (e.g., YPH499) with a deletion in the target gene (e.g.,
his3Δ,trk1Δ) [79] [81]. - Plasmids: Expression vector (e.g., pRS426-MET25) containing the gene of interest (e.g.,
EfSLS,melA) and a selectable marker (e.g.,URA3) [21] [81]. - Media:
- Reagents: Yeast transformation kit (e.g., lithium acetate/PEG method), PCR reagents for clone verification.
Step-by-Step Procedure:
- Transformation: Introduce the plasmid carrying the heterologous gene into the competent yeast mutant cells using a standard lithium acetate/polyethylene glycol (LiAc/PEG) transformation protocol [81].
- Selection and Verification: Plate the transformation mixture onto solid SC dropout medium lacking uracil (SC -Ura) to select for cells that have taken up the plasmid. Incubate at 30°C for 2-3 days until colonies form. Verify positive clones by colony PCR and sequencing.
- Complementation Test: a. Inoculate positive clones into liquid SC -Ura medium and grow overnight. b. The next day, measure the optical density at 600 nm (OD₆₀₀) and normalize the cultures to a standard density (e.g., OD₆₀₀ ≈ 0.1). c. Prepare a series of 1:5 serial dilutions for each clone. d. Spot equal volumes (e.g., 5 µL) of each dilution onto two types of plates: permissive medium (e.g., SC -Ura) to confirm general cell viability, and selective complementation medium (e.g., SC -Ura -His) where growth rescue is tested.
- Incubation and Analysis: Incubate the plates at 30°C for 3-4 days. Document the growth phenotypes by photographing the plates. Compare the growth of the mutant strain expressing the gene of interest to positive and negative controls (e.g., wild-type strain and empty vector control) [81].
Protocol 2: Quantitative Complementation in Liquid Culture
For more precise quantification of complementation efficiency, the assay can be performed in liquid culture, allowing for growth curve analysis.
Procedure:
- Inoculation: Inoculate verified clones from Protocol 1 into liquid selective complementation medium.
- Growth Monitoring: Grow the cultures in a microplate reader or shake flasks at 30°C with continuous shaking. Monitor the OD₆₀₀ at regular intervals (e.g., every 15-30 minutes) for 24-48 hours.
- Data Analysis: Calculate quantitative growth parameters, such as maximum growth rate (μₘₐₓ) and final biomass yield, from the growth curves. The degree of functional complementation is proportional to the restoration of growth kinetics towards the wild-type profile.
Data Presentation and Analysis
Quantitative Analysis of Complementation Efficiency
Data from functional complementation assays, particularly when used for variant effect profiling, can be quantitatively analyzed to compare the functional impact of different alleles. The table below summarizes hypothetical data for different mutants of a potassium channel gene, illustrating how growth and electrophysiological data can be compared [79] [82].
Table 1: Representative data from a functional complementation study of K+ channel mutants, showing the complex relationship between growth rescue and electrophysiological properties.
| Variant | Growth Rescue (%) | Unitary Conductance (pS) | Open Probability | Functional Score |
|---|---|---|---|---|
| Wild-Type | 100 ± 5 | 58 ± 3 | 0.95 ± 0.03 | 1.00 |
| L94A | 88 ± 7 | 45 ± 4 | 0.91 ± 0.05 | 0.82 |
| L94D | 15 ± 4 | 12 ± 2 | 0.10 ± 0.02 | 0.05 |
| L94F | 95 ± 3 | 40 ± 3 | 0.98 ± 0.01 | 0.89 |
| L94R | 2 ± 1 | 5 ± 1 | 0.05 ± 0.01 | 0.01 |
| Empty Vector | 0 ± 1 | N/A | N/A | 0.00 |
Key Analysis: The data often reveals that growth rescue (a complex cellular phenotype) does not always have a simple 1:1 correlation with specific biophysical parameters measured in vitro (e.g., conductance). A variant like L94F shows strong growth rescue and high open probability but reduced conductance, indicating that the cell may compensate for lower conductance through other mechanisms, such as increased channel expression in the plasma membrane [79] [82].
Integration with High-Throughput Functional Data
Functional complementation data is increasingly used to benchmark computational variant effect predictors (VEPs). The correlation between assay results and VEP scores is a key metric of predictor performance.
Table 2: Correlation between functional complementation data and computational predictions for missense variants in different genes, demonstrating the utility of experimental data for benchmarking. [80]
| Gene | Type of Functional Assay | Number of Variants Tested | Spearman's ρ vs. Top VEP |
|---|---|---|---|
| HMBS | Indirect Yeast Complementation | ~2,500 | 0.78 |
| GCK | Indirect Yeast Complementation | ~3,100 | 0.72 |
| PTEN | Direct Protein Abundance Assay | ~7,500 | 0.85 |
| TP53 | Direct Transcriptional Assay | ~9,000 | 0.89 |
Advanced Applications: Enhancing Heterologous Pathway Efficiency
Functional complementation is not limited to single genes. In yeast cell factory development, a major challenge is the inefficient folding and low activity of heterologous enzymes. A powerful extension of the complementation principle is the use of chaperone co-expression to boost the functional output of a biosynthetic pathway.
Concept: A chaperone overexpression library is mated with a query strain producing a heterologous small molecule. Diploid strains are screened for improved production, identifying chaperone combinations that enhance the folding and stability of pathway enzymes [21].
Workflow for Chaperone-Assisted Strain Engineering:
Application Example: In the production of the small molecule aspulvinone E, this approach identified the combined overexpression of chaperones Ydj1 (Hsp40) and Ssa1 (Hsp70) as the best hit, which increased the final titer by 84% in batch fermentations. The mechanism was attributed to increased levels of the functional MelA synthetase, the key enzyme in the pathway [21].
The Scientist's Toolkit
Table 3: Essential research reagents and materials for conducting functional complementation assays in yeast.
| Reagent / Material | Function / Description | Example / Source |
|---|---|---|
| Specialized Yeast Mutants | Engineered haploid strains with deletions in specific metabolic genes; the foundation for the assay. | TRK1/TRK2 knockout for K+ uptake [79], HIS3 knockout for histidine auxotrophy [81]. |
| Expression Vectors | Plasmids for cloning and expressing genes of interest in yeast, featuring inducible/const. promoters and selectable markers. | pRS426-MET25 (URA3 marker) [81], integration vectors for genomic insertion (e.g., at site XII-5) [21]. |
| Selective Growth Media | Defined media formulations that select for plasmid maintenance and impose metabolic pressure for complementation. | Synthetic Complete (SC) Dropout Media (e.g., -Ura, -His) [81], YPG with galactose for induction [21]. |
| Chaperone Library | A pre-constructed collection of yeast strains overexpressing individual or combinations of molecular chaperones. | Used to identify chaperones that improve the folding and function of heterologous pathway enzymes [21]. |
| Phosphopantetheinyl Transferase | An essential activator for type I polyketide synthases (PKS) and non-ribosomal peptide synthetases (NRPS) expressed in yeast. | Aspergillus nidulans NpgA, often co-expressed with large synthetases like MelA [21]. |
The engineering of Saccharomyces cerevisiae as a microbial cell factory for sustainable bioproduction represents a cornerstone of modern industrial biotechnology. A critical challenge in this field involves the efficient expression and assembly of heterologous metabolic pathways. The success of these engineering efforts hinges on the ability to accurately quantify the functional expression of pathway enzymes. Reliable quantification directly determines the capacity to diagnose bottlenecks in synthetic pathways, optimize metabolic flux, and ultimately achieve high titers of target compounds such as pharmaceutical natural products, biofuels, and food additives [69]. Within this context, three principal methodologies form the essential toolkit for researchers: fluorescent reporter systems like GFP for rapid screening, Western blotting for direct protein detection, and enzymatic activity assays for functional validation. This application note provides detailed protocols and frameworks for implementing these critical techniques, specifically tailored for researchers developing yeast-based cell factories.
GFP Reporter Systems for Rapid Screening
Green Fluorescent Protein (GFP) and its variants serve as versatile molecular biosensors, enabling non-invasive monitoring of gene expression, protein localization, and protein solubility in living yeast cells. The application of GFP is particularly valuable in bioprocess engineering for rapidly assessing transfection efficiency and, when fused to a target protein, for tracking the expression and stability of heterologous enzymes [83].
Key Research Reagent Solutions
Table 1: Essential Reagents for GFP-Based Analysis.
| Item | Function | Example |
|---|---|---|
| Anti-GFP Antibody | Immunodetection of native GFP and GFP-fusion proteins via Western blot or immunoprecipitation. | Rabbit Polyclonal (A11122) or Mouse Monoclonal (A11120) [84]. |
| Recombinant GFP Protein | Generating standard curves for precise quantification of GFP concentration in unknown samples [83]. | Recombinant Jellyfish GFP Protein [83]. |
| GFP-Tagged Protein of Interest | A biosensor to monitor expression, solubility, and subcellular localization of a target heterologous enzyme [83]. | N/A |
| Milk-free Antibody Diluent | Prevents cross-reactivity when using anti-goat secondary antibodies in immunodetection [83]. | ProteinSimple Milk-free Antibody Diluent [83]. |
Advanced Protocol: Quantifying Contaminating GFP and GFP-Fusion Proteins Using Simple Western
The following protocol leverages automated Western blotting (e.g., ProteinSimple's Simple Western) for sensitive, quantitative analysis of GFP, which is crucial for detecting residual contaminating GFP or characterizing GFP-tagged protein products [83].
Sample Preparation:
- Prepare 0.1X Sample Buffer by diluting 10X Sample Buffer 1:100 in deionized water.
- Create a GFP Standard Curve: Prepare a two-fold serial dilution series of recombinant GFP protein in 0.1X Sample Buffer, for example, from 2 ng/mL down to 0.125 ng/mL. Include a no-protein baseline control.
- Prepare Unknown Samples: Dilute cell lysates from yeast expressing your GFP-tagged protein or contaminating GFP in 0.1X Sample Buffer. A total protein concentration of 0.3 µg per sample is often sufficient.
Antibody Preparation:
- Dilute the primary anti-GFP antibody to a concentration of 15 µg/mL in Milk-free Antibody Diluent.
- Follow the manufacturer's instructions for preparing the recommended HRP- or fluorophore-conjugated secondary antibody.
Simple Western Execution:
- Follow the default Simple Western sample preparation and assay conditions using an appropriate separation module (e.g., 12-230 kDa) and the corresponding detection module.
- Load the GFP standard curve and unknown samples in the same assay run.
Data Analysis:
- The software will generate an electropherogram, plotting signal intensity against molecular weight.
- Generate a Standard Curve: Plot the peak area of each standard against its known GFP concentration and fit a trendline.
- Quantitate Unknowns: Use the standard curve's equation to calculate the concentration of free GFP or the relative amount of the GFP-fusion protein in your test samples. This system can detect free GFP at sensitivities below 1 ng/mL [83].
Workflow for GFP Quantification using Simple Western.
Western Blotting for Direct Protein Detection
Western blotting remains a fundamental technique for confirming the expression, molecular weight, and integrity of heterologous proteins in yeast lysates. It provides critical information that fluorescent signal alone cannot, such as revealing protein degradation, aggregation, or incorrect post-translational modification.
Key Protocol: Standard Western Blotting for Yeast Lysates
STAGE 1: Sample Preparation from Yeast Culture [85]
- Harvest Cells: Pellet yeast cells from a saturated culture by centrifugation (100–500 x g, 5 min, 4°C).
- Wash Cells: Resuspend the pellet in ice-cold PBS and centrifuge again. Discard the supernatant.
- Lyse Cells: Resuspend the cell pellet in an appropriate, ice-cold lysis buffer (e.g., RIPA buffer) supplemented with protease inhibitors. Incubate for 10 minutes on ice with rocking.
- Clarify Lysate: Centrifuge the suspension at 14,000–17,000 x g for 5–10 min at 4°C. Transfer the supernatant (your lysate) to a fresh tube on ice.
- Determine Protein Concentration: Use a BCA or Bradford assay to determine the total protein concentration of the lysate.
- Denature Samples: Dilute lysate aliquots in loading buffer containing DTT to a final concentration of 1–2 mg/mL. Boil the samples at 100°C for 10 minutes.
STAGE 2: Gel Electrophoresis and Transfer [85]
- Load and Run Gel: Load an equal amount of protein (10–40 µg for lysates) into the wells of an SDS-PAGE gel alongside a molecular weight ladder. Run the gel according to the manufacturer's instructions.
- Gel Selection Guide: For proteins 10-30 kDa, use a 4-12% Bis-Tris gel with MES buffer; for 31-150 kDa, use a 4-12% Bis-Tris gel with MOPS buffer; for >150 kDa, use a 3-8% Tris-Acetate gel [85].
- Transfer to Membrane: Using a wet or semi-dry transfer system, electrophoretically transfer the separated proteins from the gel onto a nitrocellulose or PVDF membrane.
STAGE 3: Immunodetection
- Block Membrane: Incubate the membrane in a blocking solution (e.g., 5% BSA or non-fat milk in TBST) for 1 hour at room temperature to prevent non-specific antibody binding.
- Incubate with Primary Antibody: Dilute the primary antibody (e.g., anti-GFP or antibody against your heterologous protein) in blocking solution. Incubate with the membrane for 1 hour at room temperature or overnight at 4°C with gentle agitation.
- Wash Membrane: Wash the membrane 3 times for 5 minutes each with TBST to remove unbound antibody.
- Incubate with Secondary Antibody: Dilute an HRP- or fluorophore-conjugated secondary antibody, specific to the host species of the primary antibody, in blocking solution. Incubate with the membrane for 1 hour at room temperature.
- Wash Membrane: Repeat the washing steps as after the primary antibody.
STAGE 4: Detection and Imaging
- Detect Signal: For chemiluminescent detection, incubate the membrane with the appropriate substrate and image using a digital imager. For fluorescent detection, scan the membrane at the appropriate wavelength.
- Analyze: Identify bands corresponding to your target protein based on the expected molecular weight.
Enzymatic Activity Assays for Functional Validation
While Western blotting confirms protein presence, enzymatic activity assays are indispensable for determining if a heterologous enzyme is functionally folded and active in the yeast cell factory environment. This functional readout is the most direct indicator of a successful pathway integration.
Fundamentals of Enzyme Activity
- Enzyme Unit (U): The amount of enzyme that catalyzes the conversion of 1 µmol of substrate per minute under standard conditions (Definition A). A more sensitive, common definition is the conversion of 1 nmol of substrate per minute (Definition B) [86].
- Enzyme Activity: The concentration of enzyme units, typically expressed as U/mL (e.g., nmol/min/mL) [86].
- Specific Activity: A key metric of enzyme purity and quality, calculated as the number of units per mg of total protein (U/mg or nmol/min/mg) [86].
Key Protocol: Developing a Robust Enzymatic Assay
The primary goal is to measure the initial velocity of the reaction, which is the linear rate observed when less than 10% of the substrate has been converted to product [87]. Operating outside this linear range leads to inaccurate activity measurements.
Assay Development Steps:
Establish Initial Velocity Conditions [87]:
- Perform a time course experiment with multiple dilutions of your yeast lysate or purified enzyme.
- Plot the amount of product formed versus time for each enzyme dilution.
- Select a time point and enzyme concentration where the relationship is linear (i.e., the plot is a straight line) for less than 10% of substrate conversion. An example is shown in the 0.5x enzyme concentration curve below.
Determine Kinetic Parameters (Kₘ and Vₘₐₓ) [87]:
- Under the initial velocity conditions established above, measure the reaction rate at 8 or more substrate concentrations, typically spanning 0.2-5.0 times the estimated Kₘ.
- Plot the velocity (v) against substrate concentration ([S]). Fit the data to the Michaelis-Menten equation to determine Kₘ (the substrate concentration at half Vₘₐₓ) and Vₘₐₓ.
- For inhibitor screening, use a substrate concentration at or below the Kₘ value to maximize sensitivity to competitive inhibitors [87].
Validate Assay Linearity and Controls:
- Ensure the detection system (e.g., spectrophotometer, plate reader) is operating within its linear range for the product being measured [87].
- Always include appropriate controls, such as a no-enzyme control, to account for background signal.
Workflow for Enzyme Activity Assay Development.
Critical Factors Affecting Assay Performance
- Temperature: Enzyme activity is highly sensitive to temperature. A change of just 1°C can cause a 4-8% variation in measured activity. Use a temperature-controlled spectrophotometer or water bath to ensure stability [88].
- pH: Each enzyme has an optimal pH. Use a buffer with good buffering capacity at the desired pH and ensure all reagents are equilibrated to the correct pH [88].
- Substrate Concentration: Using a substrate concentration around or below the Kₘ is ideal for detecting competitive inhibitors and ensures the reaction rate is sensitive to changes in substrate concentration [87].
Table 2: Summary of Key Quantification Methods for Yeast Cell Factories.
| Method | What It Measures | Key Quantitative Outputs | Key Considerations |
|---|---|---|---|
| GFP Reporter Assays | Expression level and localization of a protein of interest when fused to GFP. | - Fluorescence intensity (A.U.)- Protein concentration (ng/mL) via standard curve [83]. | - GFP fusion may alter protein function or localization.- Provides no direct functional data. |
| Western Blotting | Presence, size, and relative abundance of a specific protein. | - Relative band intensity.- Confirmation of molecular weight.- Detection of degradation/aggregation. | - Semi-quantitative without rigorous standards.- Does not confirm protein is functional. |
| Enzymatic Activity Assay | Functional capacity of an enzyme to convert substrate to product. | - Enzyme Units (U), Activity (U/mL), Specific Activity (U/mg) [86].- Kₘ and Vₘₐₓ [87]. | - Requires careful optimization of linear range.- Highly sensitive to assay conditions (pH, T) [88]. |
The strategic integration of GFP reporters, Western blotting, and enzymatic activity assays provides a powerful, multi-faceted approach to quantifying heterologous expression in yeast cell factories. GFP systems offer high-throughput screening and localization data, Western blotting confirms protein integrity and size, and activity assays deliver the ultimate readout of functional enzyme production. By applying the detailed protocols and considerations outlined in this document, researchers can effectively diagnose and overcome expression bottlenecks, paving the way for the development of robust and efficient yeast-based production platforms for valuable chemicals and proteins. The compatibility between heterologous pathways and the host chassis can be systematically engineered at the genetic, expression, and functional levels, guided by precise quantitative data from these essential techniques [31] [69].
Within the framework of developing advanced yeast cell factories for heterologous pathway expression, the selection of an optimal microbial host is a critical determinant of success. This decision directly influences the core metrics of titer, post-translational modification fidelity, and the resultant bioactivity of the recombinant product. While the model yeast Saccharomyces cerevisiae has been a workhorse for decades, emerging non-Saccharomyces yeasts and filamentous fungi present compelling alternatives with distinct advantages. This Application Note provides a comparative analysis of several prominent fungal expression systems, benchmarking their performance in yield, glycosylation patterns, and bioactivity of produced proteins. Supported by structured data and detailed protocols, this resource is designed to assist researchers and drug development professionals in making informed host selection decisions for their specific application, from industrial enzyme production to biopharmaceutical development.
Comparative Performance of Fungal Expression Hosts
The quantitative performance of five engineered fungal hosts—spanning yeasts and filamentous fungi—is summarized in Table 1. Key metrics include the target protein, achieved titer or activity, and a critical evaluation of glycosylation patterns which directly impact bioactivity, particularly for therapeutic proteins.
Table 1: Performance Benchmarking of Fungal Expression Hosts
| Host Organism | Target Protein | Titer/Activity | Production Scale | Glycosylation & Bioactivity Notes | Citation |
|---|---|---|---|---|---|
| S. cerevisiae (Engineered) | Transferrin | 2.33 g/L | Fed-batch, 10 L bioreactor | Native high-mannose glycosylation; humanized patterns achievable via glycoengineering. | [55] |
| K. phaffii (GS115) | Acidocin 4356 (rACD) | ~20 kDa product; 34.12% yield increase post-optimization | Shake flask & Bioreactor | Resistant to AMP-mediated toxicity; produces functional antimicrobial peptide with potent activity against P. aeruginosa (58.29% growth reduction). | [89] |
| K. phaffii (X33) | Unspecific Peroxygenase (UPO) | Volumetric activity increased >60% (flask), >100% (bioreactor) | Shake flask & 5 L Bioreactor | Successfully surface-displayed; correct folding is temperature-dependent; retained regio- and stereoselective bioactivity. | [90] |
| A. niger (AnN2 chassis) | Glucose Oxidase (AnGoxM) | ~1276 - 1328 U/mL | Shake flask (50 mL) | Robust eukaryotic secretion and PTM capability; platform strain minimizes background proteolysis. | [91] |
| Pectate Lyase (MtPlyA) | ~1627 - 2106 U/mL; 18% increase with secretory engineering | Shake flask (50 mL) | [91] | ||
| Therapeutic Protein (LZ8) | 110.8 - 416.8 mg/L | Shake flask (50 mL) | [91] | ||
| O. minuta (Double Mutant) | Human Serum Albumin (HSA) & ProteinX | Process optimized for industrial scale | Industrial-scale manufacturing | Protease-deficient host (prb1::) reduces product degradation; requires careful control of feeding strategy. | [92] |
Analysis of Host-Specific Glycosylation Patterns
Protein glycosylation is a critical quality attribute with profound implications for protein stability, function, and immunogenicity. The glycosylation capabilities of fungal hosts vary significantly from human-like patterns and require careful consideration.
- S. cerevisiae: This yeast naturally produces proteins with high-mannose N-glycans, which can be immunogenic in humans and reduce the serum half-life of therapeutic proteins [55]. However, its well-characterized genetics enable glycoengineering strategies to "humanize" the glycosylation pathway, producing proteins with complex, human-compatible glycoforms such as those required for active antibodies [55] [31].
- K. phaffii and O. minuta: As methylotrophic yeasts, these hosts generally produce shorter mannan chains compared to S. cerevisiae, which can be advantageous for producing certain recombinant proteins [55] [92]. Their glycosylation pathways are also amenable to engineering to achieve more human-like glycosylation patterns [93].
- A. niger: This filamentous fungus possesses sophisticated eukaryotic secretory machinery capable of performing a range of post-translational modifications, including N- and O-linked glycosylation [91]. The specific glycan structures, however, are fungal and must be characterized for therapeutic applications, as they can differ from human patterns.
Recent advances in analytical techniques are crucial for this characterization. The Deep Quantitative Glycoprofiling (DQGlyco) method, for instance, enables unprecedented depth in glycoproteome analysis, identifying over 177,000 unique N-glycopeptides and quantifying site-specific microheterogeneity, which is essential for understanding the link between glycosylation and protein function [94]. Furthermore, functional studies in fungi like Fusarium graminearum have demonstrated that disrupting key glycosylation genes (e.g., ALG3, ALG12) leads to truncated core N-glycans, which can significantly alter the profile of secreted glycoproteins and directly impair virulence, underscoring the critical functional role of proper glycosylation [93].
Detailed Experimental Protocols
Protocol 1: High-Yield Protein Production inK. phaffiiwith RSM Optimization
This protocol details the expression and optimization of antimicrobial peptide Acidocin 4356 (ACD) in K. phaffii GS115, as described by [89].
Materials:
- Strain: Komagataella phaffii GS115.
- Vector: pPICZα-A, a methanol-inducible, Zeocin-resistant expression vector.
- Media: YPD, BMGY (growth medium), BMMY (induction medium).
- Equipment: Standard bioreactor or shaking incubator.
Method:
- Cloning and Transformation:
- Design a codon-optimized ACD gene sequence for K. phaffii.
- Clone the gene into the pPICZα-A vector and linearize with SacI.
- Integrate the linearized plasmid into the K. phaffii GS115 genome via electroporation (2500 V, 200 Ω, 25 µF).
- Select transformants on YPDS plates supplemented with 100 µg/mL Zeocin.
Fermentation and Induction:
- Inoculate a single colony into BMGY medium and incubate at 30°C with shaking until the culture reaches the desired optical density (OD600).
- Centrifuge the cells and resuspend the pellet in BMMY medium to induce expression with 1% (v/v) methanol.
- Maintain induction for 72-96 hours, feeding with methanol to a final concentration of 1% (v/v) daily.
Process Optimization via RSM:
- To maximize yield, employ Response Surface Methodology (RSM) to optimize key physicochemical parameters.
- The identified optimal conditions for rACD production are: Temperature = 21°C, pH = 6.24, Methanol Concentration = 1.089%.
- Cultivation under these optimized parameters can increase recombinant protein synthesis by over 34% compared to standard conditions (30°C, pH 6.0, 1% methanol) [89].
Purification and Analysis:
- Purify the 6xHis-tagged fusion protein using Ni–NTA affinity chromatography.
- Cleave the fusion tag using enterokinase and confirm the molecular weight and antimicrobial activity via SDS-PAGE and growth inhibition assays against P. aeruginosa, respectively.
Protocol 2: Signal Peptide Engineering for Enhanced Secretion inS. cerevisiae
This protocol leverages a high-throughput screen based on Gaussia luciferase (GLuc) to evolve improved signal peptides (SPs) for heterologous expression [95].
Materials:
- Strain: S. cerevisiae INVSc1.
- Vector: pESC-TRP for tryptophan auxotrophic selection.
- Reagent: Coelenterazine (substrate for Gaussia luciferase).
- Equipment: Luminometer, equipment for error-prone PCR.
Method:
- Construct Design:
- Fuse the gene of interest (e.g., the first folded domain of AaeUPO, 'd55') upstream of the Gaussia luciferase (GLuc) reporter gene in the pESC-TRP vector.
- The native signal peptide (SP) of the target protein is placed upstream of this fusion construct.
Signal Peptide Library Generation:
- Subject the DNA sequence encoding the native SP to error-prone PCR to create a diverse library of mutant SPs.
- Clone the mutated SP library into the construct from Step 1, replacing the native SP.
Expression and High-Throughput Screening:
- Transform the library into S. cerevisiae and culture transformants in a 96-well format.
- Induce protein expression with galactose.
- After 24-48 hours of induction, collect supernatant samples and assay for luminescence immediately upon addition of coelenterazine.
- Identify clones exhibiting the highest luminescence signals, which correspond to the most efficient SPs for secretion.
Validation:
- Isolate the top-performing mutant SP sequences from the primary screen.
- Use these optimized SPs to express the full-length target protein (without the GLuc fusion) and quantify the yield improvement. This approach has demonstrated a 13.9-fold improvement in expression over the wild-type SP [95].
Pathway and Workflow Diagrams
Multi-level Engineering of Yeast Cell Factories
The following diagram illustrates the integrated engineering strategies required to develop high-performance yeast cell factories, from transcriptional regulation to post-translational modification.
High-Throughput Signal Peptide Screening Workflow
This workflow outlines the steps for using Gaussia luciferase as a reporter to identify superior signal peptides for heterologous secretion in S. cerevisiae.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Key Reagents and Tools for Yeast Cell Factory Engineering
| Reagent/Tool | Function/Application | Example Use Case | Citation |
|---|---|---|---|
| pPICZα-A Vector | Methanol-inducible expression vector for K. phaffii; allows secretion and Zeocin selection. | Heterologous production of Acidocin 4356 (rACD) and Unspecific Peroxygenases (UPOs). | [89] [90] |
| CRISPR/Cas9 System | Enables precise genome editing, gene knock-outs, and multi-copy integration. | Engineering of A. niger chassis strain (AnN2) by deleting 13 copies of the native glucoamylase gene. | [91] |
| Gaussia Luciferase (GLuc) Reporter | High-sensitivity, secreted reporter for high-throughput screening of expression and secretion efficiency. | Screening S. cerevisiae signal peptide (SP) mutant libraries for improved secretion. | [95] |
| Response Surface Methodology (RSM) | Statistical technique for optimizing multiple process variables simultaneously. | Optimizing temperature, pH, and methanol concentration to boost rACD yield in K. phaffii by 34.12%. | [89] |
| Deep Quantitative Glycoprofiling (DQGlyco) | Advanced glycoproteomics method for deep, quantitative analysis of protein glycosylation. | Characterizing site-specific glycosylation microheterogeneity in recombinant glycoproteins. | [94] |
The strategic selection and engineering of a microbial host are paramount for the successful production of heterologous proteins with high yield, desired glycosylation, and optimal bioactivity. As demonstrated, S. cerevisiae remains a versatile and safe platform, particularly amenable to glycoengineering. The methylotrophic yeast K. phaffii excels in high-density fermentations and the production of complex proteins, including challenging antimicrobial peptides and biocatalysts. The filamentous fungus A. niger offers an exceptionally powerful secretion capacity, making it ideal for industrial enzymes. The choice between these systems must be guided by the specific requirements of the target protein. Furthermore, the integration of advanced tools—such as high-throughput screening with Gaussia luciferase, CRISPR-mediated genome editing, and sophisticated glycoprofiling—is accelerating the development of next-generation yeast cell factories. This comparative analysis provides a framework for researchers to navigate this critical decision-making process, ultimately advancing the production of valuable proteins for biopharmaceutical and industrial applications.
In the field of yeast metabolic engineering, the successful expression of heterologous pathways for the production of biofuels, organic acids, and terpenoids is often hampered by inefficiencies in protein secretion and folding [96] [97]. While transcriptomics can confirm the successful introduction and transcription of genetic constructs, it does not guarantee the corresponding production, proper folding, or secretion of the encoded enzymes and proteins [96]. The integration of transcriptomic and proteomic data provides a powerful approach to validate these pathways, revealing discrepancies between mRNA abundance and protein expression that pinpoint bottlenecks in heterologous pathway expression [98] [99]. This Application Note details a protocol for the directional integration of transcriptomics and proteomics data to comprehensively validate and optimize heterologous pathways in yeast cell factories, a critical capability for researchers and drug development professionals working to build sustainable bioeconomies [97].
Conceptual Framework for Directional Multi-Omics Integration
The core principle of this methodology is the directional integration of transcriptomic and proteomic data. This approach tests the specific biological hypothesis that mRNA expression positively correlates with protein expression, a fundamental expectation for functional heterologous pathways [98]. Directional integration prioritizes genes that show consistent, concordant changes in both transcript and protein levels, while penalizing those with significant but conflicting changes, thereby reducing false-positive findings and providing more accurate mechanistic insights [98].
The DPM (Directional P-value Merging) method is a key tool for this analysis. It uses a user-defined constraints vector (CV) to specify the expected directional relationship between datasets. For transcriptomics and proteomics, the CV is set to [+1, +1], which prioritizes genes that are either upregulated or downregulated in both datasets [98]. The method computes a directionally weighted score (XDPM) for each gene across k datasets, incorporating both P-values and directional changes, and derives a merged P-value (P'DPM) that reflects the joint significance of the gene across the input datasets given the directional information [98].
Experimental Protocols
Yeast Strain Engineering and Cultivation
Objective: To generate yeast strains expressing heterologous pathways and produce biomass for multi-omics analysis.
Materials:
- Yeast Strain: Saccharomyces cerevisiae (e.g., CEN.PK113-7D) or non-conventional yeast (e.g., Yarrowia lipolytica) [97].
- Expression Vector: CRISPR-Cas9 plasmid system for precise genome integration [97].
- Culture Medium: Appropriate defined medium (e.g., YNB) with necessary auxotrophic supplements.
Procedure:
- Strain Construction: Utilize CRISPR-Cas9 systems for precise integration of heterologous pathway genes into the yeast genome [97]. For S. cerevisiae, which has high homologous recombination efficiency, design repair templates with 40-60 bp homology arms.
- Culture Conditions: Inoculate engineered and control (wild-type) strains in triplicate in 50 mL of medium. Grow at 30°C with shaking at 250 rpm to mid-exponential phase (OD600 ~ 0.8).
- Harvesting: Centrifuge 20 mL of culture at 3,000 × g for 10 minutes at 4°C. Wash cell pellets twice with ice-cold PBS.
- Cell Lysis and Storage: Flash-freeze cell pellets in liquid nitrogen. Store at -80°C until omics analysis. Split pellets for concurrent transcriptomic and proteomic extraction.
Transcriptomic Profiling via RNA-Sequencing
Objective: To comprehensively quantify mRNA expression levels in engineered versus control yeast strains.
Materials:
- RNA Extraction Kit: Commercial kit for yeast RNA isolation (e.g., hot phenol method or column-based kits).
- RNA Quality Control Tools: Bioanalyzer or TapeStation.
- Library Prep Kit: Strand-specific mRNA library preparation kit.
Procedure:
- RNA Extraction: Extract total RNA from approximately 107 cells using a commercial kit, incorporating a DNase I digestion step to remove genomic DNA contamination.
- Quality Control: Assess RNA integrity using a Bioanalyzer. Accept samples with RNA Integrity Number (RIN) > 8.0 for library preparation.
- Library Preparation and Sequencing: Prepare stranded mRNA sequencing libraries according to the manufacturer's instructions. Perform sequencing on an Illumina platform to a depth of 20-30 million paired-end (2 × 150 bp) reads per sample.
- Bioinformatic Processing:
- Quality Control: Use FastQC to assess raw read quality.
- Alignment: Map reads to the yeast reference genome (e.g., SGD R64) using a splice-aware aligner like STAR.
- Quantification: Generate gene-level read counts using featureCounts.
- Differential Expression: Perform analysis in R using the
DESeq2package to identify significantly differentially expressed genes (adjusted p-value < 0.05, |log2FC| > 1).
Proteomic Profiling via LC-MS/MS
Objective: To identify and quantify protein expression changes in engineered versus control yeast strains.
Materials:
- Lysis Buffer: Urea-based lysis buffer (e.g., 8 M Urea in 50 mM Tris-HCl, pH 8.0) supplemented with protease inhibitors.
- Digestion Enzymes: Sequencing-grade modified trypsin.
- LC-MS/MS System: Nano-flow liquid chromatography system coupled to a high-resolution tandem mass spectrometer (e.g., Q-Exactive series).
Procedure:
- Protein Extraction: Resuspend cell pellets in 500 µL of urea lysis buffer. Disrupt cells by bead-beating (5 cycles of 1 minute beating, 1 minute on ice). Centrifuge at 16,000 × g for 15 minutes to clarify the lysate.
- Protein Digestion: Determine protein concentration by BCA assay. Digest 50 µg of protein using the standard S-Trap filter-aided sample preparation protocol: reduce with DTT, alkylate with iodoacetamide, and digest with trypsin (1:50 enzyme-to-protein ratio) overnight at 37°C.
- LC-MS/MS Analysis:
- Chromatography: Desalt peptides and separate on a nano-LC system using a C18 column with a 60-minute linear gradient from 2% to 35% acetonitrile in 0.1% formic acid.
- Mass Spectrometry: Acquire data in data-dependent acquisition (DDA) mode. Full MS scans (300-1750 m/z) at resolution 70,000, followed by MS/MS scans of the top 15 most intense ions at resolution 17,500.
- Proteomic Data Analysis:
- Identification and Quantification: Search MS/MS data against the yeast Uniprot proteome database using software such as MaxQuant. Enable the LFQ algorithm for label-free quantification.
- Differential Expression: Process protein intensities in R using the
limmapackage. Consider proteins with an adjusted p-value < 0.05 and |log2FC| > 0.585 (corresponding to a 1.5-fold change) as significantly altered.
Data Integration and Pathway Analysis Workflow
Data Preprocessing and Integration using DPM
Objective: To merge transcriptomic and proteomic datasets directionally for unified gene prioritization.
Procedure:
- Prepare Input Matrices: Create two data frames:
- P-value matrix: Rows (genes), columns (transcriptomics p-values, proteomics p-values).
- Direction matrix: Rows (genes), columns (transcriptomics log2FC direction, proteomics log2FC direction). Directions are encoded as +1 (upregulated), -1 (downregulated), based on the significance and fold-change thresholds from Sections 3.2 and 3.3.
- Define Constraints Vector (CV): Set CV = [
+1, +1] to prioritize genes with consistent directional changes in both transcript and protein levels [98]. - Run DPM Analysis: Utilize the
ActivePathwaysR package [98] to perform directional integration. The function calculates a merged p-value for each gene, representing its joint significance across both omics layers. - Generate Prioritized Gene List: Rank genes by their merged p-values. This list highlights genes with the most consistent and significant evidence across the transcriptomic and proteomic datasets.
Functional Enrichment and Pathway Mapping
Objective: To interpret the multi-omics gene list in the context of biological pathways and identify validated heterologous pathways and endogenous bottlenecks.
Procedure:
- Pathway Enrichment Analysis: Submit the top 500 prioritized genes from the DPM analysis to a functional enrichment tool such as g:Profiler [98] or Enrichr [98]. Use databases like Gene Ontology (GO) Biological Process and KEGG.
- Identify Significant Pathways: Select pathways with a false discovery rate (FDR) < 0.05. The heterologous pathway of interest should appear as a significantly enriched term, confirming its functional expression.
- Analyze Contributing Omics Layers: Use the output from
ActivePathwaysto determine which omics dataset(s) (transcriptomics, proteomics, or both) provided evidence for each significantly enriched pathway. This helps identify potential post-transcriptional bottlenecks.
Key Research Reagent Solutions
The following table details essential reagents and tools for implementing the described multi-omics validation pipeline.
Table 1: Key Research Reagent Solutions for Omics Integration in Yeast
| Item | Function/Description | Example Sources/Identifiers |
|---|---|---|
| CRISPR-Cas9 System | Precision genome editing tool for stable integration of heterologous pathways [97]. | Plasmid sets (e.g., pYES-CHERRY from Addgene). |
| RNA Extraction Kit | Isolation of high-quality, intact total RNA for transcriptomics. | RNeasy Mini Kit (Qiagen), Direct-zol RNA Miniprep (Zymo Research). |
| Stranded mRNA Prep Kit | Preparation of sequencing libraries that preserve strand information. | Illumina Stranded mRNA Prep, NEBNext Ultra II Directional RNA Library Prep Kit. |
| Trypsin, Sequencing Grade | Protease for specific digestion of proteins into peptides for LC-MS/MS analysis. | Trypsin Gold (Promega), Sequencing Grade Modified Trypsin (Promega). |
| C18 LC Columns | Reverse-phase chromatography for peptide separation prior to MS injection. | Aurora Series (Ion Opticks), PepMap (Thermo Fisher Scientific). |
| ActivePathways Software | R package for directional integration of multi-omics data using DPM and pathway enrichment [98]. | CRAN: ActivePathways. |
| Gene Ontology (GO) Database | Curated knowledgebase for functional interpretation and pathway enrichment analysis [98]. | http://geneontology.org/ |
Expected Results and Data Interpretation
Successful application of this pipeline will yield a ranked list of genes and significantly enriched pathways. The heterologous pathway(s) introduced into the yeast host should appear as significantly enriched, providing multi-omics validation of its functional expression. Furthermore, the analysis frequently reveals endogenous cellular processes that are co-regulated or perturbed.
For instance, a study aiming to enhance heterologous protein secretion in Pichia pastoris used a similar transcriptomics-driven approach and identified several novel helper factors, including Bfr2 and Bmh2 (involved in protein transport), the chaperones Ssa4 and Sse1, and the vacuolar ATPase subunit Cup5 [96]. When these genes were overexpressed, they increased antibody fragment secretion by up to 2.5-fold [96]. This demonstrates how multi-omics integration can pinpoint non-obvious engineering targets.
Table 2: Example Multi-Omics Validation Results for a Heterologous Terpenoid Pathway
| Gene/Pathway | Transcriptomics\n(log₂FC, adj. p-val) | Proteomics\n(log₂FC, adj. p-val) | DPM Merged\np-value | Status |
|---|---|---|---|---|
| Heterologous Pathway | ||||
| ERG10 (Pathway Gene 1) | +3.5, 1.2e-10 | +2.8, 5.5e-8 | 1.5e-12 | Validated |
| ERG13 (Pathway Gene 2) | +4.1, 2.3e-12 | +3.1, 1.1e-6 | 3.8e-13 | Validated |
| Endogenous Bottlenecks | ||||
| SSA4 (Chaperone) | +2.1, 4.5e-5 | +1.9, 0.003 | 2.1e-6 | Induced |
| PDI1 (Folding Enzyme) | +1.5, 0.02 | +0.8, 0.15 | 0.08 | Potential Bottleneck |
| Unrelated Process | ||||
| ADH2 (Alcohol Dehydrogenase) | -2.2, 0.001 | -0.3, 0.45 | 0.25 | Not Validated |
The table above illustrates how the multi-omics pipeline distinguishes between robustly validated pathway components, induced stress responses, potential post-transcriptional bottlenecks (like PDI1), and findings that may be artifacts of a single omics layer. This comprehensive view is critical for prioritizing the most effective metabolic engineering strategies.
The transition from small-scale screening to industrial production is a critical phase in the development of yeast-based bioprocesses for heterologous protein production. Scaling up from microtiter plates (MTPs) to stirred tank reactors (STRs) presents significant engineering and biological challenges that must be systematically addressed to maintain process performance and product quality. Within the broader context of heterologous pathway expression in yeast cell factories, successful scale-up requires careful consideration of oxygen transfer, mixing dynamics, and the cellular response to changing environmental conditions [100] [101]. This application note provides a structured framework and detailed protocols for scaling up yeast fermentation processes, with a specific focus on maintaining consistent performance across scales.
Key Engineering Parameters for Scale-Up
The scalability of bioprocesses depends on maintaining critical physiological and engineering parameters constant across different scales. The table below summarizes the key parameters and their significance in scale-up operations.
Table 1: Key Scale-Up Parameters and Their Biological Significance
| Parameter | Description | Impact on Yeast Physiology | Scale-Up Consideration |
|---|---|---|---|
| Volumetric Mass Transfer Coefficient (kLa) | Measures the oxygen transfer capacity of the system | Affects biomass yield, protein production, and metabolic pathways [100] | Keep constant to ensure equivalent oxygen supply [102] |
| Volumetric Power Input (P/V) | Power input per unit volume | Impacts hydromechanical stress, morphology, and viability [102] | Constant P/V minimizes shear stress differences |
| Oxygen Transfer Rate (OTRmax) | Maximum oxygen transfer capacity | Determines maximum cell density and metabolic activity [102] | Match OTRmax to maintain equivalent growth environments |
| Mixing Time | Time required to achieve homogeneity | Affects nutrient distribution, pH gradients, and dissolved CO₂ [101] | Shorter in small scales; can create gradients at production scale |
Experimental Protocols for Cross-Scale Evaluation
Protocol 1: Characterization of Microscale Cultivation Systems
Purpose: To determine the oxygen transfer capabilities and operating boundaries of microtiter plates prior to scale-up experiments.
Materials:
- 96-deepwell MTPs (round and square well geometries)
- Microtiter plate shaker with controlled humidity and temperature
- Gas exchange system with 5% CO₂ in air
- Sterile AreaSeal film or equivalent
- Yeast strain expressing heterologous protein of interest
- Appropriate selective medium
Procedure:
- Prepare precultures in shake flasks using standard laboratory conditions.
- Set shaking frequency to 850 rpm with 3 mm shaking diameter for MTPs [102].
- Fill MTP wells with varying working volumes (200-1000 μL) to establish kLa dependence on filling volume.
- Seal plates with gas-permeable membrane and initiate cultivation with starting OD₆₀₀ of 0.5-1.0.
- Monitor biomass growth online via scattered light measurement if available (e.g., BioLector system) [100].
- Determine kLa values for each condition using the gassing-out method with oxygen-sensitive dyes.
- Establish correlation between shaking frequency, filling volume, and kLa value.
- Identify operating conditions that provide kLa values between 100-350 h⁻¹, typical for microbial cultivations [100].
Validation: Compare growth kinetics and protein expression levels between MTP and shake flask controls at matched kLa values.
Protocol 2: Scale-Up to Stirred Tank Bioreactors
Purpose: To translate optimized cultivation conditions from microscale to stirred tank reactors while maintaining consistent process performance.
Materials:
- Laboratory-scale STR (1-5 L working volume)
- Dissolved oxygen (DO) probe and control system
- pH control system
- Exhaust gas analysis system (optional)
- Sterile sampling system
- Air or oxygen supply with mass flow controllers
Procedure:
- Calculate target kLa value for STR based on MTP characterization data.
- Determine operating conditions (agitation speed, aeration rate) that achieve target kLa using correlation equations.
- For a 1.4 L STR, typical conditions are: kLa = 370-600 h⁻¹, agitation 500-800 rpm, aeration 0.5-1.5 vvm [100].
- Implement fed-batch strategy based on carbon source consumption kinetics observed in microscale.
- Maintain dissolved oxygen above 20-30% saturation through cascade control.
- For Pichia pastoris cultivations using PAOX₁ promoter, implement methanol feed control after glycerol batch phase [103].
- Monitor key metabolites (glycerol, methanol, acetate) offline to validate metabolic state.
- Compare growth kinetics, protein titer, and product quality attributes with microscale data.
Critical Steps:
- Perform gassing-out method to verify actual kLa in STR
- Implement identical induction strategy across scales
- Maintain equivalent feed composition and feeding profiles
Scale-Up Workflow and Decision Framework
The following diagram illustrates the systematic approach for scaling up yeast fermentation processes from microtiter plates to production bioreactors.
The Scientist's Toolkit: Essential Research Reagents and Equipment
Table 2: Key Research Reagent Solutions for Yeast Fermentation Scale-Up
| Category | Specific Product/Model | Function/Application | Considerations |
|---|---|---|---|
| Microscale Cultivation | 96-deepwell MTPs (round & square) | High-throughput screening | Geometry affects oxygen transfer [102] |
| BioLector system | Online monitoring of biomass & fluorescence | Provides kinetic data from MTPs [100] | |
| μTOM device | Online oxygen transfer rate monitoring | Enables OTR-based scale-up [102] | |
| Bioreactor Systems | DASGIP or similar parallel bioreactors | Scale-down model development | Enables systematics study of process parameters |
| Standard STR (1-5 L) | Pilot-scale process development | Should have full parameter control | |
| Analytical Tools | DO and pH probes | Monitoring critical process parameters | Require proper calibration |
| HPLC/UPLC systems | Metabolite analysis | Essential for metabolic state assessment | |
| Strain Engineering | Codon-optimized genes | Enhanced translation efficiency | Critical for heterologous expression [31] |
| Improved secretion signals (pre-Ost1) | Enhanced protein secretion | Co-translational translocation [103] | |
| Protease-deficient strains | Reduced product degradation | Enhanced product stability [92] |
Addressing Scale-Up Challenges in Yeast Systems
Physiological Considerations for Yeast Cell Factories
When scaling up heterologous protein production in yeast, several host-specific factors must be considered. The metabolic burden of protein expression can alter the cellular resource allocation, potentially impacting growth and productivity at different scales [104]. Additionally, the choice of secretion signal peptide significantly influences protein translocation efficiency and final titers. Recent studies demonstrate that replacing the conventional α-MF pre-region with the Ost1 signal sequence enhances co-translational translocation, improving secretion of model proteins like E2-Crimson and lipases in Pichia pastoris [103]. Protease deficiency in production hosts such as Ogataea minuta can further enhance product yields by reducing degradation [92].
Strategies for Overcoming Oxygen Transfer Limitations
Oxygen transfer often becomes limiting at larger scales due to increased broth viscosity and longer mixing times. For yeast systems, several strategies can mitigate this constraint:
- Oxygen enrichment: Increasing oxygen partial pressure in the inlet gas
- Pressure cycling: Using increased headspace pressure to enhance oxygen solubility [101]
- Strain engineering: Expression of Vitreoscilla hemoglobin in Yarrowia lipolytica to improve oxygen utilization [101]
- Dynamic feeding strategies: Adjusting substrate feed based on dissolved oxygen signals
Quantitative Performance Comparison Across Scales
The table below summarizes typical performance metrics that can be achieved when proper scale-up methodologies are applied.
Table 3: Expected Performance Metrics Across Scales for Yeast Cultivations
| Parameter | Microtiter Plate (200 μL) | Stirred Tank Reactor (1.4 L) | Scale Factor |
|---|---|---|---|
| kLa (h⁻¹) | 100-350 [100] | 370-600 [100] | 1.1-3.5x |
| Max Biomass (OD₆₀₀) | 30-50 | 30-50 | 1x |
| Specific Growth Rate (μ, h⁻¹) | 0.15-0.25 | 0.15-0.25 | 1x |
| Heterologous Protein Titer | 90-140 mg/L (GFP) [100] | 90-140 mg/L (GFP) [100] | 1x |
| Process Duration | 24-72 h | 24-72 h | 1x |
Successful scale-up of yeast fermentation processes from microtiter plates to production bioreactors requires a systematic approach that integrates engineering principles with biological understanding. By maintaining key parameters such as kLa constant across scales and implementing the protocols described herein, researchers can achieve consistent process performance and accelerate the development of yeast cell factories for heterologous protein production. The frameworks and methodologies presented in this application note provide a foundation for robust bioprocess scale-up that can be adapted to various yeast hosts and product targets.
Conclusion
The strategic engineering of yeast cell factories for heterologous pathway expression has matured into a powerful platform for drug development and biochemical production. Success hinges on the integrated application of foundational knowledge, advanced genetic tools, systematic optimization, and rigorous validation. Key takeaways include the critical role of promoter selection tailored to fermentation stages, the emerging potential of lifespan engineering to boost production capacity, and the importance of host-specific factors for functional protein expression. Future directions point toward more sophisticated dynamic control systems, AI-assisted pathway design, and the application of these platforms to unlock novel therapeutic compounds, particularly from complex plant biosynthetic pathways. As these technologies converge, yeast cell factories are poised to become increasingly sophisticated production vehicles, accelerating the translation of genetic designs into clinically and commercially valuable bioproducts.
References
- 1. Innovative tools for customized yeast cell factory [https://www.sciencedirect.com/science/article/pii/S2693125725000111]
- 2. Recent progress on heterologous in... protein production [https://link.springer.com/article/10.1007/s11274-024-04008-9]
- 3. as a tool for Yeasts gene heterologous expression [https://pubmed.ncbi.nlm.nih.gov/22160908/]
- 4. Smart culture medium optimization for recombinant protein ... [https://www.sciencedirect.com/science/article/abs/pii/S0734975025002241]
- 5. Engineering robust Saccharomyces cerevisiae cell factory ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12408222/]
- 6. Yeast Cell Wall Extract Market Report 2025 (Global Edition) [https://www.cognitivemarketresearch.com/yeast-cell-wall-extract-market-report]
- 7. High‐Throughput Pipeline for Protein Expression and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12306927/]
- 8. Pichia pastoris: A highly successful expression system for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7228273/]
- 9. Komagataella phaffii (Pichia pastoris) as a Powerful Yeast ... [https://pubmed.ncbi.nlm.nih.gov/38939917/]
- 10. Why is Yeast Important for Recombinant Protein Expression [https://www.genscript.com/protein-news/why-is-yeast-important-for-recombinant-protein-expression.html]
- 11. Engineering Saccharomyces cerevisiae for efficient ... [https://www.sciencedirect.com/science/article/pii/S2667370323000541]
- 12. Industrial Production of Proteins with Pichia pastoris ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10046876/]
- 13. Transforming non-conventional yeasts into key players in ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1600187/full]
- 14. Saccharomyces cerevisiae and its industrial applications [https://pmc.ncbi.nlm.nih.gov/articles/PMC7099199/]
- 15. Exploring the genetic architecture of protein secretion in ... [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3001911]
- 16. Engineering genetic elements for microbial protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12629926/]
- 17. Development of an efficient heterologous protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12236015/]
- 18. Directed optimization and generation of yeast promoter ... [https://www.sciencedirect.com/science/article/abs/pii/S0141813025090622]
- 19. Identifying promoters to enhance heterologous gene ... [https://link.springer.com/article/10.1007/s00253-025-13563-6]
- 20. A toolkit for facilitating markerless integration of expression ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-025-02716-x]
- 21. Chaperone overexpression boosts heterologous small ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-025-02728-7]
- 22. Combinatorial optimization of gene expression through ... [https://www.nature.com/articles/s41467-024-44997-7]
- 23. Systematic optimization of the yeast cell factory for ... [https://www.sciencedirect.com/science/article/pii/S2405805X21000181]
- 24. Yeast Heterologous Expression Systems for the Study of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10342074/]
- 25. Plasmids 101: Yeast Vectors [https://blog.addgene.org/plasmids-101-yeast-vectors]
- 26. Yeast Vectors and Assays for Expression of Cloned Genes [https://pubmed.ncbi.nlm.nih.gov/18265101/]
- 27. Controlling heterologous gene expression in yeast cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4480987/]
- 28. Characterization of plasmid burden and copy number in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3546148/]
- 29. Posttranslational Modifications of Proteins in the Pathobiology ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3272899/]
- 30. Control of protein stability by post-translational modifications [https://www.nature.com/articles/s41467-023-35795-8]
- 31. Engineering strategies for enhanced heterologous protein ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-024-02299-z]
- 32. Post-translational modification analysis of Saccharomyces ... [https://www.sciencedirect.com/science/article/pii/S002192582000188X]
- 33. Innovative Tools and Strategies for Optimizing Yeast Cell ... [https://pubmed.ncbi.nlm.nih.gov/33008642/]
- 34. Comparative Analysis of Quantitative Mass Spectrometric ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8063867/]
- 35. A comparative review of DNA assembly strategies [https://www.sciencedirect.com/science/article/pii/S2452014425002493]
- 36. Biotechnological production of terpenoids using cell factories [https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2025.1621406/full]
- 37. Recent Developments in Heterologous Expression of ... [https://www.mdpi.com/2673-8007/5/1/22]
- 38. Identifying promoters to enhance heterologous gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12296805/]
- 39. Cloning Methods: 5 Different Ways to Assemble [https://bitesizebio.com/26961/cloning-methods-5-different-ways-to-assemble/]
- 40. Gibson Assembly vs Golden Gate: A Complete Guide to Choosing the Right Cloning Method [https://www.clyte.tech/post/gibson-assembly-vs-golden-gate-a-complete-guide-to-choosing-the-right-cloning-method]
- 41. From natural to synthetic: Promoter engineering in yeast ... [https://www.sciencedirect.com/science/article/pii/S073497502400140X]
- 42. Designing strong inducible synthetic promoters in yeasts [https://www.nature.com/articles/s41467-024-54865-z]
- 43. Controlling heterologous gene expression in yeast cell ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-015-0278-5]
- 44. Engineering heterologous core promoters to optimize the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12617796/]
- 45. Controlling gene expression in yeast by inducible site- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC115252/]
- 46. Comparison of Yeasts as Hosts for Recombinant Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6027275/]
- 47. ECO-Casein/iGEM NCHU 2025/methods [https://2025.igem.wiki/nchu/experiments/]
- 48. CRISPR Guide [https://www.addgene.org/guides/crispr/]
- 49. CRISPR-Cas9 Gene Editing in Yeast: A Molecular Biology ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8442027/]
- 50. CrEdit: CRISPR mediated multi-loci gene integration in ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-015-0288-3]
- 51. CRISPR/Cas9-based iterative multi-copy integration for ... [https://www.sciencedirect.com/science/article/pii/S2405805X2500033X]
- 52. Leveraging yeast to characterize plant biosynthetic gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10664738/]
- 53. Fine‐tuning the yeast GAL10 promoter and growth ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12012841/]
- 54. Interaction of genetic variants activates latent metabolic ... [https://www.nature.com/articles/s41467-025-63306-4]
- 55. Engineering strategies for enhanced heterologous protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10801990/]
- 56. Alleviation of ER stress via targeted genetic engineering ... [https://link.springer.com/article/10.1007/s10068-025-02044-1]
- 57. Automated strain construction for biosynthetic pathway ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12611344/]
- 58. Artificial chromosome reorganization reveals high plasticity of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12309097/]
- 59. Rewiring of the protein–protein–metabolite interactome during ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9569316/]
- 60. High-throughput metabolomics for the design and ... [https://www.nature.com/articles/s41540-023-00274-9]
- 61. The type of carbon source not the growth rate it supports ... [https://www.nature.com/articles/s42003-025-07747-z]
- 62. An Investigation of TDA1 Deficiency in Saccharomyces ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12232523/]
- 63. Switching the yeast metabolism via manipulation of sugar ... [https://www.sciencedirect.com/science/article/abs/pii/S1096717625000230]
- 64. Engineering chronological lifespan toward a robust yeast ... [https://pubmed.ncbi.nlm.nih.gov/41213019/]
- 65. Targeting metabolic pathways for extension of lifespan and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9038119/]
- 66. A quantitative yeast aging proteomics analysis reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8599547/]
- 67. Microfluidic Technologies for Yeast Replicative Lifespan Studies [https://pmc.ncbi.nlm.nih.gov/articles/PMC5035173/]
- 68. Metabolomic-based aging clocks | npj Metabolic Health ... [https://www.nature.com/articles/s44324-025-00078-x]
- 69. Compatibility engineering of synthetic metabolic pathways ... [https://www.sciencedirect.com/science/article/abs/pii/S0009250925014447]
- 70. Codon Optimization: Understanding the Basics | IDT [https://www.idtdna.com/page/support-and-education/decoded-plus/codon-optimization-the-basics-explained/]
- 71. Comparative Analysis of Codon Optimization Tools [https://pmc.ncbi.nlm.nih.gov/articles/PMC12010093/]
- 72. From genotype to phenotype with 1086 near telomere-to- ... [https://www.nature.com/articles/s41586-025-09637-0]
- 73. Deep generative optimization of mRNA codon sequences ... [https://www.nature.com/articles/s41467-025-64894-x]
- 74. Codon Optimization Tools: What to Look for in 2025 [https://synapse.patsnap.com/article/codon-optimization-tools-what-to-look-for-in-2025]
- 75. Burden Imposed by Heterologous Protein Production in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10512257/]
- 76. Metabolomic Alterations Do Not Induce Metabolic Burden in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6893308/]
- 77. Computational Modelling of Metabolic Burden and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6921056/]
- 78. Customized yeast cell factories for biopharmaceuticals [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-021-01617-z]
- 79. Yeast complementation assays provide limited information ... [https://pubmed.ncbi.nlm.nih.gov/40086750/]
- 80. Variant effect predictor correlation with functional assays is ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12016141/]
- 81. Functional complementation of yeast [https://bio-protocol.org/exchange/minidetail?id=3057723&type=30]
- 82. Yeast complementation assays provide limited information ... [https://www.sciencedirect.com/science/article/pii/S2667074725000114]
- 83. GFP Assay Using Simple Western [https://www.bio-techne.com/resources/protocols-troubleshooting/simple-western-gfp-assay]
- 84. Immunoprecipitation Protocol Using Anti-GFP Antibodies [https://www.thermofisher.com/us/en/home/references/protocols/proteins-expression-isolation-and-analysis/gfp-protocol/immunoprecipitation-protocol-using-anti-gfp-antibodies.html]
- 85. Western blot protocol [https://www.abcam.com/en-us/technical-resources/protocols/western-blot]
- 86. Guide to Enzyme Unit Definitions and Assay Design [https://resources.biomol.com/biomol-blog/guide-to-enzyme-unit-definitions-and-assay-design]
- 87. Basics of Enzymatic Assays for HTS - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK92007/]
- 88. Enzyme Assay Analysis: What Are My Method Choices? [https://www.thermofisher.com/blog/analyteguru/enzyme-assay-analysis-what-are-my-method-choices/]
- 89. Heterologous expression and optimization of the antimicrobial ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12535540/]
- 90. Optimizing Yeast Surface-Displayed Unspecific ... [https://www.mdpi.com/2306-5354/12/8/822]
- 91. Development of an efficient heterologous protein expression ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-025-02786-x]
- 92. Optimization of heterologous protein production process ... [https://www.sciencedirect.com/science/article/abs/pii/S1389172325002208]
- 93. Integrative glycomic analysis reveals the crucial role of protein ... [https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1013325]
- 94. Uncovering protein glycosylation dynamics and ... [https://www.nature.com/articles/s41594-025-01485-w]
- 95. Identification of improved signal peptides for heterologous ... [https://www.nature.com/articles/s41598-025-09669-6]
- 96. Transcriptomics-based identification of novel factors enhancing heterologous protein secretion in yeasts - PubMed [https://pubmed.ncbi.nlm.nih.gov/17766460/]
- 97. Recent advances in genetic engineering and chemical production in yeast species - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11963765/]
- 98. Directional integration and pathway enrichment analysis ... [https://www.nature.com/articles/s41467-024-49986-4]
- 99. A guide to multi-omics data collection and integration for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9747357/]
- 100. Scale-up from microtiter plate to laboratory fermenter [https://pmc.ncbi.nlm.nih.gov/articles/PMC2806293/]
- 101. Challenges and progress towards industrial recombinant ... [https://www.sciencedirect.com/science/article/abs/pii/S0734975023000289]
- 102. Scale-up of CHO cell cultures: from 96-well-microtiter plates to ... [https://jbioleng.biomedcentral.com/articles/10.1186/s13036-024-00475-8]
- 103. Bioreactor-scale cell performance and protein production ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7680431/]
- 104. Enhanced production of heterologous proteins by a synthetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7179936/]