FBA vs. MFA for E. coli Metabolic Flux Prediction: A Comprehensive Guide for Biomedical Researchers
This article provides a systematic comparison of Flux Balance Analysis (FBA) and Metabolic Flux Analysis (MFA) for predicting metabolic fluxes in Escherichia coli.
FBA vs. MFA for E. coli Metabolic Flux Prediction: A Comprehensive Guide for Biomedical Researchers
Abstract
This article provides a systematic comparison of Flux Balance Analysis (FBA) and Metabolic Flux Analysis (MFA) for predicting metabolic fluxes in Escherichia coli. Aimed at researchers and scientists in metabolic engineering and drug development, we explore the foundational principles, methodological applications, troubleshooting strategies, and validation frameworks for both techniques. By synthesizing recent advances, including hybrid machine-learning approaches and genome-scale flux analysis, this guide serves as a resource for selecting the appropriate method, interpreting results, and leveraging the complementary strengths of FBA and MFA to gain accurate insights into E. coli metabolism for biotechnological and biomedical applications.
Core Principles: Understanding FBA and MFA Fundamentals in E. coli Metabolism
Constraint-Based Modeling, and specifically Flux Balance Analysis (FBA), represents a cornerstone computational method in systems biology for predicting metabolic flux distributions in biological systems. When contrasted with Metabolic Flux Analysis (MFA), particularly in E. coli research, these frameworks provide complementary approaches for quantifying intracellular reaction rates. FBA utilizes mathematical optimization to predict flux distributions that maximize or minimize a specific cellular objective, such as biomass production, based on stoichiometric constraints and steady-state assumptions [1] [2]. In contrast, 13C-MFA employs isotopic tracer experiments and computational modeling to experimentally determine metabolic fluxes, serving as a gold standard for validating FBA predictions [3] [4].
The fundamental distinction lies in their approach: FBA is a predictive tool that relies on optimization principles, while MFA is an estimative technique grounded in experimental measurement. Both methods assume metabolic steady-state, where metabolite concentrations and reaction rates remain constant, but they differ significantly in their data requirements, scalability, and applications [3]. For E. coli researchers, understanding this dichotomy is essential for selecting the appropriate methodology based on research goals, whether for fundamental biological discovery or metabolic engineering applications.
Comparative Analysis: FBA vs. MFA
Table 1: Fundamental characteristics and applications of FBA and MFA
| Feature | Flux Balance Analysis (FBA) | Metabolic Flux Analysis (MFA) |
|---|---|---|
| Primary Approach | Prediction via linear optimization | Estimation via isotopic tracing & computational modeling |
| Core Data Required | Stoichiometric matrix, objective function, constraints | 13C-labeling patterns, extracellular fluxes |
| Model Scale | Genome-scale (thousands of reactions) | Central metabolism (dozens to hundreds of reactions) |
| Temporal Resolution | Steady-state only | Steady-state (13C-MFA) or dynamic (INST-MFA) |
| Key Assumptions | Steady-state, optimization principle | Quasi-steady-state, isotopic steady-state (for 13C-MFA) |
| Throughput | High (computational) | Low to medium (experimentally intensive) |
| Primary Validation Method | Comparison with experimental growth rates & gene essentiality | Statistical goodness-of-fit tests (e.g., χ²-test) |
| Key Applications in E. coli Research | Gene knockout prediction, growth rate prediction, network exploration | Quantitative flux mapping, pathway validation, engineering verification |
Table 2: Performance comparison for predicting E. coli fluxes
| Performance Metric | FBA | MFA | Experimental Notes |
|---|---|---|---|
| Growth Rate Prediction | High accuracy for wild-type [1] | Not applicable (measured input) | FBA successful in predicting uptake/release rates [1] |
| Gene Essentiality | High accuracy [1] | Not directly applicable | FBA successfully predicts single-gene knockout effects [1] |
| Internal Flux Prediction | Variable accuracy [1] [4] | High accuracy (<5% confidence intervals) [3] | FBA limited by solution space degeneracy [1] |
| Central Carbon Metabolism | Requires additional constraints [1] | Gold standard [4] | 13C-MFA provides precise quantification [4] |
| Computational Tractability | High (seconds to minutes) | Medium to high (hours) | 13C-MFA computation has advanced significantly [3] |
| Experimental Burden | Low (requires only stoichiometry) | High (requires isotopic labeling) | Parallel labeling experiments improve MFA precision [3] |
Methodological Frameworks and Experimental Protocols
Fundamental FBA Workflow for E. coli
The standard FBA protocol for E. coli involves several methodical steps. First, a genome-scale metabolic model (such as iML1515 or iJO1366) provides the stoichiometric matrix that encapsulates all known metabolic reactions [2] [5]. The model is constrained by defining upper and lower bounds for each reaction based on physiological data. A biologically relevant objective function (typically biomass production) is optimized using linear programming, generating a flux distribution that maximizes the objective while satisfying all constraints [1] [2].
Figure 1: The core FBA workflow for E. coli metabolism
Advanced FBA Extensions and Integration Protocols
Several sophisticated FBA extensions have been developed to enhance prediction accuracy for E. coli. corsoFBA implements a two-step optimization that first fixes the biomass objective at a predefined value, then minimizes protein cost throughout the metabolism, better predicting internal fluxes at sub-optimal growth [1]. REMI (Relative Expression and Metabolomic Integrations) incorporates gene expression, metabolite abundance, and thermodynamic data into a single optimization framework, significantly improving flux predictions compared to traditional FBA [6]. METAFlux adapts FBA for transcriptomic data, calculating metabolic reaction activity scores from gene expression levels before optimizing biomass production [7].
Enzyme-constrained FBA has emerged as a particularly powerful approach. The ECMpy workflow incorporates enzyme constraints by splitting reversible reactions, assigning kcat values, and incorporating molecular weights to create more realistic flux predictions [2]. This method has demonstrated improved accuracy for predicting E. coli metabolic behavior compared to traditional FBA.
13C-MFA Experimental Protocol
The standard 13C-MFA protocol for E. coli involves culturing cells with 13C-labeled substrates (typically glucose or glutamine), followed by precise analytical measurements and computational modeling [3] [4]. After cultivating E. coli in defined medium with 13C-labeled carbon sources, mass isotopomer distributions of intracellular metabolites are measured using GC-MS or LC-MS. These measurements, combined with extracellular flux data, are used to compute metabolic fluxes that best explain the observed labeling patterns through iterative computational fitting [3].
Figure 2: 13C-MFA workflow for experimental flux determination
Dynamic and Hybrid Approaches
Dynamic MFA extends traditional MFA to transient cultures by transforming time-series concentration measurements into flux values through polynomial smoothing and differentiation [8]. This approach has been successfully applied to E. coli cultivations shifting between carbon and nitrogen limitation, revealing lag phases and changes in maintenance energy requirements [8]. Flux sampling techniques, such as OptGP, generate statistical distributions of possible flux states rather than single solutions, providing a more comprehensive view of metabolic capabilities [5].
Research Toolkit for E. coli Flux Analysis
Table 3: Essential research reagents and computational tools for E. coli flux studies
| Category | Specific Tool/Reagent | Function/Application | Example Use Case |
|---|---|---|---|
| Metabolic Models | iML1515 [2] | Genome-scale model of E. coli K-12 MG1655 | General FBA simulations |
| iJO1366 [5] | Comprehensive E. coli metabolic network | Acetate production studies | |
| Software & Algorithms | COBRApy [2] [5] | Python package for constraint-based modeling | Implementing FBA and variants |
| ECMpy [2] | Workflow for adding enzyme constraints | Protein-cost aware flux prediction | |
| REMI [6] | Integrates multi-omics data with thermodynamics | Conditional flux comparisons | |
| METAFlux [7] | Infers fluxes from transcriptomic data | Single-cell flux analysis | |
| Experimental Reagents | 13C-labeled glucose [3] [4] | Isotopic tracer for MFA | Central carbon flux determination |
| Defined minimal media [8] | Controlled nutrient environment | Reproducible cultivation | |
| Analytical Instruments | GC-MS / LC-MS [3] | Measures mass isotopomer distributions | 13C-MFA data collection |
| Seahorse XF Analyzer [7] | Measures extracellular flux rates | Glycolytic and respiratory capacity |
Interpretation Guidelines and Validation Standards
Validation Approaches for FBA Predictions
Validating FBA predictions requires multiple complementary approaches. Comparison with experimental 13C-MFA fluxes provides the most direct validation for internal fluxes, particularly in central carbon metabolism [3] [4]. Growth rate predictions can be validated against measured growth rates in defined conditions, while gene essentiality predictions can be tested against knockout libraries like the Keio collection [4]. For methods incorporating additional constraints, such as enzyme-constrained FBA, validation should demonstrate improved accuracy over standard FBA without significant loss of predictive capacity for known physiological behavior [2].
Statistical Validation for MFA
13C-MFA relies heavily on statistical tests to validate flux estimates. The χ²-test of goodness-of-fit compares measured and simulated mass isotopomer distributions to assess model quality [3]. Additionally, flux uncertainty estimation through Monte Carlo sampling or sensitivity analysis provides confidence intervals for flux estimates [3]. For comprehensive validation, parallel labeling experiments using multiple tracer compounds can test the consistency of flux estimates across different labeling patterns [3].
Reporting Standards
Comprehensive reporting should include: sensitivity analysis for objective function selection in FBA, measures of solution space degeneracy, statistical goodness-of-fit measures for MFA, and explicit documentation of model constraints and assumptions [3] [1]. This transparency enables proper evaluation and reproduction of flux analysis results.
Flux Balance Analysis (FBA) and Metabolic Flux Analysis (MFA) represent two cornerstone methodologies for investigating metabolic networks in biological systems, each with distinct approaches and applications. FBA is a constraint-based modeling technique that predicts metabolic fluxes using a stoichiometric model of the metabolic network and an optimization principle, without necessarily requiring extensive experimental data [3]. In contrast, MFA, particularly 13C-MFA, relies on experimental measurements from isotopic labeling experiments to estimate intracellular fluxes, providing a more data-driven approach to flux determination [9]. Both methods operate under the fundamental assumption of metabolic steady-state, where reaction rates and metabolite concentrations remain constant over time [3] [10].
The selection of an appropriate objective function constitutes perhaps the most critical step in FBA, as it embodies a hypothesis about what the biological system has been evolutionarily optimized to accomplish [3]. The objective function, which is typically maximized or minimized through linear programming, directs the flow of metabolites through the network to achieve a particular biological goal. While biomass maximization has become the default objective for many FBA applications, particularly involving microbial systems, evidence suggests that organisms may not always operate at optimal growth states, necessitating the exploration of alternative objective functions [1] [10]. This comparative guide examines the performance of different objective functions in FBA relative to the experimental flux values obtained through 13C-MFA, with a specific focus on E. coli as a model organism.
Established Objective Functions in FBA
Biomass Maximization as the Standard Paradigm
The principle of biomass maximization posits that microorganisms, particularly unicellular organisms like E. coli, have evolved to maximize their growth rate under given environmental conditions. This objective function is mathematically represented as a biomass reaction that consumes cellular building blocks—including amino acids, nucleotides, lipids, and cofactors—in proportions that reflect the cellular composition [1]. The widespread adoption of biomass maximization as the primary objective function in FBA stems from its notable successes in predicting various physiological phenomena, including substrate uptake rates, growth rates under different environmental conditions, and gene essentiality in E. coli [1]. The method's computational tractability, combined with its minimal requirement for experimental input data, has made it particularly valuable for analyzing genome-scale metabolic models (GSSMs) that encompass all known metabolic reactions in an organism [3] [10].
However, the assumption of optimal growth has been increasingly questioned. Several studies suggest that unicellular organisms may not always operate at maximum growth capacity, instead functioning in sub-optimal states that necessitate exploration beyond pure biomass maximization [1]. This limitation becomes particularly evident when comparing FBA predictions with experimental flux measurements obtained through 13C-MFA, where discrepancies in internal flux distributions often emerge despite reasonable agreement with growth phenotypes [1].
Alternative Objective Functions and Constraints
The recognition that biomass maximization alone cannot fully capture the complexity of metabolic operation has spurred the development of numerous alternative objective functions. These alternatives are typically grounded in different biological principles or optimization strategies, including:
Minimization of Metabolic Adjustment (MOMA): This approach employs quadratic programming to identify a flux distribution that minimizes the metabolic adjustment between wild-type and mutant strains, proving particularly valuable for predicting the behavior of knockout mutants [3] [10].
Regulatory On/Off Minimization (ROOM): This method identifies flux distributions that minimize the number of significant flux changes compared to a reference state, using mixed-integer linear programming to effectively capture regulatory responses [3] [10].
Minimization of total flux: This strategy assumes that metabolic systems have evolved to minimize the total amount of enzyme investment, thereby representing a resource-efficient solution [3].
ATP maximization: Recent research in cancer metabolism has demonstrated that considering ATP maximization with enthalpy change limitations can better explain the preference for aerobic glycolysis observed in many cancer cell lines [11].
Protein cost optimization: Methods like corsoFBA incorporate protein molecular weight and thermodynamic penalties to minimize the overall protein cost at sub-optimal growth levels, resulting in improved predictions of internal flux distributions in E. coli central carbon metabolism [1].
These alternative approaches reflect a growing recognition that biological systems operate under multiple competing selective pressures beyond growth rate optimization alone.
Table 1: Overview of Primary Objective Functions Used in FBA
| Objective Function | Mathematical Approach | Biological Rationale | Primary Applications |
|---|---|---|---|
| Biomass Maximization | Linear Programming | Organisms evolve to maximize growth rate | Prediction of growth rates, substrate usage, gene essentiality |
| MOMA | Quadratic Programming | Metabolism minimizes redistribution after perturbation | Prediction of mutant strain behavior |
| ROOM | Mixed-Integer Linear Programming | Cells minimize significant flux changes | Prediction of regulatory responses in mutants |
| ATP Maximization | Linear Programming with enthalpy constraints | Energy efficiency under thermal constraints | Cancer metabolism, aerobic glycolysis |
| Protein Cost Minimization | Two-step optimization with molecular weight and thermodynamic penalties | Cellular resource allocation efficiency | Sub-optimal growth states, internal flux prediction |
Comparative Analysis: FBA Predictions vs. 13C-MFA Measurements in E. coli
Quantitative Performance Assessment
Direct comparisons between FBA predictions and 13C-MFA measurements in E. coli reveal significant variations in performance across different objective functions and growth conditions. A comprehensive study examining aerobic and anaerobic growth in E. coli K-12 MG1655 demonstrated that FBA could successfully predict product secretion rates in aerobic cultures when constrained with both glucose and oxygen uptake measurements [12]. However, the internal flux distributions generated through sampling the feasible solution space showed substantial discrepancies from 13C-MFA-derived fluxes, particularly through central carbon metabolism [12].
The synergy between 13C-MFA and FBA analyses revealed several physiological insights that neither method could provide independently. For instance, the 13C-MFA flux maps indicated that the fraction of maintenance ATP consumption in total ATP production was approximately 14% higher under anaerobic conditions (51.1%) compared to aerobic conditions (37.2%) [12]. FBA further elucidated that this increased ATP utilization was consumed by ATP synthase to secrete protons during fermentation processes [12]. Additionally, the TCA cycle was shown to operate in a non-cyclic manner in aerobically growing E. coli, with submaximal growth resulting from limitations in oxidative phosphorylation capacity—a finding that emerged from the combined application of both methodologies [12].
Table 2: Comparison of Experimental and FBA-Predicted Fluxes in E. coli Central Carbon Metabolism (Aerobic Conditions)
| Metabolic Pathway/Reaction | 13C-MFA Flux Value | Biomax-FBA Prediction | Protein Cost Minimization Prediction | Reference |
|---|---|---|---|---|
| Glycolysis | ||||
| Glucose uptake | 100.0 | 100.0 | 100.0 | [12] |
| PEP carboxylase | 15.8 | - | 14.2 | [1] |
| TCA Cycle | ||||
| Citrate synthase | 16.1 | 84.3 | 22.5 | [12] |
| AKG dehydrogenase | 16.1 | 84.3 | 20.8 | [12] |
| Oxidative Phosphorylation | ||||
| ATP yield (mol ATP/mol glucose) | 22.5 | 36.4 | 25.8 | [12] |
| Maintenance ATP (%-total ATP) | 37.2 | - | 35.1 | [12] |
Methodological Limitations and Validation Challenges
Several fundamental limitations affect the validation of FBA predictions against 13C-MFA measurements. A significant challenge arises from the existence of multiple optimal intracellular flux distributions that satisfy the same objective function equally well [12] [1]. This non-uniqueness of FBA solutions means that a range of flux maps can produce identical objective function values, complicating direct comparisons with experimental flux measurements [1].
Furthermore, the assumption that organisms operate at optimal growth states has been repeatedly questioned. Research indicates that E. coli central carbon metabolism behavior can be better predicted by exploring the sub-optimal FBA solution space rather than strictly optimal solutions [1]. Techniques such as Flux Variability Analysis (FVA) demonstrate that the FBA solution space expands dramatically when considering near-optimal to optimal states, accommodating substantial flux variability while maintaining similar objective function values [1].
Validation practices in FBA also vary considerably across studies. While quality control checks—such as ensuring models cannot generate ATP without an external energy source or synthesize biomass without essential substrates—represent important initial validation steps, comprehensive validation of internal flux predictions remains challenging [10]. The most robust validation approaches involve comparing FBA predictions against 13C-MFA measurements, though this requires careful consideration of model structure, constraints, and experimental conditions [3] [10].
Experimental Protocols and Methodologies
Standard 13C-MFA Protocol for E. coli
The following protocol outlines the standard methodology for conducting 13C-MFA in E. coli, which serves as the reference for validating FBA predictions:
Strain and Culture Conditions: E. coli K-12 MG1655 is cultured in defined minimal medium (e.g., M9) with uniformly labeled [U-13C] glucose (2 g/L) as the sole carbon source [12]. Both aerobic and anaerobic cultures are incubated at 37°C with appropriate agitation.
Isotopic Steady-State Achievement: Cells are harvested during mid-log phase after ensuring isotopic steady state has been reached, typically requiring multiple generations of growth on the labeled substrate [9] [12].
Metabolite Extraction and Analysis: Intracellular metabolites are extracted using appropriate quenching methods (e.g., cold methanol). Proteinogenic amino acids are hydrolyzed from cellular protein, while intracellular metabolic intermediates are directly extracted [12].
Mass Isotopomer Distribution Measurement: The 13C-labeling patterns of proteinogenic amino acids and intracellular metabolites are analyzed using GC-MS (Gas Chromatography-Mass Spectrometry) and LC-MS (Liquid Chromatography-Mass Spectrometry) [9] [12]. Additional analytical techniques including NMR spectroscopy, enzymatic assays, and gas analysis may be employed for extracellular flux measurements [12].
Flux Calculation: Computational tools such as INCA, OpenFLUX, or METRAN are used to estimate metabolic fluxes by minimizing the difference between measured and simulated mass isotopomer distributions through iterative optimization algorithms [9].
FBA with Alternative Objective Functions Protocol
The following protocol describes the implementation of FBA with alternative objective functions for comparison with 13C-MFA results:
Model Selection and Preparation: A genome-scale metabolic model of E. coli (e.g., iJR904 or iJO1366) is obtained from curated databases such as BiGG [12] [5]. Basic quality control checks are performed using tools like MEMOTE (MEtabolic MOdel TEsts) to ensure model functionality and consistency [10].
Constraint Application: Experimentally measured external fluxes (e.g., glucose uptake, oxygen consumption, product secretion rates) are applied as constraints to the model [12] [5]. Additional constraints may include thermodynamic feasibility and enzyme capacity limitations.
Objective Function Implementation:
- For biomass maximization: The biomass reaction is set as the objective to be maximized [1].
- For protein cost minimization: A two-step optimization is performed where the biomass objective is first fixed at a sub-optimal value, followed by minimization of the protein cost term incorporating enzyme molecular weights and thermodynamic penalties [1].
- For ATP maximization: The ATP maintenance or production reaction is set as the objective, potentially with additional constraints on enthalpy changes [11].
Flux Sampling and Analysis: When multiple optimal solutions exist, flux sampling techniques such as OptGP are employed to characterize the range of possible flux distributions [5]. For genome-scale models, this may involve generating multiple constraint patterns to ensure sufficient phenotypic variation [5].
Validation and Comparison: The resulting flux distributions are statistically compared with 13C-MFA measurements, with particular focus on key central metabolic pathways including glycolysis, TCA cycle, and pentose phosphate pathway [12].
Diagram 1: Integrated Workflow for Comparing FBA Objective Functions Against 13C-MFA Experimental Data. The diagram illustrates the parallel experimental and computational frameworks, their convergence through comparative validation, and the iterative refinement process that leads to biological insights.
Table 3: Essential Research Reagents and Computational Tools for FBA-MFA Comparative Studies
| Category | Specific Resource | Function/Application | Example Use Case |
|---|---|---|---|
| Strains and Culture | E. coli K-12 MG1655 | Model organism for metabolic studies | Reference strain for method comparison [12] |
| Isotopic Tracers | [U-13C] Glucose | Uniformly labeled carbon source | 13C-MFA experiments for central carbon metabolism [9] [12] |
| Analytical Instruments | GC-MS System | Measurement of mass isotopomer distributions | Quantifying 13C labeling in proteinogenic amino acids [9] [12] |
| Analytical Instruments | LC-MS System | Comprehensive metabolome analysis | Measuring labeling patterns of intracellular metabolites [9] [5] |
| Computational Tools | COBRA Toolbox | MATLAB-based FBA implementation | Constraint-based reconstruction and analysis [10] |
| Computational Tools | cobrapy | Python-based FBA implementation | Genome-scale metabolic modeling and analysis [10] |
| Computational Tools | INCA | 13C-MFA software package | Flux estimation from isotopic labeling data [9] |
| Metabolic Models | iJR904 | E. coli genome-scale model | FBA simulations and validation [12] |
| Metabolic Models | iJO1366 | E. coli genome-scale model | Large-scale flux sampling studies [5] |
| Database Resources | BiGG Models | Curated metabolic model database | Access to standardized, validated models [10] |
The comparative analysis of objective functions in FBA reveals a complex landscape where no single objective function universally outperforms others across all metabolic contexts. While biomass maximization remains valuable for predicting growth phenotypes and substrate utilization, its limitations in accurately predicting internal flux distributions highlight the need for more sophisticated approaches [1]. Alternative objective functions, particularly those incorporating protein cost minimization and thermodynamic constraints, demonstrate improved agreement with 13C-MFA measurements, especially for central carbon metabolism fluxes [1].
The integration of 13C labeling data directly with genome-scale models represents a promising frontier that may bridge the gap between comprehensive network coverage and accurate flux estimation [13]. Such integrated approaches leverage the strengths of both methodologies—the system-wide perspective of FBA and the precise flux constraints provided by 13C labeling data [13]. Furthermore, the development of more sophisticated validation frameworks and benchmark datasets will be crucial for advancing objective function selection and improving the predictive power of constraint-based models [3] [10].
As metabolic engineering and systems biology continue to tackle increasingly complex biological systems, from microbial cell factories to human diseases, the refinement of objective functions will remain essential for translating metabolic models into actionable biological insights and successful engineering outcomes.
Within the context of comparing Flux Balance Analysis (FBA) and Metabolic Flux Analysis (MFA) for E. coli flux prediction, understanding the experimental basis of 13C-MFA is paramount. Unlike FBA, which predicts fluxes from stoichiometric models and optimization principles, 13C-MFA provides an experimentally grounded approach for quantifying intracellular metabolic fluxes in vivo [3]. This guide objectively compares the performance of 13C-MFA, detailing the protocols and data that form the bedrock of this powerful technique.
At its core, 13C-MFA relies on tracking stable isotopic tracers, most commonly 13C-labeled substrates, as they propagate through the metabolic network. The resulting labeling patterns in metabolites are measured and used to compute metabolic fluxes [14]. This process provides a direct, empirical window into cellular physiology that is complementary to the theoretical predictions of FBA.
The Core Workflow of a 13C-MFA Experiment
The process of 13C-MFA integrates wet-lab experiments with computational modeling to determine flux maps. The following diagram illustrates the key stages of this workflow, from experimental design to flux validation.
Methodological Diversity in 13C-MFA
The 13C-MFA technique is not a single method but a family of approaches, classified based on the metabolic and isotopic steady state of the system. The choice of method depends on the biological question and experimental constraints.
Table 1: Classification of 13C Metabolic Flux Analysis Methods
| Method Type | Applicable System | Key Feature | Computational Complexity |
|---|---|---|---|
| Stationary MFA (SS-MFA) | Fluxes, metabolites, and their labeling are constant [14] | Relies on isotopic steady-state; most established method | Medium [14] |
| Isotopically Instationary MFA (INST-MFA) | Fluxes and metabolites are constant, but labeling is variable [14] | Uses early time-course labeling data; avoids long cultivation | High [14] |
| Metabolically Instationary MFA | Fluxes, metabolites, and labeling are all variable [14] | Captures dynamic metabolic transitions | Very High [14] |
| 13C Flux Ratios (FR) | Systems where flux, metabolites, and labeling are constant [14] | Provides local, relative flux ratios at metabolic branch points | Medium [14] |
| 13C Kinetic Flux Profiling (KFP) | Systems where flux and metabolites are constant while labeling is variable [14] | Estimates absolute flux through sequential linear reactions | Medium [14] |
Tracer Selection: A Critical Parameter for Flux Resolution
The choice of the 13C-labeled substrate (tracer) is arguably the most critical experimental design parameter, as it directly determines the information content of the labeling data and the precision of estimated fluxes [15] [16]. Different tracers illuminate different pathways, and their performance can be quantitatively compared.
Performance Comparison of Common Glucose Tracers
Table 2: Quantitative Comparison of Glucose Tracers for 13C-MFA in E. coli [17]
| Tracer | Relative Precision Score | Key Application Notes |
|---|---|---|
| [1,6-13C]Glucose | ~8.5 | Top-performing single tracer for overall network flux resolution [17]. |
| [1,2-13C]Glucose | ~7.5 | Excellent for parallel labeling experiments; synergistic with [1,6-13C]glucose [17]. |
| [U-13C]Glucose | Varies | Provides global labeling but can lead to high flux correlations; often used in mixtures [14] [17]. |
| 80% [1-13C]Glucose +\n20% [U-13C]Glucose | 1.0 (Reference) | Widely used mixture due to lower cost, but significantly less precise than optimal tracers [17]. |
The optimal tracer is not universal but depends on the pathway of interest. For instance:
- [2,3,4,5,6-13C]Glucose has been identified as optimal for resolving the oxidative pentose phosphate pathway (oxPPP) flux [18].
- [3,4-13C]Glucose is highly effective for elucidating pyruvate carboxylase (PC) flux [18].
- [U-13C]Glutamine emerges as the preferred tracer for analyzing the tricarboxylic acid (TCA) cycle in mammalian cells [16].
A rational framework for tracer design uses the Elementary Metabolite Unit (EMU) method to decouple substrate labeling from flux dependencies, allowing for an a priori selection of optimal tracers [15] [18].
Detailed Experimental Protocols for Key 13C-MFA Workflows
Protocol for Stationary 13C-MFA in Microbial Systems (e.g., E. coli)
This protocol outlines the key steps for a standard stationary 13C-MFA experiment in a microbial system like E. coli [17] [19].
Culture and Tracer Experiment:
- Grow cells in minimal medium with a single carbon source (e.g., glucose).
- At mid-exponential phase, administer the chosen 13C-tracer (e.g., [1,6-13C]glucose).
- Harvest cells rapidly once isotopic steady state is achieved, where the labeling patterns of intracellular metabolites no longer change. This typically occurs after several generations.
Metabolite Extraction and Derivatization:
- Quench Metabolism: Rapidly cool the culture using cold methanol or other quenching solutions to instantly halt metabolic activity.
- Extract Metabolites: Disrupt cells and extract polar intracellular metabolites using a solvent system like chloroform/methanol/water.
- Derivatize: Chemically modify metabolites to make them volatile for GC-MS analysis. A common procedure involves:
- Dissolving dried polar metabolites in 2% methoxyamine hydrochloride in pyridine and incubating at 37°C for 2 hours (methoximation).
- Subsequent addition of N-methyl-N-(tert-butyldimethylsilyl)trifluoroacetamide (MTBSTFA) + 1% TBDMCS and incubation at 55°C for 1 hour (silylation) [16].
Mass Spectrometry Measurement:
- Analyze derivatized samples using Gas Chromatography-Mass Spectrometry (GC-MS).
- Operate the MS in electron impact (EI) ionization mode and use Selected Ion Monitoring (SIM) to enhance sensitivity for specific metabolite fragments.
- The output is the Mass Isotopologue Distribution (MID) for each measured fragment, which reports the fractional abundances of masses M+0, M+1, M+2, etc. [16] [19].
Protocol for Isotopically Nonstationary MFA (INST-MFA)
INST-MFA is used when achieving isotopic steady state is impractical, such as in slow-growing cells or complex mammalian systems [14] [20].
Tracer Pulse and Rapid Sampling:
- Switch the carbon source from natural abundance to the 13C-labeled tracer at time zero.
- Collect multiple culture samples at short, precise time intervals (e.g., seconds or minutes) before isotopic steady state is reached.
Measurement of Pool Sizes and Labeling:
- Extract metabolites as in SS-MFA.
- Measure both the concentration (pool size) and the time-dependent MID for each metabolite. This requires calibration with known standards for absolute quantification.
Computational Flux Estimation:
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagent Solutions for 13C-MFA
| Item | Function in 13C-MFA | Example Use Cases |
|---|---|---|
| 13C-Labeled Tracers | To introduce a measurable label into metabolism, enabling flux tracing. | [1-13C]Glucose, [U-13C]Glucose, [1,2-13C]Glucose, 13C-Glutamine [16] [17]. |
| Mass Spectrometer | To accurately measure the mass isotopologue distributions (MIDs) of metabolites. | GC-MS for derivatized samples; LC-MS/MS for underivatized polar metabolites [21] [22]. |
| Metabolic Network Model | A stoichiometric model with atom mappings to simulate label propagation. | A core model of E. coli metabolism with ~70 reactions and atom transitions [19]. |
| Flux Estimation Software | To computationally estimate fluxes by fitting the model to the experimental MIDs. | Metran, 13CFLUX2, INCA, SUMOFLUX [16] [21] [22]. |
| Quenching Solution | To instantaneously halt all metabolic activity at the time of sampling. | Cold aqueous methanol solution (~60%) [19]. |
13C-MFA stands as a powerful, empirically-driven counterpart to FBA. Its strength lies in its basis in direct experimental measurement of isotope labeling, providing a high-resolution, quantitative picture of in vivo metabolic fluxes. While the choice of tracer, analytical method, and modeling framework significantly influences the precision of the results, standardized protocols and rational design frameworks have matured 13C-MFA into an indispensable tool for validating FBA predictions and uncovering the true functional state of cellular metabolism.
The Unique Strengths of FBA (Genome-Scale Prediction) and MFA (Experimental Precision)
In the field of systems biology and metabolic engineering, understanding the flow of metabolites through metabolic networks—known as metabolic flux—is crucial for deciphering cellular physiology and guiding engineering strategies. Flux Balance Analysis (FBA) and 13C-Metabolic Flux Analysis (13C-MFA) have emerged as two dominant constraint-based modeling frameworks for estimating these in vivo reaction rates [10] [3]. Both methods utilize metabolic network models operating at a metabolic steady state, where reaction rates and metabolite concentrations are assumed to be constant [10] [3]. However, they diverge fundamentally in their approaches: FBA is a predictive, genome-scale method that relies on optimization principles, while 13C-MFA is an experimental, high-precision technique typically focused on central carbon metabolism. This guide provides an objective comparison of their performance, supported by experimental data, with a specific focus on their application in E. coli research.
Core Principles and Methodologies
Flux Balance Analysis (FBA): Genome-Scale Prediction
FBA is a computational approach that predicts metabolic flux distributions by leveraging the stoichiometry of the metabolic network and an assumed cellular objective [23] [24]. It does not require experimental flux data but instead uses linear optimization to identify a flux map that maximizes or minimizes a specific objective function, such as biomass production for simulating growth [10] [24].
The core constraint is the steady-state assumption, represented mathematically as: S × v = 0 where S is the stoichiometric matrix and v is the vector of reaction fluxes [24]. The solution space defined by these constraints is often vast, and FBA identifies a single optimal solution based on the chosen objective [10].
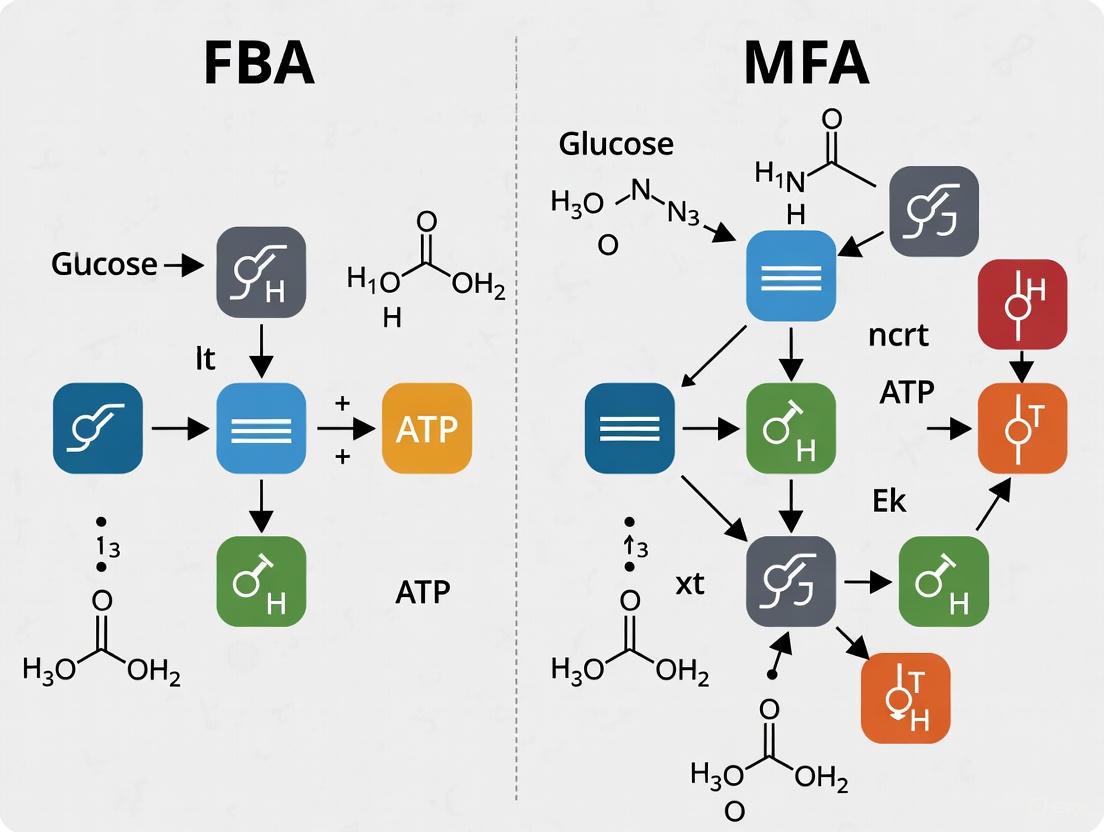
Figure 1: The FBA Workflow. FBA uses a genome-scale model, constraints, and an objective function to predict a flux distribution via linear optimization.
13C-Metabolic Flux Analysis (13C-MFA): Experimental Precision
In contrast, 13C-MFA is an experimental approach that infers metabolic fluxes by fitting network models to stable isotope labeling data [9] [23]. Cells are fed a 13C-labeled substrate (e.g., [1,2-13C]glucose), and the resulting label distribution in intracellular metabolites is measured using techniques like Mass Spectrometry (MS) or Nuclear Magnetic Resonance (NMR) spectroscopy [9] [3]. The computational process then works backward, varying flux values in a model to find the best match between the simulated and measured labeling patterns [10] [3]. This method is considered the gold standard for accurate and precise flux quantification in central carbon metabolism [23].
Figure 2: The 13C-MFA Workflow. 13C-MFA uses experimental data from isotope labeling experiments to computationally determine the most accurate flux map.
Direct Comparison: Strengths, Limitations, and Applications
The following tables summarize the fundamental characteristics and performance of FBA and 13C-MFA.
Table 1: Methodological Comparison of FBA and 13C-MFA
| Feature | Flux Balance Analysis (FBA) | 13C-Metabolic Flux Analysis (13C-MFA) |
|---|---|---|
| Core Principle | Prediction via linear optimization [24] | Estimation via experimental data fitting [9] |
| Primary Input | Stoichiometric model, constraints, objective function [24] | Measured isotope labeling patterns, external fluxes [9] [23] |
| Network Scope | Genome-scale (hundreds to thousands of reactions) [10] | Focused on central carbon metabolism [9] |
| Key Assumption | Metabolic steady state; optimal cellular growth/function [24] | Metabolic and isotopic steady state [9] |
| Output | Predicted flux distribution [10] | Estimated flux distribution with confidence intervals [10] [23] |
Table 2: Performance and Application in E. coli Research
| Aspect | Flux Balance Analysis (FBA) | 13C-Metabolic Flux Analysis (13C-MFA) |
|---|---|---|
| Quantitative Precision | Lower; often inconsistent with measured 13C-MFA fluxes [24] | High; considered the gold standard for precision [23] [4] |
| Primary Strength | Genome-scale prediction; exploration of network capabilities [23] | Experimental accuracy and validation of fluxes [23] |
| Perturbation Prediction | Less accurate for unevolved knockout mutants [25] | Used to measure actual physiological responses in knockouts [4] |
| Typical Application | Calculating theoretical yields; predicting essential genes [23] [24] | Quantifying flux rerouting in engineered strains [4] |
| Experimental Burden | Low (requires only a metabolic model) [10] | High (requires cultivation on labeled substrate and analytics) [9] |
Experimental Protocols forE. coli
A Representative FBA Workflow
A common FBA application is predicting the maximum growth rate of E. coli [24].
- Model Selection: Utilize a curated genome-scale model like iJO1366 for E. coli [5].
- Define Constraints: Set constraints based on experimental conditions, such as the maximum glucose uptake rate (
GURmax) and oxygen uptake rate (OURmax), derived from membrane transport capacity [24]. - Set Objective Function: Define the objective to be maximized, typically the reaction for biomass formation (
v_biomass) [24]. - Solve Linear Programming Problem: The model solves: Maximize
v_biomass, subject to S × v = 0 and the applied uptake constraints [24]. The solution provides a predicted growth rate and a full genome-scale flux map.
A Standard 13C-MFA Protocol
13C-MFA is used to obtain high-resolution flux maps for E. coli central metabolism, such as in studies of knockout mutants from the Keio collection [4].
- Pre-culture and Growth: Grow the E. coli strain (e.g., wild-type or knockout) in a minimal medium with unlabeled glucose until the metabolic steady state is reached [9].
- Tracer Experiment: Replace the medium with an identical one containing a specifically 13C-labeled carbon source (e.g., [1,2-13C]glucose or [U-13C]glucose). Continue cultivation until isotopic steady state is achieved, where the 13C labeling in intracellular metabolites is static [9] [4].
- Sampling and Quenching: Rapidly collect cells to quench metabolism and extract intracellular metabolites [9].
- Mass Spectrometry Analysis: Analyze the extracted metabolites using GC-MS or LC-MS to measure the Mass Isotopomer Distribution (MID)—the relative abundances of metabolite molecules with different numbers of 13C atoms [9] [10].
- Computational Flux Estimation: Use specialized software (e.g., INCA, OpenFLUX) to find the flux distribution that minimizes the difference between the simulated MID (based on the network model) and the experimentally measured MID [9] [23]. Statistical tests like the χ2-test are then used to validate the goodness-of-fit [10] [3].
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions
| Reagent / Tool | Function in FBA/MFA | Example Use Case |
|---|---|---|
| 13C-Labeled Tracers | Serve as the carbon source for 13C-MFA experiments to generate unique isotopic labeling patterns. | [1,2-13C]glucose or [U-13C]glucose for tracing carbon fate in central metabolism [9] [23]. |
| Genome-Scale Model (GSM) | Provides the stoichiometric network structure for both FBA simulations and 13C-MFA computational modeling. | The E. coli model iJO1366, used for FBA predictions and as a scaffold for 13C-MFA [5]. |
| COBRA Toolbox | A MATLAB-based software suite that implements FBA and related constraint-based modeling algorithms. | Performing FBA, Flux Variability Analysis (FVA), and flux sampling on genome-scale models [10] [24]. |
| Mass Spectrometer (MS) | The primary analytical instrument for measuring mass isotopomer distributions in 13C-MFA. | GC-MS analysis of proteinogenic amino acids or LC-MS analysis of intracellular metabolites [9] [23]. |
| INCA Software | A powerful computational platform for performing 13C-MFA, supporting both steady-state and isotopically non-stationary experiments. | Fitting a metabolic network model to experimental MS data to estimate fluxes with confidence intervals [9]. |
Integrated Workflow and Future Outlook
The strengths of FBA and 13C-MFA are highly complementary. A powerful integrated workflow uses 13C-MFA to provide high-quality validation data for refining FBA models and objective functions [3] [25]. For example, RELATCH is a constraint-based method that uses 13C-MFA data from a reference state to significantly improve the prediction of flux distributions in genetically perturbed E. coli strains, outperforming standard FBA [25]. Furthermore, methods like flux sampling (e.g., using the OptGP algorithm) can explore the space of possible flux distributions in a GSM, and the results can be compared and refined using 13C-MFA data to achieve more realistic predictions [5].
Future advancements will likely continue to bridge the gap between these two approaches, leveraging the scalability of FBA and the precision of MFA. This includes the development of more advanced validation and model selection frameworks [10] [3] and the application of 13C-MFA to more complex, non-standard systems [23]. For researchers, the choice between FBA and 13C-MFA is not a matter of selecting a superior tool, but of choosing the right tool for the specific question at hand—whether it is genome-scale prediction or experimental precision.
In the field of metabolic engineering and systems biology, understanding the flux of metabolites through biochemical networks is crucial for both fundamental research and industrial applications. Two primary methodologies have emerged: Flux Balance Analysis (FBA), which predicts metabolic capabilities based on network structure and optimization principles, and 13C-Metabolic Flux Analysis (13C-MFA), which measures in vivo metabolic activity using isotopic tracers [10]. This guide provides a objective comparison of their application in E. coli flux prediction research, detailing their principles, experimental protocols, and performance.
Core Principles and Theoretical Foundations
FBA and MFA are grounded in distinct theoretical frameworks, leading to their complementary roles in metabolic research.
Flux Balance Analysis (FBA) is a constraint-based modeling approach that predicts steady-state metabolic fluxes in a biochemical network. It requires a stoichiometric model (S) of the metabolic network, which incorporates all known biochemical reactions derived from genomic and biochemical data [26]. The core mathematical principle is the mass balance constraint, which at steady state is represented as S • v = 0, where v is the vector of metabolic fluxes [26] [27]. FBA does not require kinetic parameters. Instead, it identifies a flux distribution that maximizes or minimizes a specific biological objective function, such as biomass production or ATP yield, using linear programming [26]. It is primarily a predictive tool for exploring metabolic potential and network capabilities.
13C-Metabolic Flux Analysis (13C-MFA) is an experimental approach that measures the operational metabolic fluxes within a living cell. It works by feeding a 13C-labeled substrate (e.g., [1-13C]glucose) to a cell culture and then using mass spectrometry (MS) or nuclear magnetic resonance (NMR) to measure the resulting labeling patterns in intracellular metabolites [10] [28]. The core of 13C-MFA involves fitting a computational model of the metabolic network to the experimental mass isotopomer distribution (MID) data by varying the flux estimates [10]. The flux map that minimizes the residual between the simulated and measured labeling data is considered the most accurate representation of the in vivo metabolic state.
The following diagram illustrates the fundamental workflows of FBA and 13C-MFA, highlighting their predictive versus measurement-driven natures.
Direct Comparison of FBA and MFA
The table below summarizes the key characteristics of FBA and 13C-MFA, highlighting their complementary strengths and weaknesses.
| Feature | Flux Balance Analysis (FBA) | 13C-Metabolic Flux Analysis (13C-MFA) |
|---|---|---|
| Primary Function | Predictive simulation [26] | Experimental measurement [10] |
| Type of Output | Prediction of potential fluxes | Estimation of in vivo fluxes |
| Underlying Data | Genome annotation, stoichiometry, constraints [26] | Isotopic labeling data (MS/NMR), external fluxes [10] [28] |
| Key Assumptions | Steady-state, optimality of objective function [26] | Metabolic and isotopic steady state [10] |
| Scope | Genome-scale models (>1,000 reactions) [26] | Core metabolic networks (10-100 reactions) [10] |
| Temporal Resolution | Static (steady-state) | Static (steady-state); dynamic in INST-MFA |
| Throughput | High (seconds per simulation) [26] | Low (days to weeks per experiment) |
| Key Strengths | Genome-scale, fast, good for hypothesis generation | High accuracy and precision for core metabolism, model validation [10] [28] |
Experimental Protocols and Methodologies
Protocol for 13C-MFA inE. coli
13C-MFA is an experimental multi-step process used to determine actual intracellular fluxes.
- Strain Cultivation and Labeling: Grow the E. coli strain (e.g., wild-type or a specific knockout from the Keio collection) in a defined medium. During mid-exponential growth, introduce a 13C-labeled carbon source (e.g., [1-13C]glucose or [U-13C]glucose) for a sufficient period to achieve isotopic steady state [28].
- Metabolite Extraction and Measurement: Rapidly quench metabolism to preserve in vivo metabolite levels. Extract intracellular metabolites from the cell pellet. Analyze the extract using Gas Chromatography-Mass Spectrometry (GC-MS) or LC-MS to obtain the Mass Isotopomer Distribution (MID) for key metabolites [10] [28].
- Flux Estimation: Use dedicated software that contains a model of E. coli central metabolism. The software performs a non-linear least squares regression, iteratively adjusting the flux values in the network model until the simulated MIDs best fit the experimentally measured MIDs [10].
- Statistical Validation: Evaluate the goodness-of-fit, typically using a chi-squared (χ2) test, and perform Monte Carlo simulations to determine confidence intervals for the estimated fluxes [10].
Protocol for FBA inE. coli
FBA is a computational protocol used to predict flux distributions.
- Model Construction: Reconstruct a genome-scale metabolic network for E. coli from its annotated genome, biochemical literature, and databases (e.g., BiGG Model). This model is represented as a stoichiometric matrix (S) [26] [27].
- Application of Constraints: Define constraints on the system. This includes the steady-state mass balance constraint (S • v = 0) and capacity constraints on individual reaction fluxes (e.g., substrate uptake rates) [26] [27].
- Definition of Objective Function: Select a biologically relevant objective function to be optimized. For E. coli growing in a batch culture, this is most often the reaction representing biomass synthesis, which is maximized [26] [29].
- Linear Programming Solution: Use a linear programming solver to find the single flux distribution that satisfies all constraints and optimizes the objective function. The output is a predicted flux value for every reaction in the network [26] [27].
Performance Comparison: Experimental Data fromE. coliKnockouts
A critical test for both methods is accurately determining or predicting the metabolic phenotype of engineered strains. The following table summarizes quantitative flux data from studies on E. coli knockout mutants, comparing predictions from FBA and related algorithms against experimental measurements from 13C-MFA.
| Gene Knocked Out | Growth Condition | Key Flux Change (Enzyme/Pathway) | 13C-MFA Measured Flux (Normalized) | FBA/MOMA Predicted Flux | Accuracy & Notes |
|---|---|---|---|---|---|
| pgi (Phosphoglucose Isomerase) | Batch [28] | Citrate Synthase (TCA cycle) | 20 - 62 [28] | Under-prediction of TCA flux by FBA; MOMA improves prediction [28] | Variable accuracy. MOMA often outperforms FBA for unevolved knockouts [28]. |
| zwf (Glucose-6-phosphate Dehydrogenase) | Batch [28] | Acetate Secretion | ~44 [28] | N/A | Shows limitations. FBA cannot predict overflow metabolism without additional constraints [28]. |
| pykF (Pyruvate Kinase) | Chemostat (D=0.2 h⁻¹) [28] | PEP Carboxylase (ppc) | Major changes reported [28] | N/A | Context-dependent. Flux responses vary significantly between batch and chemostat conditions [28]. |
| Multiple Central Metabolism Genes | Aerobic, Glucose [27] | Biomass Production (in silico) | N/A | 7 genes predicted essential [27] | High predictive value. FBA successfully identifies essential genes for growth [27]. |
The relationship between prediction and measurement, and the role of model selection, can be visualized as follows.
Successful flux analysis relies on a suite of experimental and computational tools. The table below lists essential resources for conducting FBA and 13C-MFA studies in E. coli.
| Item Name | Function/Description | Relevance |
|---|---|---|
| Keio Collection [28] | A library of single-gene knockout mutants of E. coli K-12. | Provides a ready-to-use resource for systematic perturbation studies using MFA or for validating FBA predictions. |
| 13C-Labeled Substrates (e.g., [1-13C]Glucose) | Carbon sources with specific atoms replaced with the 13C isotope. | The essential tracer input for 13C-MFA experiments to track metabolic activity [10] [28]. |
| GC-MS / LC-MS Instrumentation | Gas or Liquid Chromatography coupled to Mass Spectrometry. | Used to measure the mass isotopomer distribution (MID) of metabolites, which is the primary data for 13C-MFA [10]. |
| COBRA Toolbox [10] | A MATLAB-based software suite for constraint-based modeling. | The standard toolkit for building metabolic models and performing FBA, gene deletion studies, and other analyses. |
| MEMOTE [10] | (MEtabolic MOdel TEsts) | A pipeline for quality control and testing of genome-scale metabolic models to ensure basic functionality and consistency. |
| 13C-MFA Software (e.g., INCA, OpenFLUX) | Specialized software packages for simulation and fitting of 13C labeling data. | Used to estimate metabolic fluxes from experimental MID data by fitting the network model [10]. |
Practical Implementation: Applying FBA and MFA to E. coli Metabolic Engineering
A Step-by-Step Workflow for Performing 13C-MFA in E. coli
Metabolic Flux Analysis (MFA) using 13C-labeling has established itself as a cornerstone technique for quantifying intracellular reaction rates (fluxes) in living E. coli cells. Unlike constraint-based methods like Flux Balance Analysis (FBA), which predict fluxes based on assumed optimality principles, 13C-MFA utilizes experimental data from isotope labeling experiments (ILEs) to infer metabolic activity, providing a more direct and empirical measurement of in vivo flux distributions [30] [31]. This guide provides a detailed, step-by-step workflow for implementing 13C-MFA in E. coli, objectively compares the performance of different methodologies and software, and situates the discussion within the broader thesis of comparing FBA and MFA for E. coli flux prediction.
FBA, while powerful for predicting gene essentiality and growth capabilities, relies on a predefined cellular objective—typically biomass maximization—which may not always hold true, especially in engineered strains or complex environments [31]. In contrast, 13C-MFA does not presuppose an objective function, instead deriving fluxes from measured isotopic patterns, making it particularly valuable for characterizing mutant strains, validating model predictions, and identifying non-obvious metabolic bottlenecks [32] [33]. The following sections outline the complete experimental and computational pipeline for a successful 13C-MFA study.
A Step-by-Step 13C-MFA Workflow for E. coli
The following workflow is adapted from large-scale studies and best practices in the field [34] [32].
Step 1: Experimental Design and Tracer Selection
The foundation of a successful 13C-MFA experiment is a well-considered design, primarily focusing on the choice of isotopic tracer.
- Tracer Selection: No single tracer is optimal for resolving all fluxes in the E. coli metabolic network. The choice depends on the pathways of interest.
- For upper glycolysis and pentose phosphate pathway fluxes, a mixture of 75% [1-13C]glucose and 25% [U-13C]glucose has been shown to be highly effective [34].
- For fluxes in the lower part of metabolism (TCA cycle, anaplerotic reactions), [4,5,6-13C]glucose or [5-13C]glucose provide superior resolution [34].
- The COMPLETE-MFA approach, which uses multiple parallel labeling experiments, has emerged as the gold standard. It significantly improves flux precision and observability, especially for exchange fluxes, by integrating data from several complementary tracers [34]. Example tracers for a parallel study include [1,2-13C]glucose, [2,3-13C]glucose, and [1-13C]glucose + [U-13C]glucose mixtures [34].
- Culture Conditions: E. coli cells (e.g., strain K-12 MG1655) are grown in defined minimal medium (e.g., M9) with the chosen 13C-labeled tracer as the sole carbon source. Cultivation in controlled, aerated mini-bioreactors is recommended to ensure reproducible growth and metabolic steady-state conditions [34].
Step 2: Cultivation and Metabolite Labeling
- Inoculum and Growth: A single colony is used to start a pre-culture in unlabeled medium. This culture is then used to inoculate the main culture containing the 13C-tracer. The initial optical density (OD600) should be low (~0.03) to minimize dilution of the label from carry-over carbon [34].
- Sampling: Cells are harvested during the mid-exponential growth phase for two purposes:
- Extracellular Fluxes: Measurements of substrate uptake (e.g., glucose consumption) and product secretion (e.g., acetate formation) rates are crucial for constraining the model [32].
- Intracellular Labeling: Cells are rapidly quenched to halt metabolism, and metabolites are extracted for analysis of Mass Isotopomer Distributions (MIDs) [34].
Step 3: Analytical Measurement of Isotopic Labeling
- Mass Spectrometry (MS): Gas Chromatography-Mass Spectrometry (GC-MS) is commonly used to measure the MIDs of proteinogenic amino acids or intracellular metabolites. The MID represents the fractional abundance of molecules with a given number of heavy isotopes (e.g., M+0, M+1, M+2) [34] [35]. These MIDs are the primary data input for flux calculation.
Step 4: Computational Flux Estimation
This is the core computational phase where fluxes are inferred from the experimental data.
- Model Specification: A metabolic network model of E. coli central carbon metabolism is required. This model must include reaction stoichiometries and, critically, atom transitions for each reaction, which map how carbon atoms are rearranged [35]. The FluxML language provides a standardized, machine-readable format for defining all these aspects, ensuring reproducibility and model sharing [35].
- Flux Simulation and Fitting: Software simulates the labeling patterns expected for a given set of fluxes and then iteratively adjusts the fluxes until the simulated MIDs best match the measured MIDs. This is typically done by minimizing the residual sum of squares between measured and simulated data [36]. The use of the Elementary Metabolite Unit (EMU) framework significantly simplifies these computations [34] [36].
- Statistical Analysis: After identifying the best-fit flux map, statistical methods (e.g., Monte Carlo sampling) are used to calculate accurate confidence intervals for each estimated flux, quantifying the precision of the results [34] [33].
Step 5: Validation and Interpretation
- Goodness-of-Fit: The model fit is validated using a χ2 test to ensure the flux solution is statistically consistent with the experimental measurements [32].
- Flux Map Analysis: The final flux distribution is interpreted to identify key pathway activities, such as the split between glycolysis and pentose phosphate pathway, TCA cycle flux, and the presence of futile cycles. In metabolic engineering contexts, this analysis pinpoints bottlenecks and informs subsequent strain design strategies [32].
The entire workflow, from tracer to flux map, is summarized in the diagram below.
Comparative Performance: Tracers, Software, and Methods
The performance of 13C-MFA is highly dependent on the choices made regarding tracers, software, and statistical approaches. The data below provide a quantitative and qualitative comparison to guide researchers.
Tracer Performance for E. coli Flux Resolution
The choice of tracer directly impacts the precision and observability of specific metabolic fluxes [34].
Table 1: Performance of Different 13C-Glucose Tracers in E. coli 13C-MFA
| 13C-Tracer | Optimal For Pathway | Key Performance Findings |
|---|---|---|
| 75% [1-13C]glucose + 25% [U-13C]glucose | Upper Metabolism (Glycolysis, PPP) | Produces well-resolved fluxes in the upper part of metabolism [34]. |
| [4,5,6-13C]glucose | Lower Metabolism (TCA cycle, Anaplerotic) | Delivers optimal flux resolution in the lower part of metabolism [34]. |
| [5-13C]glucose | Lower Metabolism (TCA cycle, Anaplerotic) | Comparable performance to [4,5,6-13C]glucose for lower metabolism [34]. |
| Parallel Labeling (COMPLETE-MFA) | Entire Network | Improves flux precision and observability; resolves more independent fluxes with smaller confidence intervals than any single tracer [34]. |
Comparison of 13C-MFA Software Tools
Several software tools are available for flux simulation and estimation, each with different capabilities and performance characteristics.
Table 2: Feature Comparison of 13C-MFA Software Platforms
| Software Tool | Key Features | Supported MFA Types | Notable Advantages |
|---|---|---|---|
| 13CFLUX(v3) [36] | High-performance C++ engine; Python interface; supports FluxML. | Stationary & Nonstationary | Open-source; performance gains; supports multi-experiment integration & Bayesian inference [36]. |
| INCA [30] | User-friendly GUI; comprehensive flux analysis suite. | Stationary & Nonstationary | Widely used; implements the EMU framework [30]. |
| FluxML-Consortium Tools [35] | Standardized model exchange format. | Stationary & Nonstationary | Promotes reproducibility and model re-use; algorithm- and tool-independent [35]. |
13C-MFA vs. FBA: A Methodological Comparison
Understanding the complementary strengths and weaknesses of 13C-MFA and FBA is crucial for selecting the right tool for a given research question.
Table 3: Contrasting 13C-MFA and FBA for E. coli Flux Prediction
| Aspect | 13C-MFA | Flux Balance Analysis (FBA) |
|---|---|---|
| Basis | Empirical measurement from 13C-labeling data & extracellular rates [30]. | Theoretical optimization based on stoichiometry & assumed objective (e.g., growth maximization) [31]. |
| Requirement | Experimentally intensive (requires isotopic tracers and analytical equipment). | Requires a high-quality genome-scale model (GEM); no experimental data strictly required. |
| Flux Resolution | Provides precise, absolute fluxes for core metabolism. Limited network size. | Provides genome-scale flux distributions. Flux profiles can be less precise without additional constraints. |
| Key Strength | Direct, model-independent flux measurement; ideal for validation and characterizing mutant phenotypes [32]. | Predicts system-level capabilities (e.g., gene essentiality, growth on substrates) and enables genome-scale designs [31]. |
| Performance | High accuracy for core fluxes. COMPLETE-MFA greatly improves precision [34]. | High accuracy for metabolic gene essentiality in E. coli (~93.5%), but drops for higher organisms [31]. |
The core pathways of central carbon metabolism in E. coli that are typically resolved by 13C-MFA are illustrated below, showing the key junctions where flux splits are quantified.
Advanced Topics and Future Directions
Bayesian 13C-MFA
The standard "best-fit" approach to 13C-MFA is increasingly being supplemented by Bayesian methods. Bayesian 13C-MFA offers a unified framework for handling model selection uncertainty and enables multi-model inference through Bayesian Model Averaging (BMA). This is particularly advantageous for evaluating the evidence for or against bidirectional reaction steps (fluxes through reversible reactions), which are often difficult to resolve with conventional methods [33]. BMA acts as a "tempered Ockham's razor," automatically penalizing overly complex models that are not sufficiently supported by the data, leading to more robust flux inference [33].
Integration with Other Omics and Machine Learning
The future of flux analysis lies in integration. New machine learning frameworks like Flux Cone Learning (FCL) demonstrate this trend. FCL uses Monte Carlo sampling of the metabolic flux space (the "flux cone") defined by a genome-scale model to generate training data. A supervised learning model is then trained on this data alongside experimental fitness scores from deletion screens. This approach has been shown to outperform traditional FBA in predicting metabolic gene essentiality in E. coli and other organisms, without requiring an optimality assumption [31]. This represents a powerful synergy between mechanistic models and data-driven machine learning.
The Scientist's Toolkit: Essential Reagents and Software
Table 4: Key Research Reagent Solutions for 13C-MFA in E. coli
| Category | Item | Function and Example |
|---|---|---|
| Isotopic Tracers | 13C-labeled Glucose | Create unique labeling patterns to probe specific pathways. Examples: [1-13C]glucose, [U-13C]glucose, [4,5,6-13C]glucose [34]. |
| Strains & Media | E. coli K-12 MG1655 | A standard, well-characterized laboratory strain with established metabolic models [34] [37]. |
| Defined Minimal Medium (e.g., M9) | Provides a controlled chemical environment without unlabeled carbon sources that would dilute the tracer [34]. | |
| Analytical Instruments | GC-MS System | Measures Mass Isotopomer Distributions (MIDs) of metabolites, the primary data for flux calculation [34] [35]. |
| Computational Tools | 13CFLUX(v3), INCA | Software platforms for simulating isotope labeling and estimating metabolic fluxes [30] [36]. |
| FluxML File | A standardized file format for unambiguously defining a 13C-MFA model, ensuring reproducibility [35]. | |
| Metabolic Models | iML1515, iCH360 | Genome-scale (iML1515) and compact, curated core models (iCH360) for E. coli provide the stoichiometric framework for flux analysis [31] [37]. |
This guide has detailed a comprehensive workflow for performing 13C-MFA in E. coli, from careful experimental design with optimal tracers to computational flux estimation using modern software. The comparative data clearly shows that while FBA remains a powerful tool for genome-scale prediction, particularly in microbes, 13C-MFA provides an unmatched, empirical window into the actual operational fluxes of core metabolism. The emergence of advanced methodologies like COMPLETE-MFA, Bayesian flux inference, and integrated machine-learning approaches is steadily enhancing the resolution, robustness, and scope of 13C-MFA. For researchers aiming to understand and engineer E. coli metabolism with high quantitative precision, 13C-MFA is an indispensable and constantly evolving technology.
Constraint-based metabolic modeling has become an indispensable tool for systems biologists and metabolic engineers, providing a computational framework to predict cellular behavior under various genetic and environmental conditions. For Escherichia coli, a cornerstone organism in biotechnology and metabolic engineering, two primary methodologies have emerged: Flux Balance Analysis (FBA) and 13C-Metabolic Flux Analysis (13C-MFA). FBA uses optimization principles, typically maximizing biomass production, to predict flux distributions through a genome-scale metabolic network, requiring only the stoichiometry of the metabolic network and uptake/secretion rates [3] [38]. In contrast, 13C-MFA utilizes isotopic tracer experiments and mass balancing to empirically determine intracellular fluxes, providing high-resolution data for core metabolism but at a significant experimental cost [28] [19]. This guide systematically compares these approaches, providing researchers with a practical framework for model selection, experimental validation, and computational optimization specific to E. coli flux prediction.
Comparative Analysis of FBA and MFA
Core Methodological Differences and Applications
The fundamental distinction between these approaches lies in their methodology and scope. FBA is inherently predictive, based on the hypothesis that metabolism has been evolutionarily optimized for efficiency, often simulated by maximizing biomass yield or ATP production [3] [38]. Its strength is the ability to rapidly interrogate genome-scale networks and predict outcomes of genetic manipulations. 13C-MFA, however, is descriptive, using experimental data from isotopic labeling to calculate the actual, in vivo flux state, providing a gold standard for flux measurements in central carbon metabolism but lacking genome-scale coverage [3] [19]. The choice between them is not mutually exclusive; rather, they form a powerful synergistic loop where MFA data validates and refines FBA models [3] [28].
Quantitative Performance Comparison
The table below summarizes the performance characteristics of FBA and 13C-MFA based on validation studies in E. coli.
Table 1: Performance Comparison of FBA and 13C-MFA for E. coli
| Feature | Flux Balance Analysis (FBA) | 13C-Metabolic Flux Analysis (13C-MFA) |
|---|---|---|
| Model Scope | Genome-scale (e.g., 1,445 genes, 2,286 reactions in EcoCyc-18.0-GEM) [38] | Core metabolism (typically 50-100 reactions) [19] |
| Primary Input | Stoichiometric matrix, uptake/secretion rates, objective function [38] | Isotopic labeling data, extracellular fluxes [3] |
| Key Output | Predicted flux distribution | Estimated flux distribution with confidence intervals [3] |
| Gene Essentiality Prediction Accuracy | Up to 95.2% (EcoCyc-18.0-GEM) [38] | Not a direct output (used for validation) [28] |
| Nutrient Utilization Prediction Accuracy | 80.7% (on 431 conditions) [38] | Not a direct output (used for validation) |
| Typical Validation Method | Comparison to gene essentiality & growth phenotyping data [38] | Statistical goodness-of-fit tests (e.g., χ²-test) [3] |
| Major Strengths | Genome-scale scope; high-throughput; predicts knockout phenotypes [28] [38] | High precision and accuracy in core metabolism; captures regulatory effects [28] |
| Major Limitations | Relies on assumed objective function; may not capture regulation [3] [28] | Experimentally intensive; limited to core metabolism [3] |
Computational Optimization and Model Selection
Advanced FBA Formulations for Knockout Strains
Standard FBA, which assumes optimal growth, often fails to accurately predict fluxes in unevolved knockout strains. To address this, several advanced algorithms have been developed:
- Minimization of Metabolic Adjustment (MOMA): Postulates that flux distributions in knockout strains are as close as possible (by Euclidean distance) to the wild-type FBA optimum, favoring solutions with many small flux changes [28].
- Regulatory On/Off Minimization (ROOM): Minimizes the number of significant flux changes from the wild-type solution, which can be more consistent with regulatory constraints than MOMA [28].
- RELATCH (RELATive CHange): Uses experimental flux data from a reference strain and aims to minimize regulatory and distribution pattern changes before activating latent pathways [28].
These methods generally show improved accuracy in predicting the fluxome of E. coli knockouts compared to standard FBA, though a comprehensive assessment is limited by inconsistencies in available experimental data [28].
Integrating Proteomic Constraints
A significant advancement in FBA has been the incorporation of proteomic constraints to better model overflow metabolism, such as acetate production in fast-growing E. coli. A proteome-allocation theory can be incorporated into FBA by adding a concise constraint with parameters representing the differential proteomic cost of respiration versus fermentation pathways [39]. This approach successfully predicts the onset and extent of overflow metabolism across different E. coli strains, revealing that the proteomic cost of fermentation is consistently lower than that of respiration, explaining its activation under rapid growth [39].
Workflow for Integrated FBA and MFA Analysis
The following diagram illustrates a pipeline that integrates MFA data with kinetic model parameterization, ensuring consistency between experimental data and model predictions.
Diagram Title: FBA-MFA Kinetic Model Pipeline
Model Selection and Validation Framework
Robust model selection is critical for reliable flux predictions. For 13C-MFA, the χ²-test of goodness-of-fit is widely used but has limitations; it should be complemented with checks for overfitting and consistency with metabolite pool size data where available [3]. For FBA, the most robust validation is direct comparison against 13C-MFA estimated fluxes [3]. Key performance metrics include:
- Gene Essentiality Prediction: The accuracy of predicting whether a gene knockout will prevent growth [38].
- Nutrient Utilization: The accuracy of predicting growth on different carbon sources [38].
- Quantitative Flux Prediction: The ability to match the magnitude of fluxes measured by 13C-MFA, particularly for key nodes in central carbon metabolism [28] [19].
Experimental Protocols for Flux Determination
Protocol for 13C-MFA in E. coli
Objective: To empirically determine intracellular metabolic fluxes in E. coli central carbon metabolism.
Materials:
- Biological Material: E. coli strain of interest (e.g., from Keio collection for knockouts) [28].
- Culture System: Controlled bioreactor (e.g., chemostat for steady-state cultures) [28].
- Labeled Substrate: 13C-labeled glucose (e.g., [1-13C] glucose or [U-13C] glucose) [19].
- Analytical Instrumentation: GC-MS or LC-MS for measuring mass isotopomer distributions (MIDs) of proteinogenic amino acids or intracellular metabolites [3] [19].
Methodology:
- Cultivation: Grow the E. coli strain in a defined minimal medium with the 13C-labeled substrate as the sole carbon source. Maintain metabolic and isotopic steady-state, typically in a chemostat [28].
- Sampling & Quenching: Harvest cells during mid-exponential growth and rapidly quench metabolism to preserve isotopic labeling patterns.
- Metabolite Extraction & Derivatization: Extract intracellular metabolites or hydrolyze cellular protein to release proteinogenic amino acids. Derivatize samples for MS analysis [19].
- Mass Spectrometry Analysis: Measure the MIDs of the target metabolites or amino acid fragments. These distributions reflect the labeling state of their precursor metabolites in central metabolism.
- Computational Flux Estimation: Use a computational software package to solve a least-squares optimization problem, minimizing the difference between the measured MIDs and the MIDs simulated by a stoichiometric model of the metabolic network. The outputs are the estimated flux values and their confidence intervals [3] [19].
Protocol for FBA Model Validation Using Gene Essentiality Data
Objective: To validate an FBA model's predictive accuracy against a gold-standard experimental dataset.
Materials:
- Computational Model: A genome-scale model of E. coli metabolism (e.g., EcoCyc-18.0-GEM or iJO1366) [38].
- Experimental Dataset: A comprehensive gene essentiality dataset, such as data for the Keio collection of E. coli single-gene knockouts [28] [38].
Methodology:
- Simulation: For each gene in the model, simulate a knockout by constraining the flux through the associated reaction(s) to zero.
- Growth Prediction: Solve the FBA problem (e.g., maximizing biomass) for each in silico knockout. A growth rate above a defined threshold (e.g., >1% of wild-type) predicts viability; below predicts non-viability.
- Comparison: Compare the predicted growth phenotype (viable/non-viable) for each gene to the experimental observation.
- Accuracy Calculation: Calculate the prediction accuracy as the percentage of genes for which the model's prediction matches the experimental result. High-performing models like EcoCyc-18.0-GEM achieve accuracies >95% [38].
The Scientist's Toolkit: Essential Research Reagents and Solutions
The table below lists key resources for conducting FBA and MFA research in E. coli.
Table 2: Essential Research Reagents and Computational Tools for E. coli Flux Analysis
| Item Name | Function/Application | Relevant Use Case |
|---|---|---|
| Keio Collection [28] | A library of all viable E. coli single-gene knockouts. | Systematic investigation of metabolic network responses to genetic perturbations. |
| 13C-Labeled Substrates (e.g., Glucose, Glycerol) [19] | Tracers to follow carbon fate through metabolic networks. | Experimental input for 13C-MFA to determine in vivo flux distributions. |
| EcoCyc Database [38] | A curated bioinformatics database of E. coli biology. | Source for generating and visualizing high-quality, curated genome-scale metabolic models (GEMs) via MetaFlux. |
| EcoCyc-18.0-GEM Model [38] | A highly accurate, genome-scale metabolic model for E. coli K-12. | Benchmark FBA model for predicting gene essentiality and nutrient utilization. |
| MOMA/ROOM Algorithms [28] | Alternative FBA formulations for sub-optimal states. | Predicting flux distributions in un-evolved gene knockout strains. |
| GC-MS or LC-MS Instrumentation [3] [19] | Measuring mass isotopomer distributions (MIDs) of metabolites. | Generating the primary experimental data for 13C-MFA flux elucidation. |
The selection between FBA and MFA for E. coli flux prediction is guided by the research objective: use FBA for genome-scale, predictive simulations of genetic engineering interventions, and employ 13C-MFA for obtaining high-resolution, empirical flux maps of core metabolism under defined conditions. The future of accurate flux prediction lies in the continued integration of these approaches—using MFA data to validate and refine FBA models, and incorporating additional biological constraints, such as proteomic efficiency, to enhance predictive power. By leveraging the protocols, models, and datasets outlined in this guide, researchers can effectively navigate the complexities of metabolic flux analysis in E. coli to advance both basic science and biotechnological applications.
Flux Balance Analysis (FBA) and related constraint-based approaches provide powerful mathematical frameworks for predicting metabolic behavior in biological systems. Within the broader context of comparing Flux Balance Analysis (FBA) and Metabolic Flux Analysis (MFA) for E. coli flux prediction research, a critical application lies in predicting the metabolic phenotypes of genetically engineered knockout strains. While FBA operates on the premise that metabolism achieves an optimal state through evolutionary selection, this assumption often fails for laboratory-generated mutants that haven't been subjected to long-term evolutionary pressure [40]. This limitation motivated the development of complementary algorithms specifically designed for perturbed metabolic networks, most notably Minimization of Metabolic Adjustment (MOMA) and Regulatory On/Off Minimization (ROOM) [40] [41]. This guide provides an objective comparison of these three fundamental algorithms—FBA, MOMA, and ROOM—for analyzing knockout strains in E. coli, detailing their underlying principles, performance characteristics, and appropriate application contexts.
Theoretical Foundations and Mathematical Formulations
Flux Balance Analysis (FBA)
FBA predicts metabolic flux distributions at steady state by using linear programming to optimize a cellular objective, typically the maximization of biomass production [40] [26]. This approach is justified by the premise that prokaryotes such as E. coli have maximized their growth performance through evolution [40]. The mathematical foundation of FBA consists of mass balance constraints and an optimization objective:
Mass Balance Constraints: The system is represented by the stoichiometric matrix ( S ), where ( S_{ij} ) represents the stoichiometric coefficient of metabolite ( i ) in reaction ( j ). At steady state, the net sum of all production and consumption fluxes for each metabolite is zero: ( S \cdot v = 0 ), where ( v ) is the flux vector [40] [26].
Flux Constraints: Additional inequalities constrain flux values: ( αj ≤ vj ≤ β_j ), representing thermodynamic irreversibility, enzyme capacity, or substrate uptake limitations [40].
Objective Function: FBA typically maximizes biomass production: ( \max Z = c^Tv ), where ( c ) is a vector of coefficients defining the biological objective [26].
For knockout strain analysis, FBA implements gene deletions by constraining the corresponding reaction flux to zero and re-optimizing to find a new optimal growth state [40] [26].
Minimization of Metabolic Adjustment (MOMA)
MOMA introduces a fundamentally different hypothesis for knockout strains: that metabolic fluxes undergo minimal redistribution with respect to the wild-type flux configuration [40]. Rather than assuming immediate optimality in mutants, MOMA identifies a flux distribution that is closest to the wild-type point while satisfying the gene deletion constraint:
Objective Function: MOMA employs quadratic programming to minimize the Euclidean distance between wild-type and mutant flux distributions: ( \min ║v{wt} - v{mt}║ ), where ( v{wt} ) is the wild-type flux vector and ( v{mt} ) is the mutant flux vector [40] [42].
Mathematical Implementation: The minimization of the Euclidean distance function ( D = ║v{wt} - v{mt}║ ) is equivalent to minimizing ( f(x) = \frac{1}{2}x^Tx - w^Tx ), where ( Q ) is an ( N × N ) unit matrix and ( L = -w ) [40].
MOMA relaxes the optimal growth assumption for gene deletions, recognizing that laboratory-generated mutants likely don't possess immediate regulatory mechanisms to achieve new flux optima [40].
Regulatory On/Off Minimization (ROOM)
ROOM represents an alternative hypothesis for mutant metabolic states, proposing that knockout metabolic fluxes minimize the number of significant flux changes relative to the wild type [41]:
Objective Function: ROOM minimizes the number of significant flux changes from the wild-type flux distribution, using mixed-integer linear programming or related techniques [41].
Theoretical Basis: Unlike MOMA, which prevents large modifications in single fluxes, ROOM allows significant rerouting through alternative pathways when necessary, which has been observed in experimental studies [41].
ROOM is particularly valuable when large flux modifications are required for rerouting metabolic flux through alternative pathways, a scenario where MOMA's distance-minimization may be less effective [41].
Table 1: Core Mathematical Principles of FBA, MOMA, and ROOM
| Algorithm | Optimization Type | Objective Function | Key Constraints |
|---|---|---|---|
| FBA | Linear Programming | Maximize biomass production: ( \max c^Tv ) | ( S·v = 0 ), ( αj ≤ vj ≤ β_j ) |
| MOMA | Quadratic Programming | Minimize Euclidean distance: ( \min ║v{wt} - v{mt}║ ) | ( S·v = 0 ), ( v_j = 0 ) for knockout reactions |
| ROOM | Mixed-Integer Linear Programming | Minimize number of significant flux changes | ( S·v = 0 ), ( v_j = 0 ) for knockout reactions, flux change thresholds |
The following diagram illustrates the conceptual relationships and workflow between these three algorithms in the context of knockout strain analysis:
Performance Comparison and Experimental Validation
Quantitative Comparison of Prediction Accuracy
Extensive studies have compared the performance of FBA, MOMA, and ROOM against experimental flux data. The foundational MOMA paper demonstrated its superior performance for predicting fluxes in an E. coli pyruvate kinase mutant (PB25), where MOMA displayed a significantly higher correlation with experimental flux data than FBA [40]. Subsequent research has further refined our understanding of the relative strengths of each method:
Table 2: Algorithm Performance Comparison for E. coli Knockout Strains
| Algorithm | Prediction Accuracy | Computational Complexity | Best Application Context |
|---|---|---|---|
| FBA | Lower for un-evolved knockouts | Low (Linear Programming) | Wild-type strains, evolved mutants |
| MOMA | Higher for recent knockouts | Medium (Quadratic Programming) | Laboratory-generated knockouts without evolutionary history |
| ROOM | Superior when pathway rerouting occurs | High (Mixed-Integer Programming) | Knockouts requiring significant flux rerouting |
A comprehensive comparison with yeast experimental epistasis data revealed limitations for all constraint-based methods. The tested methods (FBA, MOMA, and FBA with molecular crowding) collectively predicted only one-third of experimentally observed epistatic interactions, with each method generating largely non-overlapping sets of correct predictions [43]. For negative epistatic interactions, the best precision achieved was approximately 45% with only 2.8% recall, indicating that while predictions can be accurate when made, most real interactions are missed [43].
Experimental Protocols for Validation
Validating flux predictions requires integrating multiple experimental techniques. For E. coli knockout strain analysis, the following methodological approach provides robust validation:
Strain Construction: Utilize E. coli MG1655 or derivative strains (e.g., JM101 for wild-type studies, PB25 for pyruvate kinase mutants). Gene knockouts are implemented through complete gene deletion, constraining corresponding fluxes to zero in silico, with validation via PCR and sequencing [40] [8].
Culture Conditions: Employ defined minimal media with controlled carbon sources (e.g., glucose at 16.5-33 g/L) and limiting nutrients (ammonium sulfate at 2.5-5 g/L for N-limited conditions). Maintain steady-state growth in chemostats with dilution rates typically between 0.142-0.155 h⁻¹, ensuring metabolic steady-state by waiting at least five residence times after the batch phase before sampling [8].
Flux Measurement: Determine extracellular flux rates by measuring metabolite concentrations in the reactor broth using HPLC. Calculate exchange fluxes based on concentration changes, applying polynomial smoothing to reduce noise from differentiation [8].
Intracellular Flux Determination: Use ¹³C metabolic flux analysis (¹³C-MFA) with isotopic labeling to measure intracellular fluxes. Feed ¹³C-labeled substrates and measure mass isotopomer distributions via mass spectrometry or NMR [3].
Data Integration: Implement computational pipelines (e.g., in Python with SciPy library) to process concentration data, compute derivatives, and calculate metabolic fluxes using stoichiometric models [8].
The following research reagents table outlines essential materials for implementing these experimental protocols:
Table 3: Essential Research Reagents for E. coli Flux Analysis Experiments
| Reagent/Category | Specific Example | Function/Application |
|---|---|---|
| Bacterial Strain | E. coli MG1655 | Model organism for metabolic studies |
| Culture Media | Minimal medium with (NH₄)₂SO₄ and Glucose | Defined growth conditions with specific nutrient limitations |
| Analytical Instrument | HPLC system with appropriate detectors | Quantification of metabolite concentrations |
| Isotopic Tracer | ¹³C-labeled glucose (e.g., [1-¹³C]glucose) | Tracing metabolic pathways via ¹³C-MFA |
| Computational Tool | COBRApy, GNU Linear Programming Kit | Implementing FBA/MOMA/ROOM algorithms |
Advanced Applications and Hybrid Approaches
Dynamic Extensions and Metaheuristic Integration
The core FBA, MOMA, and ROOM algorithms have been extended to dynamic conditions and integrated with optimization metaheuristics for enhanced predictive capabilities:
Dynamic FBA (DFBA): Extends FBA to transient conditions using either static optimization (SOA) or dynamic optimization (DOA) approaches. DOA formulates the problem as: ( \max \int{t0}^{tf} f(x)dx ) subject to ( \frac{dX}{dt} = S·v ), ( v{min} ≤ v ≤ v{max} ), ( x{min} ≤ x ≤ x{max} ), and ( X(t0) = X_0 ) [41].
Dynamic MOMA (M-DFBA) and ROOM (R-DFBA): Combine MOMA or ROOM principles with DFBA to predict metabolic dynamics under perturbation. These approaches incorporate hypotheses about minimal fluctuation of metabolic profiles over time [41].
Metaheuristic Hybrids: Swarm intelligence algorithms (PSO, ABC, CS) have been hybridized with MOMA to identify optimal gene knockout strategies for metabolic engineering. These approaches (PSOMOMA, ABCMOMA, CSMOMA) efficiently search the high-dimensional space of possible genetic modifications to maximize target metabolite production [42].
Addressing Mathematical Degeneracy with Advanced Formulations
A significant challenge in FBA is solution degeneracy—multiple flux distributions can achieve identical optimal growth. The PSEUDO (Perturbed Solution Expected Under Degenerate Optimality) method addresses this by explicitly accounting for regions of degenerate near-optimality [44]. Rather than assuming metabolism achieves a single optimal point, PSEUDO identifies a region of flux space allowing nearly optimal growth (e.g., ≥90% maximal growth rate), then finds mutant fluxes minimally distant from this region [44]. This approach outperformed both FBA and MOMA in predicting central carbon flux redistribution in E. coli metabolic mutants [44].
FBA, MOMA, and ROOM represent complementary approaches for knockout strain analysis, each with distinct strengths and appropriate application contexts. FBA performs best for wild-type strains and evolved mutants where optimality assumptions hold. MOMA provides superior predictions for recent laboratory knockouts without evolutionary optimization. ROOM excels when knockouts require significant flux rerouting through alternative pathways. Experimental validation remains challenging, with current methods collectively predicting only a minority of empirically observed genetic interactions, highlighting the need for continued method development and multi-factorial validation approaches.
For researchers investigating E. coli knockout strains, the following recommendations emerge from experimental comparisons:
Validate predictions against ¹³C-MFA data wherever possible, as this provides the most direct measurement of intracellular fluxes [3].
Consider hybrid approaches that leverage metaheuristic optimization with MOMA for metabolic engineering applications targeting metabolite overproduction [42].
Account for solution degeneracy through methods like FVA (Flux Variability Analysis) or PSEUDO when precise flux predictions are required beyond growth rate alone [45] [44].
Temper expectations for epistasis prediction, as current constraint-based methods capture only a fraction of experimentally observed genetic interactions [43].
The field continues to evolve with integrations of proteomic constraints, kinetic information, and multi-scale modeling approaches promising to enhance the predictive power of these foundational algorithms for knockout strain analysis.
Metabolic Flux Analysis (MFA) is a cornerstone technique for quantifying intracellular reaction rates in living cells, providing critical insights into metabolic network functionality and engineering potential [30] [3]. For the model organism Escherichia coli, precise flux quantification is essential for both basic physiology research and industrial bioprocess optimization [12]. While classical 13C-MFA relies on isotopic steady-state measurements, two advanced methodologies have emerged to address its limitations: Isotopically Nonstationary MFA (INST-MFA) and Genome-Scale MFA (GS-MFA). INST-MFA utilizes time-resolved labeling data before the system reaches isotopic steady state, enabling flux estimation in systems where stationary labeling provides insufficient information [30]. GS-MFA expands the scope of traditional MFA from core metabolic pathways to genome-scale networks, offering a comprehensive view of cellular metabolism [3]. This guide objectively compares the performance, data requirements, and applications of INST-MFA and GS-MFA within the broader context of constraint-based modeling for E. coli flux prediction research.
Core Methodological Principles
Isotopically Nonstationary MFA (INST-MFA)
INST-MFA is specifically designed for systems where the isotopically stationary state is uninformative, such as autotrophic plant metabolism where all metabolites become fully labeled [30]. The fundamental principle involves tracking the incorporation of a labeled substrate (e.g., 13C or 15N) into metabolic intermediates over time, before the system reaches isotopic equilibrium [30]. The computational core of INST-MFA involves solving ordinary differential equations (ODEs) that describe the temporal evolution of mass isotopomer distributions (MIDs) with reaction fluxes as parameters optimized to fit the experimental measurements [30].
INST-MFA implementations can be categorized into global and local approaches. Global INST-MFA estimates all identifiable fluxes in a network simultaneously, requiring substantial computational power and comprehensive labeling data [30]. In contrast, local INST-MFA approaches focus on estimating fluxes for specific sub-networks or reactions, reducing data demands and computational complexity [30]. Key local approaches include:
- Kinetic Flux Profiling (KFP): Utilizes only the unlabeled (M+0) isotopomer fraction and can estimate total flux through a metabolite [30].
- Non-stationary Metabolic Flux Ratio Analysis (NSMFRA): Estimates relative local fluxes at metabolic branch points and can simulate labeling data for metabolites without measurements [30].
- ScalaFlux: Designed to estimate fluxes for any reaction or sub-network with sufficient labeling data, automating ODE construction from network structure [30].
Genome-Scale MFA (GS-MFA)
GS-MFA extends the principles of traditional MFA to genome-scale metabolic models (GEMs), which incorporate all known metabolic reactions in an organism based on genome annotation and manual curation [3] [10]. The primary challenge in GS-MFA is the underdetermined nature of GEMs, where the number of fluxes exceeds the constraints from available measurements [3] [46].
To address this underdetermination, GS-MFA often integrates with Flux Balance Analysis (FBA), a constraint-based approach that predicts fluxes by assuming the cell optimizes an objective function, most commonly biomass yield [3] [12]. This synergy allows researchers to combine experimental labeling data with computational predictions to obtain genome-scale flux maps [12]. Advanced variants like parsimonious FBA (pFBA) incorporate additional biological principles, such as minimizing total enzyme burden, to improve flux predictions [47]. Recent innovations like complex-balanced FBA (cbFBA) further refine predictions by maximizing multi-reaction dependencies inherent in network stoichiometry [47].
Table 1: Core Characteristics of INST-MFA and GS-MFA
| Characteristic | INST-MFA | GS-MFA |
|---|---|---|
| Isotopic State | Nonstationary (time-resolved) | Stationary (snapshot) |
| Network Scope | Typically sub-networks or specific pathways | Genome-scale |
| Primary Data | Time-course mass isotopomer distributions (MIDs) | Stationary MIDs, exchange fluxes |
| Computational Core | ODE optimization fitting labeling kinetics | Large-scale inverse problem, often integrated with FBA |
| Key Applications | Systems with uninformative stationary labeling (e.g., autotrophic metabolism); nitrogen/carbon flux studies in plants [30] | Comprehensive flux mapping; integration with multi-omics data; strain design [3] [48] |
Performance Comparison inE. coliFlux Prediction
Direct comparisons between INST-MFA and GS-MFA are scarce in the literature, as their applications often target different biological questions. However, their performance can be evaluated based on accuracy, precision, scope, and practical implementation in E. coli studies.
Accuracy and Precision of Flux Estimates
INST-MFA excels in providing precise estimates for specific pathways, particularly when global approaches are applied to well-defined networks. For example, a synergy study between 13C-MFA and FBA in E. coli revealed detailed flux maps under aerobic and anaerobic conditions, uncovering a non-cyclic TCA operation that contradicted some previous models [12]. This demonstrates MFA's power to correct erroneous assumptions about pathway operation.
GS-MFA, particularly when enhanced with advanced FBA techniques, shows improving accuracy in predicting intracellular fluxes. A systematic evaluation of cbFBA against pFBA demonstrated superior agreement with experimentally measured fluxes from 17 E. coli strains [47]. cbFBA also produced more precise predictions due to a smaller space of alternative solutions, a key advantage for metabolic engineering applications where target identification is crucial [47].
Scope of Flux Information
The fundamental trade-off between these approaches lies in network coverage versus flux detail:
- INST-MFA provides high-resolution flux estimates, including exchange fluxes (forward and reverse reaction rates) for reversible reactions, which are crucial for understanding metabolic efficiency and regulation [12]. However, its scope is typically limited to central carbon metabolism due to analytical and computational constraints.
- GS-MFA offers comprehensive network coverage, enabling discovery of systemic effects and interactions between distant pathways [3]. However, it may lack the resolution to quantify exchange fluxes accurately and often relies on optimality assumptions that may not hold in all genetic or environmental contexts [12].
Table 2: Performance Comparison for E. coli Flux Prediction
| Performance Metric | INST-MFA | GS-MFA |
|---|---|---|
| Pathway Resolution | High (including exchange fluxes) [12] | Medium (net fluxes typically) |
| Network Coverage | Limited to core metabolism | Comprehensive (genome-scale) [3] |
| Quantitative Accuracy | High for measured pathways [12] | Variable; improved with methods like cbFBA [47] |
| Data Requirements | High (time-course labeling) | Lower (stationary labeling or no labeling) |
| Computational Demand | High (ODE optimization) | Medium-High (Linear Programming, sampling) |
Experimental Protocols
Protocol for INST-MFA in E. coli
- Labeling Experiment: Grow E. coli (e.g., K-12 MG1655) in minimal medium (e.g., M9) with a single carbon source (e.g., 2 g/L glucose). Introduce a 13C-labeled substrate (e.g., [U-13C]glucose) at mid-log phase [12].
- Time-Course Sampling: Rapidly collect cell aliquots at multiple time points (seconds to minutes) after tracer introduction. Quench metabolism immediately (e.g., using cold methanol) [30].
- Metabolite Extraction: Extract intracellular metabolites and measure absolute pool sizes where required for absolute flux estimation [30].
- Mass Spectrometry Analysis: Analyze mass isotopomer distributions (MIDs) of targeted metabolites (e.g., from central metabolism) using GC-MS or LC-MS. The unlabeled (M+0) fraction is essential for KFP, while all fractions are used by NSMFRA and ScalaFlux [30].
- Flux Estimation: Optimize reaction fluxes by fitting the parameters of the ODE system to the time-course MID data. For local approaches, this is done only for the sub-network of interest [30].
Protocol for GS-MFA Integrated with FBA
- Strain and Culture: Use a defined E. coli strain (e.g., K-12 MG1655). Cultivate under controlled conditions (e.g., 37°C in M9 glucose medium) to metabolic steady-state [12].
- External Flux Measurements: Quantify substrate uptake (e.g., glucose) and product secretion (e.g., acetate, CO2) rates. Measure the specific growth rate [12] [46].
- Stationary Labeling (Optional): If using labeling data, feed a 13C-labeled substrate and measure the stationary MIDs of metabolites or proteinogenic amino acids [12].
- Flux Prediction: Solve the FBA problem: maximize biomass objective function subject to stoichiometric constraints ( S \cdot v = 0 ) and measured exchange flux bounds [3] [12].
- Flux Refinement: Apply additional biological constraints to reduce solution space. For example, use pFBA to minimize total flux or cbFBA to maximize multi-reaction dependencies [47]. Integrate stationary MIDs if available to further constrain fluxes.
Workflow Visualization
The following diagram illustrates the core workflows for INST-MFA and GS-MFA, highlighting their key differences in data input and processing logic.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents and Tools for Advanced MFA
| Reagent / Tool | Function / Application | Relevance |
|---|---|---|
| [U-13C] Glucose | Uniformly labeled carbon tracer for probing glycolytic and TCA cycle fluxes. | Fundamental for both INST-MFA (time-course) and GS-MFA (stationary) labeling experiments in E. coli [12]. |
| GC-MS / LC-MS Systems | Analytical instruments for measuring Mass Isotopomer Distributions (MIDs) of metabolites. | Core analytical technology for experimental flux determination in both MFA types [30] [12]. |
| Genome-Scale Model (e.g., iML1515) | A stoichiometric matrix of all known metabolic reactions in E. coli. | Essential framework for GS-MFA and FBA; provides structural constraints for flux prediction [49]. |
| COBRA Toolbox | MATLAB-based software suite for constraint-based modeling and FBA. | Widely used platform for implementing GS-MFA and related FBA simulations [3]. |
| INCA Software | A widely used software platform for INST-MFA. | Enables global INST-MFA by simulating labeling patterns and estimating fluxes [30]. |
| Machine Learning Models (e.g., for kcat prediction) | Predicts enzyme kinetic parameters (turnover numbers) from features like protein structure. | Used to parameterize mechanistic genome-scale models, improving proteome allocation predictions in GEMs [49] [48]. |
INST-MFA and GS-MFA represent complementary paradigms in advanced metabolic flux analysis. INST-MFA is the method of choice when high-resolution quantification of fluxes in specific pathways is required, particularly in labeling scenarios where stationary approaches fail. Its reliance on time-course data, however, imposes significant experimental and computational burdens. GS-MFA, particularly when integrated with FBA and machine learning, provides a systems-level view of metabolism, enabling genome-scale predictions that are invaluable for strain design and systems biology. The accuracy of its internal flux predictions continues to improve with methods like cbFBA [47] and hybrid modeling approaches like MINN [48]. The selection between INST-MFA and GS-MFA ultimately depends on the research question: INST-MFA for precise, pathway-specific flux elucidation, and GS-MFA for comprehensive, network-wide flux prediction and integration with multi-omics data. Future advancements will likely focus on further integrating these approaches to leverage their respective strengths.
Flux Balance Analysis (FBA) and Metabolic Flux Analysis (MFA) represent two powerful computational frameworks for quantifying metabolic fluxes in biological systems, each with distinct methodological foundations and applications. FBA is a constraint-based modeling approach that predicts flow of metabolites through biochemical networks by utilizing mathematical optimization, typically maximizing or minimizing an objective function such as biomass production or ATP yield [50]. This method relies on stoichiometric models of metabolism that contain all known metabolic reactions for an organism and requires minimal experimental input, making it particularly valuable for predicting metabolic capabilities and conducting in silico simulations of genetic and environmental perturbations [50] [23]. The core principle of FBA involves solving a system of linear equations representing mass balance constraints under steady-state assumptions, where the concentration of internal metabolites remains constant over time [50].
In contrast, 13C-Metabolic Flux Analysis (13C-MFA) is an experimentally driven methodology that quantifies intracellular metabolic fluxes by employing stable isotope tracing and advanced analytical techniques [23]. This approach utilizes 13C-labeled substrates fed to growing cells, with subsequent measurement of label incorporation into metabolic products using mass spectrometry or NMR spectroscopy [12] [23]. The resulting labeling patterns enable precise estimation of metabolic reaction rates through optimized fitting of internal fluxes to the experimental measurements [3]. Unlike FBA, 13C-MFA does not assume optimal cellular performance and provides direct empirical quantification of fluxes, establishing it as the gold standard for accurate flux quantification in metabolic engineering [23].
For Escherichia coli metabolism specifically, both methods offer complementary insights. FBA leverages genome-scale metabolic models to predict system-level capabilities, while 13C-MFA delivers high-precision validation of actual metabolic operation under defined conditions [51] [12]. This case study examines how their synergistic application elucidates the complex metabolic adaptations of E. coli during transition from aerobic to anaerobic conditions, revealing insights that neither method could provide independently.
Comparative Analysis of FBA and MFA
Methodological Foundations and Limitations
The fundamental differences between FBA and MFA establish their complementary nature for metabolic flux studies. FBA operates primarily as a prediction tool based on biochemical constraints and optimization principles, while MFA functions as a measurement approach grounded in experimental data from isotopic labeling [3] [23]. This distinction creates a reciprocal relationship where FBA can generate testable hypotheses about metabolic network operation, and MFA provides empirical validation and refinement of these predictions [12].
Key limitations of FBA include its inability to predict metabolite concentrations, its restriction to steady-state conditions, and its general lack of incorporation of regulatory effects such as enzyme activation or gene expression control [50]. Additionally, FBA solutions may not be unique, with multiple flux distributions potentially satisfying the optimization criteria equally well [12]. The predictive accuracy of FBA is heavily dependent on appropriate selection of objective functions and constraint boundaries, which, if poorly chosen, can lead to biologically irrelevant flux predictions [3].
MFA, while providing greater empirical accuracy, faces its own constraints. The method is primarily limited to describing metabolic activities related to carbon flow, with non-carbon metabolism and transport receiving less attention [12]. MFA also requires the system to maintain metabolic steady state throughout the labeling experiment, making it challenging to apply to transient conditions or heterogeneous cell populations [23]. Furthermore, technical limitations include the need for specialized analytical equipment and expertise in isotope measurement techniques, which can present barriers to implementation [3].
Quantitative Comparison of Aerobic and Anaerobic Fluxes in E. coli
The synergistic application of FBA and 13C-MFA to E. coli K-12 MG1655 grown under aerobic and anaerobic conditions in glucose-minimal medium revealed profound metabolic adaptations. Quantitative flux data derived from these complementary methods provides a comprehensive view of how E. coli reshapes its metabolic network in response to oxygen availability.
Table 1: Comparative Metabolic Flux Distributions in E. coli under Aerobic and Anaerobic Conditions
| Metabolic Parameter | Aerobic Conditions | Anaerobic Conditions | Measurement Method |
|---|---|---|---|
| Glucose uptake rate | Baseline | ~70% increase | Experimental measurement [12] |
| Acetate secretion | Not detected | 31% increase | Enzyme assay & ¹H NMR [12] |
| TCA cycle operation | Non-cyclic, 16.1% of glucose uptake | Significantly reduced | ¹³C-MFA [51] [12] |
| Maintenance ATP fraction | 37.2% of total ATP production | 51.1% of total ATP production | ¹³C-MFA [51] [12] |
| Predicted growth rate | 1.65 hr⁻¹ | 0.47 hr⁻¹ | FBA simulation [50] |
| ATP synthase activity | Standard oxidative phosphorylation | Increased usage for proton secretion | FBA prediction [51] |
The data reveal several key metabolic adaptations. Under anaerobic conditions, E. coli increases glucose uptake substantially while shifting ATP metabolism toward higher maintenance costs. The TCA cycle operates in a non-cyclic mode during aerobic growth, with significantly reduced flux under anaerobic conditions. FBA predictions of growth rates corresponded well with experimental measurements, validating the modeling approach [50].
Table 2: Methodological Comparison of FBA and MFA for E. coli Flux Analysis
| Characteristic | Flux Balance Analysis (FBA) | 13C-Metabolic Flux Analysis (13C-MFA) |
|---|---|---|
| Basis | Stoichiometric constraints & optimization | Isotopic labeling & mass balance |
| Experimental requirements | Minimal (uptake/secretion rates) | Extensive (isotope tracing, MS/NMR) |
| Network scale | Genome-scale (e.g., iJR904 with 906 genes) | Core metabolism (central carbon pathways) |
| Key assumptions | Steady state, optimal growth | Metabolic & isotopic steady state |
| Regulatory incorporation | Limited without extensions | Captures operational regulation |
| Output precision | Predictive capabilities | High precision for central metabolism |
| Unique capabilities | Gene knockout predictions, network capabilities | Exchange flux estimates, pathway validation |
Experimental Protocols for Synergistic Flux Analysis
Integrated FBA-MFA Workflow for E. coli Anaerobiosis
The synergistic application of FBA and MFA follows a structured workflow that leverages the strengths of both methodologies. The integrated approach begins with cultivation of E. coli K-12 MG1655 in defined M9 minimal medium with glucose (2g/L) as sole carbon source, under both aerobic and anaerobic conditions at 37°C [12]. Cells are harvested during mid-log phase growth for subsequent analysis, ensuring metabolic steady state required for both FBA and MFA.
For MFA, the experimental protocol involves feeding 13C-labeled glucose tracers to the cultures, followed by precise measurement of substrate uptake and product secretion rates using enzymatic assays and ¹H NMR [12]. The labeling patterns of proteinogenic amino acids and intracellular metabolites are determined via GC-MS and LC-MS, providing the isotopic data required for flux calculation [12] [23]. For FBA, the genome-scale metabolic model iJR904—containing 906 genes, 931 metabolites, and 1075 reactions—serves as the computational framework [12]. Flux predictions are generated by applying constraints based on measured glucose uptake rates and applying biomass maximization as the objective function.
The synergy emerges when MFA-validated flux maps are compared with FBA predictions, enabling identification of discrepancies that reveal important physiological insights. For instance, when FBA predictions diverge from MFA measurements, this often indicates gaps in model formulation or the presence of regulatory constraints not captured in the stoichiometric model [51] [12]. This iterative refinement process enhances both the predictive power of FBA and the interpretive context for MFA results.
Analytical Techniques for Flux Determination
The experimental protocol for synergistic flux analysis employs multiple analytical techniques to generate complementary data sets. For quantitative extracellular flux measurements, enzymatic assays provide precise determination of metabolite concentrations in the culture medium, while ¹H NMR spectroscopy enables identification and quantification of metabolic products such as acetate, lactate, succinate, formate, and ethanol [12]. Gas analysis methods measure CO₂ levels, particularly important for verifying metabolic pathways under anaerobic conditions where formate accumulation may occur instead of CO₂ release [12].
For isotopic labeling analysis, gas chromatography-mass spectrometry (GC-MS) measures mass isotopomer distributions of proteinogenic amino acids, which serve as proxies for intracellular metabolite labeling [12] [23]. Liquid chromatography-mass spectrometry (LC-MS) provides complementary labeling data for intracellular metabolic intermediates. In some cases, nuclear magnetic resonance (NMR) spectroscopy offers additional positional labeling information through 13C-detected experiments, which is particularly valuable for resolving certain metabolic fluxes [12].
Computational flux estimation involves fitting the experimental labeling data to the metabolic network model using specialized software tools. The model parameters are optimized to minimize the difference between simulated and measured isotopic labeling patterns, resulting in a statistically validated flux map [3]. For FBA, the COBRA (Constraints-Based Reconstruction and Analysis) Toolbox implements the linear programming algorithms needed to solve the optimization problem and predict flux distributions [50]. The convergence of these experimental and computational approaches provides a robust framework for flux quantification.
Metabolic Pathway Adaptations to Anaerobiosis
Visualizing E. coli Central Metabolic Flux Rewiring
The transition from aerobic to anaerobic conditions triggers substantial reorganization of E. coli central metabolism, as revealed by the integrated FBA-MFA approach. The flux maps generated through 13C-MFA provide empirical validation of long-hypothesized metabolic adaptations while uncovering unexpected features of anaerobic metabolism.
Key Metabolic Findings from Integrated Analysis
The synergistic application of FBA and MFA to E. coli anaerobiosis has yielded several fundamental insights into bacterial metabolic adaptation. First, the TCA cycle operates in a non-cyclic manner during aerobic growth, with minimal flux through oxidative reactions, contradicting the traditional view of a fully cyclic TCA cycle in aerobically growing cells [12]. This finding was confirmed through labeling measurements of intracellular CO₂/HCO₃⁻ based on terminal carbons of arginine, providing direct evidence for incomplete TCA cycle operation [12].
Second, anaerobic conditions trigger a significant increase in ATP maintenance requirements, with the fraction of maintenance ATP consumption rising from 37.2% under aerobic conditions to 51.1% of total ATP production during anaerobic growth [51] [12]. FBA simulations revealed that this increased ATP utilization is consumed by ATP synthase to secrete protons generated during fermentation, representing an important energy burden not previously quantified [51].
Third, submaximal growth under aerobic conditions appears due to limitations in oxidative phosphorylation capacity rather than carbon uptake or central metabolic functions [51]. This insight emerged from the discrepancy between FBA predictions assuming optimal ATP yield and MFA measurements showing lower actual fluxes, highlighting a constraint not captured in standard FBA formulations.
Finally, the integrated analysis demonstrated that FBA successfully predicts product secretion rates in aerobic cultures when constrained with both glucose and oxygen uptake measurements, but the most frequently predicted values of internal fluxes obtained through sampling the feasible solution space often differ substantially from MFA-derived fluxes [12]. This indicates significant flexibility in E. coli's metabolic network, with multiple flux distributions capable of achieving similar growth outcomes.
Essential Research Tools and Reagents
The Scientist's Toolkit for Bacterial Flux Analysis
Successful implementation of synergistic FBA-MFA studies requires specific research tools and reagents spanning biological, analytical, and computational domains. The following table catalogues essential resources for conducting integrated flux analyses of E. coli metabolism.
Table 3: Essential Research Reagents and Computational Tools for FBA-MFA Studies
| Category | Item | Specification/Example | Application Note |
|---|---|---|---|
| Biological Materials | E. coli strain | K-12 MG1655 (ATCC 47076) | Wild-type reference strain [12] |
| Culture medium | M9 minimal medium | Defined composition enables precise flux measurements [12] | |
| Isotopic tracers | 13C-glucose (e.g., [1,2-13C]) | Enables MFA through isotopic labeling [23] | |
| Analytical Instruments | GC-MS system | Gas chromatograph-mass spectrometer | Measures mass isotopomer distributions [12] |
| LC-MS system | Liquid chromatograph-mass spectrometer | Analyzes labeling of intracellular metabolites [23] | |
| NMR spectrometer | ¹H NMR capability | Quantifies extracellular metabolites & positional labeling [12] | |
| Computational Tools | COBRA Toolbox | MATLAB-based package | Performs FBA simulations & constraint-based modeling [50] |
| Metabolic modeling software | ClusterFLUX, INCA | Performs 13C-MFA flux estimation [12] | |
| Genome-scale model | iJR904 (906 genes) | E. coli metabolic reconstruction for FBA [12] | |
| Reference Databases | Metabolic database | EcoCyc | E. coli genes, metabolism, & regulatory information [2] |
| Enzyme kinetics database | BRENDA | Comprehensive enzyme kinetic parameters [2] |
The implementation of these tools follows a structured workflow. Biological materials establish the experimental system under controlled conditions, with careful attention to maintaining metabolic steady state throughout isotopic labeling experiments. Analytical instruments generate the quantitative data required for both constraining FBA models and calculating MFA fluxes. Computational tools then integrate these data sets to generate and validate flux predictions, with reference databases providing essential biochemical context for model construction and refinement.
Particularly important for FBA-MFA integration is the use of consistent metabolic network models. The iJR904 genome-scale model used for FBA contained 906 genes and was adapted to create the 13C-MFA network model with complete carbon rearrangements for central metabolism [12]. This model consistency enables direct comparison between FBA predictions and MFA measurements, facilitating identification of discrepancies that reveal novel physiological insights. Specialized software tools like the COBRA Toolbox for FBA and ClusterFLUX for MFA implement the complex numerical algorithms required for flux estimation while providing user-friendly interfaces accessible to biological researchers [50] [12].
The synergistic application of Flux Balance Analysis and Metabolic Flux Analysis provides a powerful framework for elucidating the complex metabolic adaptations of E. coli to anaerobic conditions. FBA offers genome-scale predictive capabilities and hypothesis generation, while MFA delivers high-precision empirical validation of intracellular fluxes. Together, these methods have revealed key insights including the non-cyclic operation of the TCA cycle during aerobic growth, increased ATP maintenance demands during anaerobiosis, and limitations in oxidative phosphorylation capacity constraining aerobic growth.
The integrated approach demonstrates that neither methodological paradigm alone can fully capture the complexity of metabolic adaptation. FBA successfully predicts optimal metabolic strategies and external flux phenotypes but often fails to accurately recapitulate internal flux distributions measured by MFA. Conversely, MFA provides precise quantification of operational fluxes but offers limited predictive capability for novel genetic or environmental conditions. Their combination creates a virtuous cycle where FBA predictions guide MFA experimental design, and MFA results refine and validate FBA models.
For researchers investigating microbial metabolism, this case study establishes a template for leveraging the complementary strengths of constraint-based modeling and experimental flux analysis. The continued development of both methodologies promises enhanced capacity to engineer microbial systems for biomedical and industrial applications, from drug development to sustainable bioproduction. As both FBA and MFA methodologies advance, their integration will remain essential for translating metabolic network knowledge into predictive understanding of cellular physiology.
Overcoming Challenges: Improving the Accuracy of Flux Predictions
Flux Balance Analysis (FBA) and Metabolic Flux Analysis (MFA) are cornerstone methods in constraint-based metabolic modeling, both operating on the fundamental premise of metabolic steady-state where reaction rates and metabolite levels remain constant [3]. While these methods are widely used to investigate biochemical networks in biological and biotechnological research, they approach flux estimation from fundamentally different angles. FBA uses linear optimization to predict flux distributions that maximize or minimize a specified cellular objective, typically biomass production, without requiring experimental labeling data [3] [12]. In contrast, 13C-MFA works backward from experimentally measured isotopic label distributions in metabolites to estimate intracellular fluxes, providing a more data-driven approach to flux determination [3] [12].
The synergy between these approaches is particularly valuable in Escherichia coli flux prediction research, where MFA can describe the actual metabolic status through intracellular carbon flow, while genome-scale constraint-based models reveal the theoretical metabolic capacities of the system [12]. However, several persistent pitfalls undermine the reliability of FBA predictions, with incorrect gene-essentiality predictions and gap-filling errors representing two critical challenges that can significantly impact downstream applications in metabolic engineering and drug development.
The Gene Essentiality Prediction Problem in FBA
Fundamental Limitations in Prediction Accuracy
A central challenge in FBA is the accurate prediction of gene essentiality, which refers to identifying genes whose impairment would prevent cell survival. The standard FBA approach for this task operates on a critical assumption: both wild-type and gene deletion strains optimize the same fitness objective, typically growth rate [52]. While this optimality assumption may hold for wild-type microbial strains under evolutionary pressure, deletion strains often display suboptimal growth phenotypes and are not subject to the same long-term evolutionary pressures [52].
This fundamental limitation manifests clearly in model organisms like Escherichia coli, where FBA has shown reasonable prediction accuracy, but produces mixed and often unsatisfactory results for eukaryotes and higher-order organisms [52]. The accuracy of these predictions varies substantially across different metabolic models and organisms, reflecting both limitations in model quality and the inherent shortcomings of the optimality assumption for knockout strains [52].
Emerging Solutions: Hybrid FBA-Machine Learning Approaches
To address these limitations, researchers have developed innovative hybrid approaches that combine FBA with machine learning. The FlowGAT framework represents one such advancement, using a graph-structured representation of metabolic fluxes predicted by FBA where nodes correspond to enzymatic reactions and edges quantify metabolite mass flow between reactions [52]. This information is integrated into a graph neural network trained on knock-out fitness assay data, allowing the model to predict gene essentiality directly from wild-type metabolic phenotypes without assuming optimality for deletion strains [52].
This approach demonstrates that essentiality of enzymatic genes can be predicted by exploiting the inherent network structure of metabolism, achieving prediction accuracy close to FBA gold standards for E. coli across multiple growth conditions [52]. The method highlights the benefits of combining mechanistic insights from genome-scale models with the pattern recognition capabilities of deep learning to overcome fundamental limitations in traditional FBA.
Gap-Filling Errors and Thermodynamic Infeasibilities
The Challenge of Thermodyamically Infeasible Cycles
A pervasive issue in genome-scale metabolic models (GEMs) is the presence of thermodynamically infeasible cycles (TICs), which represent a significant source of uncertainty in FBA predictions [53]. These cycles function analogously to perpetual motion machines, violating the second law of thermodynamics by cycling metabolites indefinitely without any real change or nutrient input [53]. The presence of TICs can severely compromise predictive capabilities by distorting flux distributions, producing erroneous growth and energy predictions, generating unreliable gene essentiality predictions, and undermining multi-omics integration efforts [53].
The root of this problem often lies in incomplete knowledge or model curation errors that result in blocked reactions - those that cannot carry flux due to either dead-end metabolites or thermodynamic infeasibility [53]. Traditional gap-filling approaches frequently introduce or overlook these thermodynamic inconsistencies, creating cascading errors throughout the model.
Computational Frameworks for Thermodynamic Consistency
Recent computational advances have produced comprehensive solutions for addressing TICs in metabolic models. The ThermOptCOBRA framework incorporates four specialized algorithms that integrate thermodynamic constraints directly into model construction and analysis [53]. This approach leverages network topology to efficiently identify TICs across thousands of published models, determine thermodynamically feasible flux directions, and detect blocked reactions to yield more refined models with fewer thermodynamic inconsistencies [53].
A key innovation in ThermOptCOBRA is its ability to construct thermodynamically consistent context-specific models that are more compact than those generated by traditional algorithms like Fastcore in 80% of cases [53]. By incorporating TIC removal constraints directly into the model construction process, this framework ensures that resulting models contain no blocked reactions arising from thermodynamic infeasibility, significantly improving the biological realism of predictions.
Comparative Analysis: Quantitative Assessment of FBA Limitations
Performance Comparison of Flux Prediction Methods
Table 1: Comparison of Flux Prediction Methods Against Experimental Data
| Method | Principle | Accuracy vs Experimental E. coli Fluxes | Precision (Solution Space Size) | Key Limitations |
|---|---|---|---|---|
| Standard FBA | Maximizes biomass objective function | Variable; poor internal flux prediction [12] | Low (large alternative solution space) [54] | Multiple optimal solutions; inaccurate internal fluxes [12] |
| Parsimonious FBA (pFBA) | Minimizes total enzyme usage after growth optimization [54] | Less accurate than cbFBA [54] | Moderate [54] | Poor agreement with experimentally measured fluxes [54] |
| Complex-Balanced FBA (cbFBA) | Maximizes multi-reaction dependencies [54] | Better agreement with experimental data from 17 E. coli strains [54] | High (smaller space of alternative solutions) [54] | Computational complexity |
| FlowGAT | Hybrid FBA-graph neural network using wild-type fluxes [52] | 接近FBA gold standard for E. coli [52] | N/A | Requires training data; complex implementation |
Gene Essentiality Prediction Accuracy Across Organisms
Table 2: Gene Essentiality Prediction Performance Across Organisms
| Organism Type | FBA Prediction Accuracy | Major Contributing Factors | Potential Solutions |
|---|---|---|---|
| Model microbes (E. coli) | Successful predictions [52] | Well-curated models; optimality assumption may hold [52] | Hybrid machine learning approaches [52] |
| Eukaryotes & higher-order organisms | Mixed results [52] | Model gaps/errors; suboptimal deletion strain phenotypes [52] | Thermodynamic constraint integration; improved model curation [53] |
| Pan-genomic applications | Variable quality [52] | Mapping between enzymatic genes and metabolic reactions [52] | Advanced gap-filling algorithms [53] |
Experimental Protocols for Method Validation
Protocol 1: Validation of Gene Essentiality Predictions
Strain Cultivation: Grow wild-type E. coli K-12 MG1655 in defined minimal medium (M9) with glucose (2 g/L) as sole carbon source at 37°C with continuous shaking at 250 rpm [12].
Knockout Strain Generation: Create single-gene deletion mutants using standard genetic engineering techniques, ensuring complete gene disruption.
Growth Phenotyping: Measure growth parameters and substrate uptake/secretion rates for both wild-type and knockout strains during mid-log phase using spectrophotometry and analytical methods like GC/MS or NMR [12].
Essentiality Determination: Classify genes as essential when deletion results in complete growth arrest or significantly impaired growth (<10% of wild-type growth rate) under defined conditions.
Computational Prediction: Perform FBA simulations constraining model with measured substrate uptake rates and comparing predicted growth of deletion strains to experimental results [52].
Accuracy Assessment: Calculate prediction accuracy metrics (precision, recall, F1-score) by comparing computational predictions with experimental essentiality calls.
Protocol 2: Detection and Resolution of Thermodynamically Infeasible Cycles
Model Preprocessing: Obtain genome-scale metabolic model in standard SBML format and convert to stoichiometric matrix representation [53].
TIC Identification: Apply ThermOptEnumerator algorithm to efficiently identify all thermodynamically infeasible cycles by leveraging network topology without requiring external experimental data [53].
Blocked Reaction Detection: Use ThermOptCC method to identify reactions blocked due to dead-end metabolites or thermodynamic infeasibility, more efficiently than traditional loopless-FVA approaches [53].
Model Correction: Implement thermodynamic constraints to eliminate TICs through reaction directionality constraints, removal of duplicate reactions, and correction of cofactor usage [53].
Validation: Compare flux variability before and after TIC removal, confirming elimination of thermodynamically infeasible flux loops while maintaining biological functionality [53].
Visualization of Key Concepts and Workflows
FBA Pitfalls and Solutions Diagram
Gap-Filling Error Propagation
Essential Research Reagent Solutions
Table 3: Key Research Reagents and Computational Tools
| Reagent/Tool | Function | Application Context |
|---|---|---|
| 13C-labeled substrates | Tracing carbon fate in metabolic networks [3] | 13C-MFA experiments for flux validation [3] [12] |
| ThermOptCOBRA | Detects and resolves thermodynamically infeasible cycles [53] | Model curation and refinement |
| FlowGAT | Hybrid FBA-graph neural network for essentiality prediction [52] | Gene essentiality prediction without optimality assumption |
| cbFBA Algorithm | Predicts fluxes by maximizing multi-reaction dependencies [54] | Improved intracellular flux prediction |
| GC/MS and NMR | Measures isotopic labeling in metabolites [12] | Experimental flux determination for method validation |
| COBRA Toolbox | Constraint-based reconstruction and analysis [53] | Metabolic model simulation and gap-filling |
The comparative analysis of FBA and MFA for E. coli flux prediction research reveals significant limitations in traditional FBA approaches, particularly concerning gene essentiality predictions and gap-filling errors. The fundamental issue stems from incorrect biological assumptions, such as the optimality of deletion strains, and technical shortcomings in model construction that introduce thermodynamic inconsistencies.
Moving forward, the integration of hybrid approaches that combine mechanistic modeling with machine learning, along with rigorous thermodynamic validation during model construction, represents the most promising path toward more accurate metabolic predictions. Frameworks like ThermOptCOBRA that systematically address TICs and methods like cbFBA that leverage multi-reaction dependencies rather than simple parsimony demonstrate tangible improvements in predictive accuracy [53] [54]. For researchers in metabolic engineering and drug development, these advanced approaches offer more reliable tools for identifying essential genes and predicting metabolic behavior across different genetic and environmental contexts.
As the field progresses, the synergy between experimental validation through MFA and computational improvements in FBA will continue to drive more accurate, biologically realistic metabolic models capable of addressing complex biological questions and biotechnological challenges.
Flux Balance Analysis (FBA) is a cornerstone constraint-based method for modeling metabolic behavior and cellular function. It predicts metabolic reaction fluxes that optimize a given objective, typically biomass production for unicellular organisms like E. coli [55]. While FBA has been highly successful at predicting growth rates under different conditions and gene essentiality, the prediction of internal cell fluxes remains a significant challenge [55]. This limitation stems from several factors: the FBA solution is often not unique, creating a solution space rather than a single output; organisms may not operate at maximum theoretical capacity; the observed metabolic state in populations is not unique; and thermodynamically infeasible loops can appear in the FBA output [55].
To address these limitations, particularly the assumption of optimal growth, the method corsoFBA (COst Reduced Sub-Optimal FBA) was developed. This innovative approach is based on the optimization of protein cost at sub-optimal objective levels, allowing researchers to explore the physiologically relevant near-optimal solution space [55]. This guide provides a comparative analysis of corsoFBA against other FBA techniques within the context of E. coli flux prediction research.
Methodological Comparison: How corsoFBA Diverges from Traditional Approaches
Traditional FBA and its common variants rely on a single optimization step, often with additional constraints applied during the optimization of the objective function. In contrast, corsoFBA implements a two-step optimization process. First, it fixes the biomass objective function at a predefined, sub-optimal value. Second, it minimizes an estimated protein cost throughout the metabolism to predict internal cell fluxes [55]. This fundamental difference in approach allows corsoFBA to explore flux distributions that are sub-optimal for growth but potentially more representative of actual cellular states.
The table below compares key features of corsoFBA against other methods that also incorporate enzymatic or protein costs.
Table 1: Comparison of FBA Methods Incorporating Enzymatic or Protein Constraints
| Method | Core Optimization Principle | Enzymatic Cost Calculation | Handling of Sub-Optimal Growth |
|---|---|---|---|
| corsoFBA [55] | Two-step: (1) Fix biomass, (2) Minimize protein cost | ∑ [J · MW · exp(α·ΔrG'°/(R·T))] | Explicitly explores sub-optimal space by fixing biomass |
| FBAwMC [55] | Single-step: Maximize biomass with crowding constraint | ∑ (aᵢ Jᵢ) ≤ 1 | Constrains solution space but does not explore sub-optimal |
| MOMENT [55] | Single-step: Maximize biomass with enzyme budget | ∑ (gᵢ · MWᵢ) ≤ C | Uses constraint but does not explore sub-optimal |
| Tepper et al. [55] | Single-step: Minimize sum of metabolite and enzyme levels | ∑ Mᵢ + δ · ∑ gᵢ | Minimization approach, no explicit sub-optimal exploration |
| pFBA | Single-step: Minimize flux after maximizing biomass | ∑ (Jᵢ)² | Finds minimal flux solution at optimal growth only |
The protein cost term in corsoFBA is particularly sophisticated, incorporating the net flux through a reaction (J), the enzyme molecular weight (MW), and a thermodynamic penalty for reversible reactions based on the standard Gibbs free energy (ΔrG'°) [55]. The molecular weight term represents the biosynthetic cost of producing sufficient enzyme levels, while the thermodynamic penalty accounts for the metabolite concentration changes needed to favor the reaction direction.
Experimental Framework and Performance Evaluation
Experimental Protocol for corsoFBA Validation
The development and validation of corsoFBA involved a structured computational experiment using E. coli as a model organism:
- Model Preparation: A genome-scale metabolic reconstruction of E. coli was used, represented as a stoichiometric matrix defining all metabolic reactions [55].
- Growth Condition Simulation: The method was tested under different dilution rates, mimicking various growth conditions [55].
- Flux Prediction: For each condition:
- The biomass production value was fixed at a specific sub-optimal level.
- The protein cost across all reactions was minimized using the described cost function.
- Internal metabolic fluxes were recorded [55].
- Pathway Analysis: A modified version of Extreme Pathway analysis was employed to decompose the model, quantifying energy production and overall protein cost for all possible pathways in central carbon metabolism [55].
- Validation: Predictions for key pathways (PEP Carboxylase, glyoxylate shunt, Entner-Doudoroff) were compared against experimental data at different glucose levels [55].
Comparative Performance Data
The performance of corsoFBA was evaluated against standard FBA and minimization of metabolic steps (MMS) approaches. The key metric was the accuracy in predicting the behavior of specific metabolic pathways in response to changing glucose availability.
Table 2: Predictive Performance for Pathway Usage in E. coli Central Carbon Metabolism
| Metabolic Pathway | Standard FBA | Minimization of Metabolic Steps (MMS) | corsoFBA |
|---|---|---|---|
| PEP Carboxylase | Fails to predict activity changes | Fails to predict activity changes | Correctly predicts activity across glucose levels |
| Glyoxylate Shunt | Fails to predict activity changes | Fails to predict activity changes | Correctly predicts activity across glucose levels |
| Entner-Doudoroff Pathway | Fails to predict activity changes | Fails to predict activity changes | Correctly predicts activity across glucose levels |
| Agreement with experimental data | Poor | Poor | Good agreement |
corsoFBA showed good agreement with experimental data of E. coli grown at different dilution rates. The method demonstrated that maintaining the objective function near its maximum value predicts metabolic states resembling low dilution rates, while lower biomass production values mirror higher dilution rates [55]. Furthermore, the relationship between predicted growth rate and glucose availability differed significantly between methods, with corsoFBA providing a more biologically realistic representation.
Table 3: Comparison of Predicted Growth Rate Relative to Glucose Uptake
| Method | Objective | Predicted Relationship Between Growth Rate and Glucose Uptake |
|---|---|---|
| Standard FBA | Maximize Biomass | Linear relationship, always at theoretical maximum |
| pFBA | Maximize Biomass, then Minimize Total Flux | Linear relationship, always at theoretical maximum |
| corsoFBA | Minimize Protein Cost at Fixed Biomass | Non-linear, sub-optimal relationship, varying with protein cost constraints |
Successful implementation of constraint-based metabolic models like corsoFBA requires specific computational tools and data resources.
Table 4: Essential Research Reagents and Resources for Metabolic Flux Modeling
| Resource / Reagent | Type | Primary Function in Research | Example Source / Implementation |
|---|---|---|---|
| Genome-Scale Metabolic Reconstruction | Data Structure | Provides stoichiometric representation of all known metabolic reactions in an organism [55] | BiGG Models [55] |
| COBRA Toolbox | Software Package | Provides a standardized environment for implementing Constraint-Based Reconstruction and Analysis [55] | COBRA [55] |
| Stoichiometric Matrix (S) | Mathematical Construct | Core of the model; defines metabolite relationships in reactions [55] | N/A |
| Extreme Pathway / Elementary Mode Analysis | Computational Algorithm | Decomposes network into unique, systemic pathways for functional analysis [55] | Modified Extreme Pathway Analysis [55] |
| Enzyme Kinetic Parameters (kcat, MW) | Biochemical Data | Informs enzymatic cost constraints in models like corsoFBA and MOMENT [55] | BRENDA Database |
| Thermodynamic Data (ΔrG'°) | Biochemical Data | Provides constraints on reaction directionality and calculates thermodynamic penalties [55] | eQuilibrator |
Advanced Hybrid Approaches: The Machine Learning Frontier
Beyond traditional constraint-based methods, new hybrid approaches are emerging that integrate machine learning with metabolic models. The Metabolic-Informed Neural Network (MINN) represents this advanced frontier. MINN is a hybrid model that utilizes multi-omics data to predict metabolic fluxes in E. coli under different growth rates and gene knockouts [48].
This framework embeds Genome-Scale Metabolic Models (GEMs) within a neural network architecture, combining the strengths of mechanistic and data-driven approaches [48]. MINN has demonstrated efficacy in improving prediction performances compared to parsimonious FBA (pFBA) and pure machine learning models like Random Forest (RF), particularly when handling the trade-off between biological constraints and predictive accuracy on small multi-omics datasets [48].
corsoFBA provides a significant methodological advance for exploring sub-optimal solution spaces in metabolic models. By decoupling growth optimization from protein cost minimization, it offers a more biologically realistic framework for predicting internal flux states in E. coli, especially under varying nutrient conditions. Experimental validations confirm its superior performance over standard FBA and minimization of metabolic steps in predicting pathway usage for central carbon metabolism.
The field continues to evolve with approaches like MINN demonstrating the power of integrating machine learning with mechanistic models. For researchers focusing on E. coli flux predictions, corsoFBA serves as a powerful tool for hypotheses generation regarding metabolic pathway usage under sub-optimal growth, while hybrid models represent the next frontier for leveraging multi-omics data to further refine flux predictions.
Metabolic Flux Analysis (MFA) and Flux Balance Analysis (FBA) are cornerstone techniques in systems biology for investigating the operation of biochemical networks in E. coli, a fundamental organism in both biological research and biotechnological applications [10] [3]. Both methods employ metabolic reaction network models operating at steady state, wherein reaction rates (fluxes) and metabolite levels are constrained to be constant [10]. They provide estimated (MFA) or predicted (FBA) values of intracellular fluxes, which are not directly measurable but represent an integrated functional phenotype emerging from multiple layers of biological organization [3]. Despite their widespread use, these approaches face significant challenges, particularly concerning the accuracy and scope of their predictions. Two interconnected limitations—flux range contraction and incomplete network coverage—systematically bias flux elucidation and can lead to erroneous biological conclusions. This guide objectively compares the performance of FBA and MFA for E. coli flux prediction, focusing on these inherent methodological challenges and presenting supporting experimental data.
Fundamental Techniques: FBA vs. MFA
Core Principles and Methodologies
Flux Balance Analysis (FBA) is a constraint-based modeling approach that uses linear optimization to predict flux distributions within a metabolic network [56]. It identifies a flux map that maximizes or minimizes a specified objective function (e.g., biomass growth rate) while satisfying mass-balance constraints and reaction capacity limitations [10] [56]. The typical workflow involves reconstructing a genome-scale metabolic network, mathematically representing reactions and constraints, defining an objective function, and using linear programming to calculate the optimal flux distribution [56].
13C-Metabolic Flux Analysis (13C-MFA) works inversely from experimental isotopic labeling data [10] [3]. Cells are fed 13C-labeled substrates, and the resulting label distribution in metabolites is measured via mass spectrometry or NMR [3]. A defined metabolic network model, including atom mappings that describe carbon transitions, is used to estimate intracellular fluxes by minimizing the residuals between the measured and simulated mass isotopomer distributions (MIDs) [10] [57].
The table below summarizes the core characteristics of each method.
Table 1: Fundamental Comparison Between FBA and 13C-MFA
| Feature | Flux Balance Analysis (FBA) | 13C-Metabolic Flux Analysis (13C-MFA) |
|---|---|---|
| Primary Input | Stoichiometric model, constraints, objective function | 13C-labeling experimental data, stoichiometric and atom mapping model |
| Fundamental Principle | Optimization-based prediction | Inverse estimation from experimental data |
| Key Assumption | Steady-state, cellular optimality | Metabolic and isotopic steady state |
| Typical Network Scale | Genome-scale models (GSSMs) | Core metabolic models (40-100 reactions) |
| Nature of Output | Predicted flux map | Estimated flux map |
| Key Limitation | Relies on assumed objective function | Limited network scope leads to flux range contraction |
Visualizing the Workflows and Their Key Challenge
The following diagram illustrates the fundamental workflows of FBA and 13C-MFA, highlighting the stage where limited network coverage in MFA leads to flux range contraction.
Diagram 1: Workflow comparison of FBA and 13C-MFA, highlighting the stage where MFA's limited network scope introduces flux range contraction.
The Core Challenge: Flux Range Contraction and Network Coverage
Defining the Problem
The primary challenge in traditional 13C-MFA is the use of simplified, core metabolic models that encompass only canonical central carbon pathways and lumped amino acid biosynthesis, typically comprising 40 to 100 reactions [57]. This practice introduces a systematic error known as flux range contraction, where the confidence intervals of estimated fluxes are artificially narrowed [57]. This occurs because the core model fails to account for alternative metabolic pathways and underground metabolism (reactions that occur at low rates) present in the full genome-scale metabolic network [57] [58]. Consequently, the flux solution space appears more certain and constrained than it truly is, creating a false sense of precision.
Quantitative Evidence from E. coli Studies
Experimental studies in E. coli have quantified the significant impact of network coverage on flux estimation. Research has demonstrated that projecting a flux distribution from a core model onto a genome-scale model results in a substantial contraction of flux confidence intervals.
Table 2: Quantitative Impact of Network Coverage on Flux Estimation in E. coli
| Study Focus | Model Type Used | Key Finding on Flux Ranges | Implication |
|---|---|---|---|
| General E. coli metabolism [57] | Core MFA vs. Genome-Scale MFA | 90% of flux ranges were contracted when core flux distribution was projected to genome scale. | Core MFA systematically overstates precision. |
| E. coli B and K-12 strains [59] | Genome-scale FBA model (1,369 reactions) | Model revealed strain-specific flux capacities enabling rational design of production hosts. | Comprehensive networks improve phenotypic prediction. |
| Anaerobic vs. Aerobic E. coli [51] | FBA vs. 13C-MFA | FBA-predicted internal fluxes from sampling feasible space differed substantially from MFA-derived fluxes. | Highlights inherent differences between prediction and measurement. |
Experimental Protocols for Method Comparison
Protocol for Genome-Scale 13C-MFA (GS-MFA)
GS-MFA is an advanced protocol designed to overcome the limitations of core MFA.
- Model Reconstruction: Start with a high-quality genome-scale metabolic model (GSM) for E. coli (e.g., iJO1366) [57] [58]. The model must be expanded to include an Atom Mapping Model (AMM), which details the carbon transition from reactants to products for every reaction [57].
- Tracer Experiment Design: Cultivate E. coli in a controlled bioreactor. Feed one or multiple 13C-labeled substrates (e.g., [1-13C]glucose). Harvest samples during metabolic steady state [57] [51].
- Mass Spectrometry Measurement: Quench metabolism rapidly. Extract intracellular metabolites. Measure the mass isotopomer distributions (MIDs) of proteinogenic amino acids or key intracellular metabolites using LC-MS or GC-MS [57].
- Flux Estimation: Solve an inverse problem by estimating metabolic fluxes to minimize the weighted sum of squares between the measured MIDs and those simulated by the genome-scale AMM. Use non-linear least-squares optimization [57].
- Statistical Validation: Perform a chi-squared (χ2) goodness-of-fit test to assess the model fit. Compute confidence intervals for the estimated fluxes via statistical methods like Monte Carlo sampling [10] [3].
Protocol for Integrated Multi-Omic Analysis
This protocol uses network fusion to integrate different data types for a more robust analysis.
- Data Collection: Under a defined set of environmental conditions, collect multi-omic data for E. coli: transcriptome (microarray/RNA-seq) and fluxome (from FBA or MFA) [58].
- Network Construction: Model the data as a multiplex network. Each node represents a growth condition. Create separate network layers (e.g., transcriptomic and fluxomic) where edges represent the similarity between conditions based on the respective omic data [58].
- Weighted Network Fusion: Fuse the multiple layers into a single integrated network using a weighted fusion algorithm (e.g., SNFtool). The weighting should reflect the relative reliability of each data type in predicting physiological behavior [58].
- Analysis and Prediction: Use the fused network topology to infer the position of new or untested conditions. Build condition-specific models and predict metabolic behaviors for conditions with incomplete experimental data [58].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful execution of the aforementioned protocols requires a suite of specialized reagents, software, and biological tools.
Table 3: Essential Research Reagents and Solutions for E. coli Flux Studies
| Item Name | Function/Application | Specific Example / Note |
|---|---|---|
| 13C-Labeled Substrates | Serve as metabolic tracers to track carbon fate. | [1-13C]Glucose; multiple tracers used in parallel increase flux precision [10] [57]. |
| E. coli K-12 Strains | Well-annotated model organism for foundational studies. | MG1655: wild-type; W3110: protein production; DH5α: cloning [59] [60]. |
| E. coli B Strains | Engineered for high-level protein production and metabolic engineering. | BL21(DE3): recombinant protein expression; REL606: experimental evolution studies [59] [60]. |
| Genome-Scale Models | Provide in silico representation of metabolism for FBA/GS-MFA. | iJO1366 reconstruction for E. coli K-12 MG1655 [51] [58]. |
| Metabolic Network Analysis Software | Perform FBA, FVA, and related analyses. | COBRA Toolbox, cobrapy [10]. |
| 13C-MFA Software Platforms | Estimate fluxes from labeling data. | Tools supporting FluxML format for model definition [57]. |
| LC-MS / GC-MS System | Quantify mass isotopomer distributions (MIDs) of metabolites. | Essential for generating experimental data for 13C-MFA [57]. |
Integrated FBA-MFA Approaches and Future Perspectives
Given their complementary strengths and weaknesses, a synergistic approach that integrates FBA and MFA is often most powerful. FBA can generate testable hypotheses about genome-scale metabolic capabilities, while MFA provides rigorous, data-driven validation and refinement of flux predictions in the core metabolism [51]. For instance, one study used MFA to define accurate linear constraints for FBA models, thereby improving the description of phenomena like overflow metabolism in dynamical models [46]. Another demonstrated that combining both methods provides deeper insights into metabolic adaptations, such as the substantial redirection of fluxes in E. coli between aerobic and anaerobic conditions [51].
The field is moving toward the routine use of Genome-Scale MFA (GS-MFA), which has been shown to provide a better fit to labeling data and more physiologically accurate flux distributions in E. coli and other microorganisms [57]. Key future developments will focus on automating the construction of atom mapping models, improving the computational efficiency of flux estimation algorithms for very large networks, and enhancing methods for the integrated analysis of multi-omic datasets [57] [58]. These advances will be critical for achieving a truly systems-level and reliable understanding of metabolic function in E. coli.
Integrating Omics Data and Thermodynamic Constraints for Refined Predictions
In the fields of systems biology and metabolic engineering, accurately predicting the set of biochemical reaction rates, or fluxes, within a living cell represents a significant challenge. This flux map is an integrated functional phenotype that emerges from multiple layers of biological organization and regulation, including the genome, transcriptome, and proteome [10] [3]. The primary computational frameworks for metabolic modeling are 13C-Metabolic Flux Analysis (13C-MFA) and Flux Balance Analysis (FBA). Both methods utilize metabolic reaction network models operating at steady state but differ fundamentally in their approach: 13C-MFA works backwards from experimental isotopic labeling data to estimate fluxes, whereas FBA uses linear optimization to predict fluxes based on network stoichiometry and an assumed biological objective [10] [3].
A major limitation of traditional FBA is its inability to accurately capture proteome-related limitations and thermodynamic constraints, often leading to biochemically unrealistic predictions [61]. This review objectively compares four advanced methodologies—REMI, INTEGRATE, ETFL, and MINN—that integrate multi-omics data and thermodynamic constraints to overcome these limitations, with a specific focus on their application in E. coli flux prediction research.
Comparative Analysis of Advanced Methodologies
The following table summarizes the core characteristics, data integration capabilities, and primary outputs of the four methods examined in this guide.
Table 1: Comparison of Advanced Flux Prediction Methods
| Method | Core Approach | Omics Data Integrated | Thermodynamic Constraints | Primary Output | Key Innovation |
|---|---|---|---|---|---|
| REMI [6] | Constraint-based optimization of thermodynamically curated models | Relative gene expression, relative metabolite abundance | Yes | Differential flux distributions | First method to co-integrate thermodynamics with relative expression and metabolomic data |
| INTEGRATE [62] | Pipeline using constraint-based models as a scaffold | Transcriptomics, metabolomics | Not Explicitly Stated | Reaction control (metabolic vs. transcriptional) | Discerns the level of regulation (metabolic/gene expression) controlling reactions |
| ETFL [61] | Hierarchical MILP formulation for metabolism and expression | mRNA and enzyme concentrations (absolute and relative) | Yes | Intracellular fluxes, enzyme and mRNA concentrations | Unified formulation for expression, thermodynamics, and growth-dependent parameters |
| MINN [48] | Hybrid neural network embedding a GEM | Multi-omics data | Not Explicitly Stated | Metabolic fluxes | Combines mechanistic modeling with machine learning pattern recognition |
To further elucidate the technical requirements of each method, the following table details the specific data inputs and computational frameworks employed.
Table 2: Technical Specifications and Implementation Requirements
| Method | Required Data Inputs | Model Formulation | Computational Solver | Validation Basis |
|---|---|---|---|---|
| REMI [6] | Relative gene expression, metabolite abundance, Gibbs free energy | Mixed-Integer Linear Programming (MILP) | Standard MILP solvers | Comparison with ¹³C-MFA data |
| INTEGRATE [62] | Transcriptomics, metabolomics, exo-metabolomics | Constraint-based stoichiometric model | Not Specified | Application to breast cell lines |
| ETFL [61] | Thermodynamic data, expression parameters | Mixed-Integer Linear Programming (MILP) | Common double-precision MILP solvers | Validation against characterized E. coli model |
| MINN [48] | Multi-omics data (e.g., transcriptomics, proteomics) | Neural Network with embedded GEM | Machine Learning frameworks | Prediction performance on E. coli knockout data |
Performance and Validation in E. coli Flux Prediction
Quantitative validation is crucial for establishing the predictive power of metabolic models. Among the methods discussed, REMI provides the most direct quantitative comparison against experimental fluxomic data. When applied to an E. coli GEM under wide-ranging conditions, REMI's predictions demonstrated a 32% higher Pearson correlation coefficient (r = 0.79) with fluxes determined by ¹³C-MFA compared to a similar method, GX-FBA [6]. This significant improvement highlights the benefit of integrating multiple layers of constrictive data.
ETFL has been validated for its ability to reproduce proteome-limited growth, a phenomenon where linear growth scaling with substrate uptake fails at high fluxes due to enzyme availability constraints that traditional FBA cannot capture [61]. Furthermore, the inclusion of thermodynamic constraints, as implemented in REMI, ETFL, and other frameworks, has been shown to improve predictive accuracy by enforcing biochemically realistic reaction directions and eliminating thermodynamically infeasible pathways [6] [61] [63].
Detailed Experimental Protocols
REMI (Relative Expression and Metabolomic Integrations) Workflow
REMI integrates relative gene-expression and metabolite abundance data into thermodynamically curated genome-scale models (GEMs). The protocol can be broken down into three main stages [6]:
Data Pre-processing and Model Curation:
- Convert a standard FBA model into a Thermodynamic-based Flux Analysis (TFA) model. This incorporates Gibbs free energy data of metabolites and reactions.
- Systematically convert experimental gene-expression and metabolite-level ratios between two conditions into reaction ratios.
REMI Optimization:
- The REMI framework can be applied in different modes depending on the available data: REMI-TGex (integrates thermodynamics and gene-expression), REMI-TM (integrates thermodynamics and metabolomics), or REMI-TGexM (integrates all three).
- The method uses optimization principles to maximize the consistency between the differential gene-expression levels, metabolite abundance data, and the estimated differential fluxes, all while respecting the thermodynamic constraints.
Solution Enumeration and Analysis:
- Unlike some FBA methods that return a single solution, REMI uses a mixed-integer linear programming (MILP) approach to enumerate several alternative optimal and sub-optimal flux profiles.
- A high-frequency analysis of common genes and their associated reactions across these solutions identifies the most consistently regulated components, providing more robust and biologically relevant results.
Figure 1: The REMI workflow for integrating multi-omics data with thermodynamic constraints.
ETFL (Expression and Thermodynamics Flux Models) Workflow
ETFL is an implementation of a Metabolism and Expression model (ME-model) that formulates a hierarchical MILP problem. The key steps in its formulation and application are [61]:
Formulation of the Expression Problem:
- Derive mass balances for macromolecules (mRNAs and enzymes) that link their synthesis flux, degradation flux, and concentration. The quasi-steady-state mass balance for a macromolecule is: ( v^{syn} - v^{deg} - \mu \cdot G = 0 ), where ( \mu ) is the growth rate and ( G ) is the concentration.
- Constrain metabolic reaction fluxes (( v )) by the available enzyme concentration (( E )) and its catalytic capacity: ( v \leq k_{cat} \cdot E ).
- Constrain peptide and mRNA synthesis fluxes by the catalytic capacities of ribosomes and RNA polymerase, respectively.
Integration of Thermodynamics and Growth-Dependence:
- Incorporate thermodynamic constraints to enforce reaction directionality and allow integration of metabolomics data via log-concentration variables.
- Account for growth-dependent parameters, such as the changing ratios of mRNA and protein in the cell at different growth rates, which is a simplification in classic FBA.
Model Solving and Analysis:
- The entire formulation is cast as a MILP, which is compatible with standard double-precision solvers, unlike earlier ME-models that required specialized non-linear solvers.
- The solved model simultaneously provides thermodynamics-compliant intracellular fluxes, as well as feasible concentrations of enzymes and mRNAs.
Figure 2: The ETFL workflow for unified modeling of metabolism and expression.
Successful implementation of the methods described requires a combination of computational tools, data resources, and model frameworks.
Table 3: Key Research Reagent Solutions for Advanced Flux Analysis
| Category | Item | Function in Research |
|---|---|---|
| Computational Tools | COBRA Toolbox / cobrapy [10] | Software suites providing standard functions and pipelines for constraint-based modeling, including FBA. |
| MEMOTE [10] | A test suite for ensuring the quality and consistency of genome-scale metabolic models. | |
| Standard MILP Solver (e.g., CPLEX, Gurobi) [61] | Software used to solve the optimization problems formulated by methods like ETFL and REMI. | |
| Data Types | ¹³C-MFA Fluxomic Data [6] [10] | Experimentally determined flux maps used as a gold standard for validating computational predictions. |
| Relative Gene Expression (RNA-seq) [62] [6] | Provides data on transcriptional regulation which can be integrated to refine flux predictions. | |
| Quantitative Metabolomics [62] [6] | Measurements of intracellular metabolite abundances used to inform metabolic control. | |
| Thermodynamic Data (e.g., Gibbs free energy) [6] [63] | Used to constrain reaction directions and eliminate thermodynamically infeasible fluxes. | |
| Model Frameworks | Genome-Scale Model (GEM) [6] [48] | A stoichiometric model of all known metabolic reactions in an organism, serving as the core scaffold. |
| Thermodynamic-based Flux Analysis (TFA) Model [6] [61] | A GEM augmented with thermodynamic constraints to ensure biochemically realistic solutions. |
The integration of multi-omics data and thermodynamic constraints represents a paradigm shift in metabolic modeling, moving from simplistic stoichiometric calculations toward biochemically realistic and context-specific simulations. For researchers focusing on E. coli, methods like REMI and ETFL offer validated paths to superior flux predictions by systematically incorporating gene expression, metabolomics, and thermodynamic principles. While REMI excels in providing highly accurate differential flux profiles validated against ¹³C-MFA data, ETFL offers a unique, unified framework that explicitly models the costs and constraints of the expression machinery.
The emerging trend of hybrid approaches, exemplified by MINN, suggests a future where deep mechanistic understanding from constraint-based models is powerfully augmented by the pattern recognition capabilities of machine learning. As validation and model selection practices continue to mature [10] [3], the confidence in these refined predictions will grow, accelerating their impact in both basic research and industrial drug and bio-product development.
Flux Balance Analysis (FBA) stands as a cornerstone in computational systems biology for predicting metabolic behaviors. This constraint-based approach uses genome-scale metabolic models (GEMs) to predict steady-state reaction fluxes by optimizing a cellular objective, typically biomass growth [64] [65]. A common application involves predicting gene essentiality—identifying metabolic genes whose knockout would impair cellular growth. However, traditional FBA faces a fundamental limitation: it assumes that both wild-type and knockout strains optimize the same fitness objective, an assumption often violated when deletion strains undergo metabolic reprogramming to meet alternative survival objectives [64] [66].
The integration of machine learning (ML) with mechanistic metabolic models represents a paradigm shift, addressing core FBA limitations while leveraging its network-based predictive capabilities. These hybrid approaches harness ML's pattern recognition strengths while preserving biochemical constraints embedded in GEMs, creating a new class of predictive biological tools with enhanced accuracy. This review examines these emerging hybrid methodologies, with particular focus on their application to gene essentiality prediction in E. coli, comparing their performance, methodological innovations, and practical implementation requirements.
Fundamental Limitations of Traditional FBA in Essentiality Prediction
Traditional FBA predicts gene essentiality by simulating gene knockout and evaluating the impact on biomass production. Despite its widespread use, this approach suffers from several theoretical and practical shortcomings:
- Optimality Assumption Failure: FBA assumes knockout strains optimize the same objective (typically growth) as wild-type strains. However, experimental evidence shows that deletion mutants often undergo substantial physiological adaptations and may optimize different objectives for survival [64] [25].
- Network Context Neglect: Standard FBA does not fully account for the network context of reactions, particularly how disruption of one enzyme affects interconnected metabolic processes through metabolite mass flow and pathway redundancy [64].
- Quantitative Prediction Challenges: FBA often provides quantitatively inaccurate flux predictions unless labor-intensive measurements of media uptake fluxes are performed [65].
- Solution Space Ambiguity: FBA frequently identifies multiple alternative optimal solutions (flux distributions) with identical objective values but different underlying flux patterns, creating uncertainty in specific predictions [6].
These limitations have motivated the development of hybrid approaches that complement FBA's mechanistic foundation with machine learning's adaptability.
Hybrid FBA-ML Approaches: Methodological Frameworks
FlowGAT: Graph Neural Networks for Essentiality Prediction
FlowGAT represents a sophisticated hybrid methodology that combines FBA with graph attention networks (GATs) to predict gene essentiality directly from wild-type metabolic phenotypes [64]. The approach leverages the inherent network structure of metabolism through several innovative steps:
Methodological Workflow:
- Wild-Type FBA Solution: Generate a reference flux distribution for the wild-type strain using standard FBA.
- Mass Flow Graph Construction: Project the FBA solution onto a directed graph structure where:
- Nodes represent enzymatic reactions
- Edges quantify the propagation of metabolite mass flow between a reaction and its neighbors
- Edge weights are proportional to the mass transfer between connected reactions
- Graph Neural Network Processing: Train a graph neural network on the mass flow graph representation, using known knock-out fitness assay data for supervision.
- Essentiality Classification: Use the trained network to predict whether specific gene knockouts would be lethal based on the wild-type flux distribution and network topology [64] [66].
This approach circumvents the need to assume optimality for deletion strains by exploiting the informational content embedded in the wild-type flux distribution and its network representation.
Figure 1: FlowGAT combines FBA-predicted wild-type fluxes with graph neural networks. The metabolic network structure and flux distribution are transformed into a mass flow graph, which is processed by a graph attention network (GAT) trained on experimental knockout fitness data.
Alternative Hybrid Architectures
Beyond FlowGAT, several complementary hybrid architectures have emerged:
Artificial Metabolic Networks (AMNs): This framework embeds FBA within artificial neural networks, creating trainable hybrid models. AMNs use a neural preprocessing layer to predict uptake flux bounds from extracellular concentrations, effectively capturing transporter kinetics and resource allocation effects. The mechanistic layer then computes steady-state fluxes respecting stoichiometric constraints [65].
RELATCH (RELATive CHange): While not strictly an ML approach, RELATCH introduces a relative optimality concept that minimizes relative flux changes from a reference state when predicting perturbation responses. This method demonstrates how reference flux patterns can guide predictions without assuming identical optimization objectives across strains [25].
REMI (Relative Expression and Metabolomic Integrations): This method integrates multi-omics data (gene expression, metabolite abundance) with thermodynamic constraints into FBA models, improving flux prediction accuracy by incorporating additional biological layers [6].
Comparative Performance Analysis
Quantitative Performance Metrics
Table 1: Comparative Performance of FBA and Hybrid Methods for Gene Essentiality Prediction
| Method | Core Approach | Key Assumptions | Prediction Accuracy | Experimental Data Requirements | Computational Complexity |
|---|---|---|---|---|---|
| Traditional FBA | Linear optimization of biomass production | Knockout strains optimize same objective as wild-type | Moderate (varies with model and conditions) [4] | Minimal (only substrate uptake rates) | Low |
| FlowGAT | Graph neural networks + FBA flux graphs | Essentiality predictable from wild-type flux network topology | Near state-of-the-art [64] [66] | Knockout fitness data for training | Moderate to high (GNN training) |
| AMN Hybrid | Neural network preprocessing + FBA constraints | Relationship between medium composition and uptake fluxes is learnable | Systematically outperforms FBA [65] | Flux distributions for training | Moderate (neural network training) |
| RELATCH | Relative flux change minimization | Relative metabolic flux pattern maintained post-perturbation | Up to 100-fold decrease in SSE vs. FBA [25] | Reference state flux distribution | Moderate (quadratic programming) |
Method-Specific Advantages and Limitations
FlowGAT demonstrates particular strength in leveraging the inherent network structure of metabolism without requiring explicit assumptions about knockout strain optimality. By training directly on experimental fitness data, it captures complex, non-intuitive relationships between reaction connectivity and gene essentiality [64].
AMN Hybrid Models excel in quantitative phenotype prediction, particularly in translating extracellular conditions to intracellular flux states. These models require training set sizes orders of magnitude smaller than classical machine learning methods, effectively addressing the curse of dimensionality in whole-cell modeling [65].
Traditional FBA maintains advantages in interpretability and computational efficiency, requiring no training data and providing directly interpretable flux predictions based on biochemical constraints.
Experimental Protocols and Validation Frameworks
FlowGAT Implementation Protocol
Data Requirements and Preprocessing:
- Genome-Scale Metabolic Model: A well-curated GEM such as iML1515 for E. coli [65]
- Wild-Type Flux Distributions: FBA solutions across multiple growth conditions
- Training Data: Experimental gene essentiality data from knock-out fitness assays
- Graph Construction: Convert FBA solutions to mass flow graphs with reactions as nodes and metabolite mass transfer as edges
Model Training and Validation:
- Network Architecture: Implement graph attention networks with appropriate depth and attention heads
- Cross-Validation: Use k-fold cross-validation to assess prediction robustness
- Benchmarking: Compare against FBA predictions using statistical measures (precision, recall, F1-score)
- Condition-Specific Testing: Evaluate performance across diverse growth environments
Validation with 13C-Metabolic Flux Analysis (13C-MFA)
13C-MFA provides experimental validation for computational flux predictions by measuring intracellular fluxes through isotopic tracer experiments [4] [57]. This technique serves as a gold standard for validating FBA and hybrid method predictions:
- Tracer Experiments: Cells are fed 13C-labeled substrates (e.g., [1,2-13C]glucose)
- Mass Isotopomer Measurement: Mass spectrometry quantifies labeling patterns in intracellular metabolites
- Flux Estimation: Computational algorithms determine fluxes that best fit measured labeling data
- Validation Metrics: Compare predicted vs. measured fluxes using statistical measures (Pearson correlation, sum of squared errors)
Studies consistently show that hybrid approaches like REMI demonstrate approximately 32% higher Pearson correlation coefficients with 13C-MFA data compared to traditional FBA [6].
Table 2: Key Research Reagents and Computational Tools for Hybrid FBA-ML Research
| Resource Category | Specific Tools/Reagents | Function/Purpose | Implementation Considerations |
|---|---|---|---|
| Genome-Scale Models | E. coli iML1515, iAF1260 | Provides biochemical network structure for FBA | Quality of curation significantly impacts prediction accuracy [25] [65] |
| Machine Learning Frameworks | PyTorch, TensorFlow with Graph Nets | Implementation of graph neural networks | GPU acceleration recommended for large networks |
| Constraint-Based Modeling | Cobrapy, COBRA Toolbox | FBA simulation and model manipulation | Enables integration with ML pipelines [65] |
| Experimental Validation | 13C-labeled substrates, Mass spectrometry | 13C-MFA flux validation | Costly but essential for ground-truth flux data [4] [57] |
| Omics Data Integration | RNA-seq, Metabolomics platforms | Data for REMI and similar multi-omics methods | Normalization and quality control critical [6] |
| Reference Datasets | Keio collection (E. coli knockouts) | Training and validation data for essentiality prediction | Standardized resources enable benchmarking [4] |
Hybrid FBA-ML approaches represent a significant advancement over traditional constraint-based modeling for gene essentiality prediction. By combining mechanistic biochemical constraints with data-driven pattern recognition, these methods address fundamental limitations while preserving biological interpretability.
The evidence indicates that FlowGAT and related hybrid methods consistently outperform traditional FBA in prediction accuracy, particularly in challenging cases where knockout strains undergo significant physiological adaptation. These approaches demonstrate that wild-type FBA solutions contain sufficient information to predict essentiality without assuming optimality of deletion strains [64] [66].
Future development directions include:
- Multi-omics Integration: Incorporating transcriptomic, proteomic, and metabolomic data layers
- Cross-Organism Generalization: Extending hybrid approaches to non-model organisms
- Dynamic Flux Prediction: Incorporating temporal dynamics into hybrid models
- Automated Pipeline Development: Creating user-friendly tools for broader research community adoption
As hybrid methodologies mature, they promise to enhance both fundamental biological understanding and biotechnological applications, including drug target identification and metabolic engineering strategies.
Benchmarking Performance: Validating and Comparing FBA and MFA Results
Quantifying intracellular metabolic fluxes is fundamental for advancing metabolic engineering and understanding cellular physiology. Two dominant computational frameworks for this task are Flux Balance Analysis (FBA) and 13C Metabolic Flux Analysis (13C-MFA), each with distinct philosophical and methodological approaches [10]. FBA is a constraint-based modeling approach that predicts reaction rates (fluxes) in genome-scale metabolic models (GEMs) by assuming the cell optimizes a biological objective, such as maximizing growth rate, under steady-state and nutritional constraints [2] [67]. In contrast, 13C-MFA is a primarily experimental approach that estimates fluxes in core metabolic models by fitting simulations to data from 13C isotope labeling experiments, thereby identifying the flux map that best explains the experimental isotopic distribution [10] [67]. For researchers, scientists, and drug development professionals, selecting the appropriate method hinges on the specific research question, available data, and required level of quantitative accuracy. This guide provides a structured comparison of their performance, grounded in quantitative metrics and experimental validation protocols.
The evaluation of these models extends beyond simple point estimates of fluxes. A critical challenge in the field is robust uncertainty quantification and model selection [10]. Traditional FBA provides a single flux solution, while 13C-MFA typically reports confidence intervals derived from frequentist statistics. However, Bayesian methods are emerging as powerful alternatives for both FBA and MFA, capable of characterizing the full probability distribution of possible fluxes compatible with experimental data, thus providing a more nuanced view of flux uncertainty [68] [67]. This article will objectively compare the performance of FBA and MFA, with a focus on E. coli as a model organism, leveraging precision-recall concepts and statistical tests to guide method selection.
Core Methodologies and Experimental Protocols
Understanding the fundamental workflows of FBA and 13C-MFA is essential for interpreting their performance metrics. The following diagram illustrates the logical relationships and key outputs of these primary flux analysis methods.
Figure 1: A workflow diagram comparing the core methodologies of FBA and 13C-MFA, highlighting their inputs, computational processes, and outputs, including the emerging role of Bayesian methods.
Flux Balance Analysis (FBA) Protocol
The standard FBA protocol involves the following steps [2]:
- Model Construction: Begin with a Genome-Scale Metabolic Model (GEM). A common, well-curated model for E. coli is iML1515, which encompasses 1,515 genes, 2,719 reactions, and 1,192 metabolites [2].
- Application of Constraints: Impose constraints based on the experimental condition.
- Steady-State Constraint: The system of equations is defined as
Sv = 0, whereSis the stoichiometric matrix andvis the flux vector, ensuring metabolite concentrations remain constant [68] [2]. - Physiological Bounds: Set lower and upper bounds (
lbandub) for each reaction flux, particularly for substrate uptake and product secretion rates, based on measured data [2].
- Steady-State Constraint: The system of equations is defined as
- Define Objective Function: Identify a reaction to optimize, most commonly the biomass reaction, to simulate maximal growth [2].
- Solve Linear Program: Use a linear programming solver (e.g., via the COBRA Toolbox or cobrapy) to find the flux distribution
vthat maximizes the objective function while satisfying all constraints [2].
Advanced FBA with Enzyme Constraints (ECMpy): To improve realism, the ECMpy workflow adds enzyme capacity constraints. This involves splitting reversible reactions, incorporating enzyme molecular weights, kcat values (from databases like BRENDA), and protein abundance data (e.g., from PAXdb). A total enzyme capacity constraint is added, ensuring the cumulative flux-weighted enzyme demand does not exceed the cell's proteomic budget [2].
13C Metabolic Flux Analysis (13C-MFA) Protocol
The standard 13C-MFA protocol involves these key stages [10] [67]:
- Tracer Experiment: Grow cells (e.g., E. coli) on a defined medium containing a 13C-labeled carbon source (e.g., [1-13C]glucose or [U-13C]glucose).
- Data Collection: At metabolic steady-state, harvest cells and measure:
- Exchange Fluxes: Extracellular substrate uptake and product secretion rates.
- Isotopic Labeling: Measure the Mass Isotopomer Distribution (MID) of intracellular metabolites using Mass Spectrometry (MS) or NMR.
- Model Definition: Construct a stoichiometric model of central carbon metabolism, including atom transition mappings for each reaction.
- Flux Estimation: Use a non-linear optimization algorithm to find the set of fluxes that minimizes the difference between the simulated MID and the experimentally measured MID. This is often a weighted least-squares problem.
- Statistical Evaluation and Uncertainty Analysis:
- Goodness-of-Fit: Evaluate the model fit using a χ2-test, where the sum of squared residuals is compared to a χ2-distribution [10].
- Confidence Intervals: Determine confidence intervals for the estimated fluxes, typically via parameter continuation or Monte Carlo sampling, to quantify flux uncertainty [10] [67].
Quantitative Performance Comparison
The performance of FBA and MFA can be evaluated based on their scope, data requirements, inherent strengths, and quantitative accuracy. The table below summarizes these aspects for direct comparison.
Table 1: A systematic comparison of FBA and 13C-MFA characteristics and performance.
| Evaluation Criterion | Flux Balance Analysis (FBA) | 13C Metabolic Flux Analysis (13C-MFA) |
|---|---|---|
| Model Scope | Genome-Scale (e.g., >2,000 reactions) [2] | Core Metabolism (typically 50-150 reactions) [67] |
| Primary Data Used | Stoichiometry, Exchange fluxes, Growth objective [2] | Exchange fluxes, 13C Labeling data (MID) [10] |
| Key Assumption | Steady-state & Optimal growth (or other objective) [2] | Metabolic and Isotopic Steady-state [10] |
| Computational Approach | Linear Programming [2] | Non-linear Optimization [67] |
| Primary Output | Point estimate of fluxes [68] | Point estimate with confidence intervals [67] |
| Uncertainty Quantification | Requires separate methods (e.g., FVA, sampling) [68] | Integral part of the method (e.g., χ2-test, confidence intervals) [10] |
| Quantitative Accuracy | Good for growth/precursor yields under optimal conditions; may lack precision for internal fluxes without constraints [10] [48] | High precision for central carbon fluxes, considered the "gold standard" for empirical measurement [67] |
| Throughput | High (computational) [10] | Low (requires wet-lab experiment) [10] |
The Role of Bayesian Methods and Advanced Metrics
Emerging Bayesian methods like BAMFA (Bayesian Metabolic Flux Analysis) and BayFlux offer a paradigm shift in flux uncertainty quantification for both FBA and MFA [68] [67]. These approaches model the full posterior probability distribution of fluxes, p(v|y), where v is the flux vector and y represents the observed data (e.g., exchange fluxes, 13C labeling data, or objective assumptions) [68]. This provides several advantages:
- Robust Uncertainty Characterization: They reveal the complete space of flux solutions compatible with the data, which is particularly valuable when multiple, distinct flux regions fit the data equally well (non-gaussian posteriors) [67].
- Model Flexibility: They allow probabilistic relaxation of strict steady-state and optimal growth assumptions by placing prior distributions on metabolite changes and fluxes [68].
- Informed Decision-Making: The full posterior distribution enables more robust predictions for genetic interventions. For instance, P-13C MOMA and P-13C ROOM are Bayesian versions of classic knockout prediction methods that quantify the uncertainty in the predicted flux change [67].
A key finding from Bayesian 13C-MFA is that genome-scale models can produce narrower flux distributions (i.e., reduced uncertainty) compared to smaller core models, as the larger network imposes additional constraints through interconnected reactions [67].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful execution of flux analysis requires a combination of computational tools and experimental reagents. The following table details key solutions used in the featured experiments.
Table 2: Key research reagents, software, and databases essential for conducting FBA and 13C-MFA studies.
| Item Name | Type | Function / Application | Example Sources / Notes |
|---|---|---|---|
| iML1515 Model | Computational Model | A genome-scale metabolic model of E. coli K-12 MG1655; serves as a base for FBA simulations and engineering. [2] | Contains 2,719 reactions and 1,192 metabolites; well-curated. [2] |
| 13C-Labeled Substrate | Chemical Reagent | Provides the isotopic tracer for 13C-MFA experiments to infer intracellular fluxes. | e.g., [1-13C]glucose, [U-13C]glucose; purity is critical. |
| COBRA Toolbox / cobrapy | Software Package | Provides a standardized framework for constraint-based modeling, including FBA, FVA, and sampling. [68] [2] | Open-source tools for MATLAB and Python, respectively. |
| BRENDA Database | Database | A primary resource for enzyme kinetic data, specifically kcat values, used in enzyme-constrained FBA. [2] |
Used in the ECMpy workflow to set catalytic constraints. [2] |
| Mass Spectrometer | Analytical Instrument | Measures the mass isotopomer distribution (MID) of metabolites, the primary data type for 13C-MFA. | GC-MS or LC-MS instruments are standard. |
| BAMFA / BayFlux | Software Package | Implements Bayesian inference for flux analysis, providing full flux distributions for FBA and MFA. [68] [67] | BAMFA is COBRA-compatible; BayFlux uses MCMC sampling. [68] [67] |
The choice between FBA and 13C-MFA is not a matter of identifying a single superior tool, but of selecting the right tool for the specific research objective. FBA excels in high-throughput, predictive modeling at genome-scale, guiding strain design for metabolic engineering under the assumption of fitness optimization. In contrast, 13C-MFA provides a high-precision, empirical measurement of fluxes in core metabolism, serving as a gold standard for validating model predictions and understanding physiological states where optimality may not hold [10] [67].
The future of flux analysis lies in the integration of these approaches and the adoption of more sophisticated statistical frameworks. Bayesian methods, such as BAMFA and BayFlux, are bridging the gap by offering a unified probabilistic framework that naturally incorporates diverse data types and provides comprehensive uncertainty quantification [68] [67]. Furthermore, hybrid approaches that embed GEMs into machine learning models, such as Metabolic-Informed Neural Networks (MINNs), show promise for integrating multi-omics data to improve flux predictions [48]. As the field moves forward, robust validation and model selection practices, coupled with these advanced computational techniques, will be crucial for enhancing confidence in flux predictions and accelerating their application in biotechnology and drug development.
Systematic Comparison of FBA Predictions vs. Experimental 13C-MFA Flux Maps
The accurate prediction and measurement of intracellular metabolic fluxes are fundamental to advancing systems biology and metabolic engineering. Two of the most prominent methodologies in this domain are Flux Balance Analysis (FBA), a constraint-based modeling approach that predicts fluxes, and 13C-Metabolic Flux Analysis (13C-MFA), an experimental approach that estimates fluxes using isotopic tracers [3] [23]. While FBA leverages genome-scale models and optimization principles to predict metabolic capabilities, 13C-MFA provides a quantitative snapshot of the actual metabolic state by integrating stable isotope labeling data [12] [57]. Framing this comparison within the context of E. coli flux prediction research is particularly instructive, as E. coli serves as a model organism with well-annotated metabolic networks and extensive experimental data available for both aerobic and anaerobic growth conditions [12]. This guide provides an objective, data-driven comparison of these two powerful techniques, highlighting their respective strengths, limitations, and synergies for the research community.
Methodology Comparison: FBA vs. 13C-MFA
Fundamental Principles and Technical Approaches
Flux Balance Analysis (FBA) is a constraint-based computational method that predicts steady-state metabolic flux distributions. It operates on a genome-scale metabolic model (GEM), which contains the stoichiometry of all known metabolic reactions in an organism [3] [1]. The core of FBA involves solving a linear optimization problem to find a flux distribution that maximizes or minimizes a specified cellular objective, most commonly biomass production [3] [1]. The solution is constrained by mass balance, reaction directionality, and substrate uptake rates [23]. FBA does not require extensive experimental data beyond basic constraints and can analyze genome-scale networks, but it relies heavily on the assumed optimality principle [57].
13C-Metabolic Flux Analysis (13C-MFA) is an experimentally-driven methodology that quantifies intracellular fluxes by leveraging stable isotope tracing. Cells are fed with 13C-labeled substrates (e.g., [1,2-13C]glucose), and the resulting labeling patterns in intracellular metabolites are measured using mass spectrometry (MS) or NMR [3] [23]. Metabolic fluxes are estimated by fitting a metabolic network model to the experimental labeling data, minimizing the difference between measured and simulated labeling distributions [23] [57]. This approach provides high-resolution flux maps for core carbon metabolism but has traditionally been limited to smaller network models [57].
The table below summarizes the core methodological differences:
Table 1: Fundamental Methodological Differences Between FBA and 13C-MFA
| Feature | Flux Balance Analysis (FBA) | 13C-Metabolic Flux Analysis (13C-MFA) |
|---|---|---|
| Primary Basis | Computational prediction based on constraints and optimization | Experimental measurement based on isotope labeling |
| Network Scale | Genome-scale models (hundreds to thousands of reactions) | Core metabolic models (typically 40-100 reactions), with growing use of genome-scale [57] |
| Key Inputs | Stoichiometric model, constraints (e.g., uptake rates), objective function | Stoichiometric model, extracellular fluxes, 13C-labeling data |
| Key Assumptions | Metabolic steady-state, optimal cellular behavior (e.g., growth rate maximization) | Metabolic and isotopic steady-state |
| Measured Output | Predicted flux distribution | Estimated flux distribution with confidence intervals |
Experimental and Computational Workflows
The following diagram illustrates the typical workflows for FBA and 13C-MFA, highlighting their complementary nature:
Quantitative Comparison of Flux Predictions inE. coli
Aerobic vs. Anaerobic Growth Conditions
A direct comparative study of wild-type E. coli (K-12 MG1655) grown aerobically and anaerobically on glucose provides robust quantitative data for evaluating FBA performance against 13C-MFA derived fluxes [12]. The study utilized a genome-scale model (iJR904) for FBA and a consistent network model for 13C-MFA, allowing for a fair comparison.
Table 2: Comparison of Key Metabolic Fluxes in E. coli under Different Growth Conditions (flux values in mmol/gDCW/h)
| Metabolic Flux | Aerobic Condition | Aerobic Condition | Anaerobic Condition | Anaerobic Condition |
|---|---|---|---|---|
| 13C-MFA | FBA Prediction | 13C-MFA | FBA Prediction | |
| Glucose Uptake | 5.8 | 5.8 (constrained) | 9.9 | 9.9 (constrained) |
| Growth Rate (h⁻¹) | 0.43 | 0.43 (constrained) | 0.38 | 0.38 (constrained) |
| TCA Cycle Flux (CS) | 16.1% of glucose uptake | Variable (multiple optima) | N/A (incomplete) | N/A (incomplete) |
| Pentose Phosphate Pathway | ~20% of glucose uptake | Underestimated | Increased relative to aerobic | Variable predictions |
| Acetate Secretion | Minimal | Accurately predicted when constraints applied | Significant (~31% higher than aerobic) | Accurately predicted when constraints applied |
| ATP Maintenance (fraction) | 37.2% | Implicit in biomass objective | 51.1% | Implicit in biomass objective |
Analysis of Agreement and Discrepancies
The comparative data reveals several critical points:
- FBA excels at predicting exchange fluxes: When FBA is constrained with measured uptake rates and growth rates, it can accurately predict the secretion rates of by-products like acetate under both aerobic and anaerobic conditions [12].
- Internal flux predictions show significant deviations: The most frequently predicted values of internal fluxes from sampling the FBA solution space often differ substantially from 13C-MFA derived fluxes [12]. A key finding was that the TCA cycle operates in a non-cyclic mode under aerobic conditions in wild-type E. coli, a phenomenon correctly identified by 13C-MFA but not predicted by standard FBA assuming optimality [12].
- Energy metabolism differences: 13C-MFA revealed that the fraction of ATP spent on maintenance is significantly higher under anaerobic conditions (51.1%) compared to aerobic conditions (37.2%). FBA, which typically incorporates maintenance into the biomass objective function, does not directly resolve these metabolic inefficiencies [12].
Technical Protocols for Comparative Studies
Key Experimental Protocol for 13C-MFA inE. coli
For the generation of reliable experimental flux maps against which FBA predictions can be validated, the following protocol, derived from the cited literature, provides a robust framework [12]:
- Strain and Culture Conditions: Use E. coli K-12 MG1655 (or other relevant strain) cultured in defined minimal medium (e.g., M9) with labeled glucose as the sole carbon source.
- Tracer Experiment: Feed cells with a specifically 13C-labeled substrate (e.g., [1,2-13C]glucose) during mid-log-phase growth. Conduct parallel experiments for aerobic (shaken flasks or bioreactor with aeration) and anaerobic (sealed cultures with inert atmosphere) conditions.
- Metabolite Measurement: Quantify substrate uptake and product secretion rates (e.g., acetate, lactate, succinate, formate, ethanol) using techniques like HPLC, enzymatic assays, and gas analysis for CO₂.
- Isotope Labeling Measurement: Harvest cells and measure the 13C-labeling patterns of proteinogenic amino acids or intracellular metabolites. This is typically done using GC-MS (Gas Chromatography-Mass Spectrometry) and/or NMR (Nuclear Magnetic Resonance) spectroscopy.
- Flux Estimation: Use a computational software tool (e.g., INCA, Cosmos) to fit the metabolic network model to the measured extracellular fluxes and isotopic labeling data. The optimization involves minimizing the residual sum of squares (RSS) between the measured and simulated labeling patterns.
- Statistical Validation: Perform a χ²-test of goodness-of-fit to validate the model and estimate confidence intervals for the calculated fluxes to assess their precision [3].
Computational Protocol for FBA
To perform a comparable FBA study:
- Model Selection: Obtain a curated genome-scale metabolic model for E. coli (e.g., iJR904, iML1515).
- Application of Constraints: Constrain the model with experimentally measured uptake and secretion rates (e.g., glucose, oxygen, acetate) and the measured growth rate from the parallel 13C-MFA experiment.
- Objective Function: Set the objective function, most commonly to maximize biomass production.
- Flux Prediction: Solve the linear optimization problem using a computational toolbox like the COBRA Toolbox or cobrapy to obtain a flux distribution [3].
- Solution Space Analysis: Since the optimal solution may not be unique, use Flux Variability Analysis (FVA) or Flux Sampling to characterize the range of possible fluxes that satisfy the constraints and achieve near-optimal growth [3] [69] [1].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 3: Key Research Reagents and Computational Tools for FBA and 13C-MFA
| Reagent / Tool | Function / Application | Examples / Specifications |
|---|---|---|
| 13C-Labeled Substrates | Tracers for 13C-MFA to track carbon fate | [1,2-13C]glucose, [U-13C]glucose; >99% isotope purity |
| Mass Spectrometry (MS) | Measure mass isotopomer distributions (MIDs) for 13C-MFA | GC-MS, LC-MS; high mass resolution and accuracy |
| NMR Spectroscopy | Alternative/complement to MS for measuring positional isotopomers | 1H, 13C NMR |
| Genome-Scale Model (GEM) | Stoichiometric matrix for FBA | E. coli models: iJR904, iML1515; from databases like BiGG |
| COBRA Toolbox | MATLAB-based suite for constraint-based modeling | Includes functions for FBA, FVA, and sampling [3] |
| cobrapy | Python-based package for constraint-based modeling | Alternative to COBRA Toolbox for FBA [3] |
| INCA | Software platform for 13C-MFA | Supports both steady-state and instationary MFA [30] |
| MEMOTE | Test suite for quality control of metabolic models | Checks stoichiometric consistency and format standards [3] |
Integrated Analysis and Synergistic Applications
The relationship between FBA and 13C-MFA is not merely comparative but inherently synergistic. The integration of these methods can significantly enhance the reliability of metabolic models and their predictions.
- Validation of FBA Predictions: 13C-MFA provides the most robust experimental validation for internal flux predictions generated by FBA. Discrepancies can reveal gaps in model annotation, incorrect optimality assumptions, or the presence of unaccounted-for regulatory mechanisms [3] [12].
- Refinement of FBA Models: 13C-MFA data can be used to refine FBA models by identifying and correcting incorrect network gaps, testing alternative objective functions, and providing realistic constraints on internal fluxes, thereby improving the model's predictive power [3] [12].
- Guiding Tracer Experiments: FBA can be used in the design of 13C-MFA experiments. For instance, FBA can predict which carbon tracer will be most informative for resolving specific fluxes of interest, optimizing the experimental design for maximum flux resolution [23].
- Exploration of Sub-Optimal States: Techniques like corsoFBA demonstrate how FBA can be extended to explore sub-optimal solution spaces by incorporating protein cost minimization, leading to flux predictions that more closely align with 13C-MFA data, especially under conditions where growth is not maximal [1].
The following diagram summarizes this synergistic cycle of model improvement:
The accurate prediction of metabolic fluxes is a central challenge in systems biology and metabolic engineering. Two primary computational approaches, Flux Balance Analysis (FBA) and 13C-Metabolic Flux Analysis (13C-MFA), offer distinct methodologies for estimating intracellular reaction rates. FBA uses optimization of an objective function (e.g., biomass maximization) within stoichiometrically constrained genome-scale models to predict fluxes [23]. In contrast, 13C-MFA leverages data from stable-isotope labeling experiments to estimate fluxes, often with high precision, but typically for a smaller core metabolic network [10] [23]. A critical step in advancing these methodologies is rigorous validation and model selection, for which systematic genetic perturbation data is indispensable [10] [70].
The E. coli Keio Knockout Collection provides a foundational resource for this validation workflow. This collection comprises 3,985 single-gene, in-frame deletions of non-essential genes in E. coli K-12 strain BW25113, with two independent mutants saved for each gene [71] [72]. By providing a comprehensive set of defined genetic perturbations, the Keio collection enables researchers to benchmark the predictive power of FBA and the estimation accuracy of 13C-MFA against a consistent experimental background, thereby facilitating model corroboration and refinement [28].
The Keio Collection: A Primer for Validation Studies
Collection Specifications and Design
The Keio collection was constructed using a precise, high-throughput method to ensure consistency and reliability, which is paramount for validation studies. Table 1 summarizes its key characteristics.
Table 1: Specifications of the E. coli Keio Knockout Collection
| Feature | Specification | Significance for Validation |
|---|---|---|
| Strain Background | E. coli K-12 BW25113 | Provides a uniform genetic context, eliminating background effects. |
| Number of Mutants | 7,970 strains (two independent mutants for each of 3,985 genes) | Enables biological replication and controls for secondary mutations. |
| Genetic Modification | In-frame deletion, marker flanked by FRT sites | Minimizes polar effects on downstream genes; allows for cassette excision. |
| Antibiotic Resistance | Kanamycin | Provides selectable marker for strain maintenance. |
The design of the collection is particularly suited for functional genomics and reverse genetics. Each deleted gene is replaced with a kanamycin resistance cassette that can be excised by FLP recombination, leaving a precise, in-frame deletion [71] [72]. This meticulous design ensures that the observed phenotypic changes in the mutants are directly attributable to the deletion of the target gene.
Accessing the Keio Collection for Research
The collection is accessible to the scientific community through several distributors. Individual clones are typically provided as live cultures in LB medium with glycerol and kanamycin, shipped at room temperature, and should be stored at -80°C upon receipt [71]. Bulk orders or the entire collection are shipped in 96-well microtiter plates on dry ice [71]. The E. coli Genetic Resource Center (ECGRC) also provides a straightforward platform for ordering individual Keio strains [73].
Computational Prediction of Knockout Fluxes: FBA and Its Variants
Flux Balance Analysis and its related algorithms provide a suite of computational tools to predict how gene knockouts alter metabolic fluxes. These predictions can be directly tested against experimental data from the Keio collection.
Core Methodologies and Algorithms
The primary algorithms used for predicting knockout phenotypes include:
- Standard FBA: Often assumes the cell optimizes for biomass production. While successful for wild-type predictions under competitive growth, its evolution-based objective function may be less accurate for unevolved knockout strains immediately after perturbation [28].
- Minimization of Metabolic Adjustment (MOMA): This algorithm posits that the flux distribution of a knockout mutant will be as close as possible (in Euclidean distance) to the wild-type FBA solution. It favors solutions with many small flux changes rather than a few large ones [28].
- Regulatory On/Off Minimization (ROOM): An alternative to MOMA, ROOM minimizes the number of significant flux changes from the wild-type FBA solution, which may better reflect regulatory adaptation costs [28].
Advanced Frameworks: Integrating Uncertainty
Recent advances have introduced more sophisticated statistical frameworks for these predictions. The BayFlux method, for example, uses Bayesian inference and Markov Chain Monte Carlo (MCMC) sampling to quantify the full distribution of fluxes compatible with experimental data, rather than providing a single point estimate [67]. This approach enables the development of methods like P-13C MOMA and P-13C ROOM, which improve upon traditional MOMA and ROOM by explicitly quantifying prediction uncertainty, a crucial aspect for rigorous model validation [67].
Experimental Flux Measurement: 13C-MFA as a Validation Benchmark
13C-Metabolic Flux Analysis is considered the "gold standard" for experimentally measuring intracellular metabolic fluxes [67] [23]. It provides the benchmark data against which computational predictions like FBA are validated.
The 13C-MFA Workflow
The following diagram illustrates the standard experimental workflow for using 13C-MFA to validate model predictions with Keio knockout strains.
Experimental Protocol for Knockout Flux Validation
A detailed methodology for generating validation data using Keio knockouts and 13C-MFA is as follows:
- Strain Selection & Cultivation: Select the target knockout strain(s) from the Keio collection and the parental wild-type BW25113 as a control. Cultivate the strains in a defined minimal medium with a single carbon source (e.g., glucose). For the experimental culture, replace the natural-abundance carbon source with an equivalent 13C-labeled substrate (e.g., [1-13C]glucose or [U-13C]glucose) [23].
- Steady-State Achievement: Grow the cells in a controlled bioreactor (e.g., a chemostat) to ensure a metabolic and isotopic steady state is reached. This is critical for accurate flux determination [10] [23].
- Metabolite Harvesting and Extraction: Rapidly harvest cells and quench metabolism to preserve the instantaneous isotopic labeling state. Extract intracellular metabolites using a suitable solvent system like cold methanol/water [23].
- Mass Spectrometry Analysis: Analyze the extracted metabolites using Gas Chromatography-Mass Spectrometry (GC-MS) or Liquid Chromatography-MS (LC-MS) to measure the Mass Isotopomer Distribution (MID) of key metabolic intermediates [23].
- Flux Calculation: Use specialized software to compute the flux map that best fits the measured MID data and extracellular uptake/secretion rates, while satisfying stoichiometric constraints of the metabolic network model [23].
A Framework for Direct Model Corroboration
The quantitative flux data obtained from 13C-MFA experiments on Keio knockouts serve as the ground truth for evaluating the performance of different computational models. The logical relationship between the models, data, and validation outcomes is shown below.
Performance Comparison of Modeling Approaches
Table 2 provides a comparative summary of how different computational methods perform against 13C-MFA data from knockout studies, highlighting their core principles, strengths, and limitations in the context of validation.
Table 2: Comparison of Computational Models for Predicting E. coli Knockout Fluxes
| Method | Core Principle | Performance & Use Case | Key Limitations |
|---|---|---|---|
| FBA | Maximizes/Kinetic Models (e.g., growth rate) [28] [23]. | Often accurate for wild-type and evolved strains under selection; less accurate for immediate knockout responses [28]. | Relies on a potentially incorrect optimality assumption for knockouts. |
| MOMA | Finds flux distribution with minimal Euclidean distance to wild-type FBA solution [28]. | Improved prediction for unevolved knockouts; assumes a "sub-optimal" but minimally adjusted state [28]. | May not capture all regulatory constraints; performance varies. |
| ROOM | Minimizes the number of large flux changes from the wild-type solution [28]. | Can be more accurate than MOMA when regulatory on/off switches govern the response [28]. | Like MOMA, is a static prediction that may miss dynamic adaptations. |
| BayFlux | Uses Bayesian inference to sample all flux distributions compatible with data [67]. | Quantifies prediction uncertainty; enables probabilistic versions of MOMA/ROOM (P-13C MOMA/ROOM) [67]. | Computationally intensive; requires specialized statistical expertise. |
Interpretation of Validation Results
A robust validation framework must account for the fact that a model's failure to predict knockout fluxes is not merely a setback but an opportunity for discovery. Discrepancies between predicted and measured fluxes can:
- Reveal unknown regulatory mechanisms not captured in the model [28].
- Indicate the presence of alternative metabolic pathways or promiscuous enzyme activities [28].
- Highlight incorrect network topology or gaps in the genome-scale model [67].
- Challenge the validity of the chosen objective function in FBA for specific genetic or environmental contexts [28].
Essential Research Reagents and Tools
Successful implementation of this validation framework relies on a set of key reagents and computational tools. The following table lists essential components.
Table 3: The Scientist's Toolkit for Knockout Flux Validation
| Category | Item | Function & Application |
|---|---|---|
| Biological Reagents | Keio Collection Knockout Strains [71] [72] | Source of defined genetic perturbations for systematic validation. |
| 13C-Labeled Substrates (e.g., [U-13C] Glucose) [23] | Tracers for 13C-MFA experiments to measure in vivo fluxes. | |
| Defined Minimal Media | Ensures controlled and reproducible cultivation conditions. | |
| Analytical Tools | GC-MS or LC-MS Instrumentation [23] | Measures mass isotopomer distributions for 13C-MFA. |
| MEMOTE Suite [10] | Quality control and consistency testing for genome-scale metabolic models. | |
| Computational Software | COBRA Toolbox / cobrapy [10] | Standard platforms for constraint-based modeling (FBA, MOMA, ROOM). |
| 13C-MFA Software (e.g., INCA, OpenFLUX) | Fits metabolic network models to 13C-labeling data to calculate fluxes. | |
| BayFlux [67] | Bayesian framework for flux sampling and uncertainty quantification. |
The E. coli Keio knockout collection provides an unparalleled biological resource for the rigorous validation of metabolic models. By integrating systematic gene deletions with quantitative 13C-MFA flux measurements, researchers can move beyond correlative studies and perform direct, causal tests of model predictions. This corroboration framework is essential for advancing the predictive accuracy of both FBA and 13C-MFA, ultimately leading to more reliable in silico models for fundamental biological discovery and applied metabolic engineering. The continued development of methods like BayFlux, which explicitly handle uncertainty, and the generation of consistent, genome-scale knockout flux datasets will further solidify the foundation for model selection and validation in the field [28] [67].
Metabolic flux represents the integrated functional phenotype of a living cell, emerging from multiple layers of biological organization and regulation [3]. For researchers working with E. coli in both basic research and biotechnological applications, accurately predicting or measuring these fluxes is crucial for understanding cellular behavior and optimizing metabolic engineering strategies. Two primary approaches have emerged for flux analysis: constraint-based Flux Balance Analysis (FBA) and experimental 13C-Metabolic Flux Analysis (13C-MFA). More recently, integrated approaches that combine these methods have shown significant promise. This guide provides an objective comparison of these methodologies, supported by experimental data and detailed protocols, to help researchers select the appropriate tool for their specific research context in E. coli flux prediction.
Core Principles and Methodologies
Flux Balance Analysis (FBA): A Constraint-Based Predictive Approach
Flux Balance Analysis is a mathematical constraint-based approach that uses genome-scale metabolic models (GEMs) to predict metabolic fluxes without requiring extensive experimental data [3]. FBA operates on the principle of metabolic steady-state, where metabolite concentrations and reaction rates remain constant, and uses linear optimization to identify flux distributions that optimize specific cellular objectives [2]. The most commonly used objective function is the maximization of biomass production, based on the hypothesis that microorganisms like E. coli have evolved to maximize growth under given conditions [3] [12].
The FBA workflow begins with constructing a stoichiometric matrix from a genome-scale metabolic model containing all known metabolic reactions in an organism. For E. coli, well-curated models like iML1515 or iJO1366 are typically used [2] [5]. This matrix, combined with reaction bounds and constraints, defines a solution space containing all possible flux distributions. Linear optimization is then applied to identify the specific flux distribution that maximizes the objective function while satisfying all constraints [2].
Key advantages of FBA include its computational tractability and ability to analyze genome-scale models without requiring kinetic parameters [3]. However, its predictions depend heavily on the chosen objective function and constraints, which may not always accurately reflect cellular priorities [3].
13C-Metabolic Flux Analysis (13C-MFA): An Experimental Measurement Approach
13C-Metabolic Flux Analysis is an experimentally based method that determines internal carbon fluxes by tracking atom rearrangements in metabolic networks [3] [12]. This approach uses 13C-labeled substrates fed to cells, followed by measurement of the resulting labeling patterns in metabolic products using mass spectrometry or NMR techniques [3]. Fluxes are estimated by optimizing the fit between simulated and measured labeling patterns, providing a quantitative description of carbon flow through metabolism [12].
A significant strength of 13C-MFA is its ability to estimate exchange fluxes through reversible reactions, reporting on substrate cycling and metabolic regulation [12]. However, the method is technically challenging, requires specialized equipment and expertise, and primarily describes carbon-related metabolism while ignoring non-carbon metabolic processes [12]. It also typically focuses on central carbon metabolism rather than genome-scale networks [3].
Integrated Approaches: Combining Strengths
Hybrid approaches that integrate mechanistic models with data-driven methods have emerged as powerful platforms for metabolic flux analysis [48]. The Metabolic-Informed Neural Network (MINN) represents one such framework, embedding GEMs within neural networks to combine the strengths of both FBA and omics data integration [48]. These models can handle the trade-off between biological constraints and predictive accuracy, outperforming traditional FBA and pure machine learning methods on multi-omics datasets [48].
Other integrated strategies include incorporating enzyme constraints into FBA models using workflows like ECMpy, which caps fluxes based on enzyme availability and catalytic efficiency without altering the fundamental GEM structure [2]. Such approaches avoid unrealistic flux predictions by accounting for proteomic limitations [74].
Comparative Performance Analysis
Quantitative Comparison of Method Capabilities
Table 1: Comparative analysis of FBA, MFA, and integrated approaches for E. coli flux prediction
| Feature | FBA | 13C-MFA | Integrated Approaches |
|---|---|---|---|
| Principle | Constraint-based optimization using stoichiometric models [3] | Experimental tracking of 13C label distribution [3] | Hybrid of mechanistic and data-driven models [48] |
| Data Requirements | Stoichiometric matrix, constraints, objective function [2] | 13C labeling data, extracellular fluxes [3] | Multi-omics data, GEMs, often 13C-MFA data for validation [48] |
| Network Scale | Genome-scale (e.g., iML1515: 2,719 reactions) [2] | Central carbon metabolism (limited number of reactions) [3] | Variable, can incorporate genome-scale models [48] |
| Primary Output | Predicted flux distribution maximizing objective [3] | Estimated intracellular fluxes fitting experimental data [3] | Flux predictions balancing constraints and data patterns [48] |
| Key Strengths | Genome-scale coverage; No need for experimental flux data; Computationally efficient [3] [12] | Direct experimental basis; Estimates exchange fluxes; High confidence in core metabolism [12] | Improved accuracy over pure methods; Integration of multiple data types; Balanced trade-offs [48] |
| Key Limitations | Dependent on objective function; May predict unrealistic fluxes [3] [2] | Experimentally intensive; Limited to carbon metabolism; Technical expertise required [3] [12] | Complexity in implementation; Potential conflicts between objectives [48] |
| Validation Approach | Comparison with experimental growth/secretion rates [3] | Statistical goodness-of-fit tests (e.g., χ²-test) [3] | Comparison against MFA fluxes and other omics data [48] |
| Computational Demand | Low to moderate [3] | High (parameter estimation) [3] | High (model training and optimization) [48] |
Experimental Evidence and Case Studies
Anaerobic Adaptations in E. coli
A synergistic study combining 13C-MFA and FBA provided insights into E. coli metabolic adaptation to anaerobiosis [12]. The 13C-MFA flux maps revealed that the fraction of maintenance ATP consumption in total ATP production was approximately 14% higher under anaerobic (51.1%) than aerobic conditions (37.2%) [12]. FBA complemented these findings by showing that increased ATP utilization was consumed by ATP synthase to secrete protons during fermentation [12]. The study also demonstrated that the TCA cycle is incomplete in aerobically growing cells, and submaximal growth results from limited oxidative phosphorylation [12].
While FBA successfully predicted product secretion rates in aerobic culture when constrained with glucose and oxygen uptake measurements, sampling of the feasible solution space showed that the most frequently predicted internal fluxes differed substantially from 13C-MFA-derived fluxes [12]. This highlights that while FBA can capture input-output relationships, its internal flux predictions may not always match experimental measurements.
Overflow Metabolism Prediction
Incorporating proteomic constraints into FBA improved quantitative prediction of acetate overflow metabolism in various E. coli strains [74]. The Proteome Allocation Theory (PAT) suggests that overflow metabolism originates from differential proteomic efficiencies between fermentation and respiration pathways [74]. By constraining proteome allocation sectors in FBA, researchers achieved improved prediction of acetate production rates across different growth conditions [74].
This approach implemented a concise proteomic constraint:
[ wfvf + wrvr + b\lambda = 1 - \phi_0 ]
Where (wf) and (wr) represent proteomic costs per unit fermentation and respiration flux ((vf) and (vr)), (b) quantifies proteome fraction per unit growth rate ((\lambda)), and (\phi_0) represents growth-independent proteome fraction [74]. This modification enabled quantitative prediction of both the onset and extent of overflow metabolism [74].
Hybrid Model Performance
The Metabolic-Informed Neural Network (MINN) demonstrated improved performance compared to traditional pFBA and random forest models when integrating multi-omics data from E. coli single-gene knockout mutants grown in minimal glucose medium [48]. MINN implementations addressed conflicts between data-driven and mechanistic objectives while enhancing interpretability through coupling with pFBA [48].
Decision Framework and Experimental Protocols
Method Selection Guide
Table 2: When to use each flux analysis method for E. coli research
| Research Goal | Recommended Method | Rationale | Key Implementation Considerations |
|---|---|---|---|
| Genome-scale flux prediction | FBA | Computational efficiency at large scale; Comprehensive network coverage [3] | Carefully select objective function; Incorporate relevant constraints [3] |
| Quantitative flux measurement in core metabolism | 13C-MFA | Experimental basis; Higher confidence in core fluxes [12] | Prepare 13C-labeled substrates; Access to MS/NMR instrumentation [3] |
| Integrating multiple omics data types | Integrated approaches (e.g., MINN) | Combines strengths of both methods; Handles multi-omics data [48] | Requires both modeling expertise and experimental data [48] |
| Predicting response to genetic perturbations | FBA with enzyme constraints | Accounts for proteomic limitations; More realistic flux predictions [2] [74] | Incorporate enzyme abundance and Kcat values [2] |
| Metabolic engineering design | FBA initially, validated with 13C-MFA | Rapid screening with experimental validation [3] [12] | Use FBA for strain design, verify with targeted 13C-MFA [12] |
| Characterizing unknown metabolism | 13C-MFA | Data-driven without pre-specified objectives [3] | Can reveal unexpected fluxes and pathway activities [12] |
Detailed Experimental Protocol: Integrated FBA and 13C-MFA
Objective: To synergistically combine FBA and 13C-MFA for understanding E. coli metabolic adaptation to different growth conditions [12].
Materials and Strains:
- E. coli K-12 MG1655 (or other appropriate strain)
- Defined minimal medium (e.g., M9) with labeled or unlabeled glucose
- 13C-labeled glucose (e.g., [1-13C] glucose or [U-13C] glucose)
- GC-MS or LC-MS instrumentation for isotopic labeling measurement
- Genome-scale metabolic model (e.g., iML1515 or iJO1366)
Procedure:
Culture Conditions:
- Grow E. coli in defined minimal medium with labeled (for 13C-MFA) or unlabeled (for FBA constraints) glucose as sole carbon source under targeted conditions (aerobic/anaerobic, specific growth rates) [12].
- Harvest cells at mid-log phase for metabolite analysis and measure extracellular fluxes (substrate uptake, product secretion, growth rates) [12].
13C-MFA Flux Determination:
- Supply 13C-labeled substrates to cells and measure isotopic labeling of proteinogenic amino acids and intracellular metabolites using GC-MS or LC-MS [12].
- Use computational tools (e.g., ClusterFLUX) to estimate intracellular fluxes by optimizing the fit between measured and simulated labeling patterns [12].
- Validate flux maps using statistical goodness-of-fit tests and estimate confidence intervals for flux values [3].
FBA Model Construction and Simulation:
- Use a curated genome-scale model (e.g., iML1515 for E. coli K-12) [2].
- Constrain the model with measured uptake and secretion rates [12].
- Implement appropriate objective function (e.g., biomass maximization) and solve using linear optimization [3].
- Optionally, incorporate additional constraints based on proteomic limitations if enzyme abundance data is available [74].
Integrated Analysis:
Workflow Visualization
Flowchart Title: Decision Framework for Selecting Flux Analysis Methods
Table 3: Key research reagents and computational resources for E. coli flux analysis
| Resource Type | Specific Examples | Function/Role | Availability |
|---|---|---|---|
| E. coli GEMs | iML1515 [2], iJO1366 [5] | Genome-scale metabolic models for FBA | Publicly available (BioModels, etc.) |
| Computational Tools | COBRApy [2], ECMpy [2] | Implement FBA and enzyme constraints | Open-source Python packages |
| 13C-Labeled Substrates | [1-13C] glucose, [U-13C] glucose [12] | Tracers for 13C-MFA experiments | Commercial suppliers |
| Analytical Instruments | GC-MS, LC-MS, NMR [12] | Measure isotopic labeling patterns | Core facilities or specialized labs |
| Enzyme Kinetics Databases | BRENDA [2] | Source of Kcat values for enzyme constraints | Public database |
| Protein Abundance Data | PAXdb [2] | Enzyme abundance for constraint-based modeling | Public database |
| Metabolic Databases | EcoCyc [2] | Curated E. coli metabolic knowledge | Public database |
The choice between FBA, 13C-MFA, and integrated approaches for E. coli flux prediction depends primarily on research objectives, available resources, and required precision. FBA offers genome-scale predictive capability with minimal experimental input, making it ideal for initial screening and hypothesis generation. 13C-MFA provides experimentally validated fluxes in core metabolism with higher confidence, suited for quantitative analysis of central carbon metabolism. Integrated approaches leverage the strengths of both methods, offering improved accuracy at the cost of increased complexity.
For metabolic engineers, we recommend an iterative approach: using FBA for initial strain design and 13C-MFA for experimental validation of key strains. For basic research characterizing metabolic adaptations, 13C-MFA provides gold-standard flux measurements, while FBA can explore network capabilities beyond immediate experimental conditions. As hybrid methods continue to develop, they promise to further bridge the gap between predictive modeling and experimental measurement in E. coli metabolism research.
In the field of metabolic engineering, constraint-based modeling approaches like Flux Balance Analysis (FBA) have become indispensable tools for predicting cellular behavior. FBA uses optimization principles, typically maximizing biomass production or other cellular objectives, to predict flux distributions through genome-scale metabolic networks [3] [12]. However, these predictions are inherently based on computational optimizations rather than direct experimental measurements, creating a critical need for robust validation methodologies. This is where 13C-Metabolic Flux Analysis (13C-MFA) emerges as the gold standard for validating and refining these genome-scale models, particularly for the model organism Escherichia coli [3] [51].
The fundamental challenge in metabolic modeling lies in the fact that in vivo fluxes cannot be directly measured, necessitating modeling approaches to estimate or predict them [3] [10]. While FBA provides a powerful framework for exploring metabolic capabilities, its predictions require experimental validation to ensure biological relevance. 13C-MFA serves this critical function by providing empirically determined flux maps based on isotopic labeling patterns, enabling researchers to test the reliability of FBA predictions and identify areas where model refinements are necessary [51] [12]. This synergistic relationship between the two methodologies has become increasingly important as metabolic engineering efforts grow more ambitious in scale and complexity.
Quantitative Comparison: 13C-MFA vs. FBA Flux Predictions in E. coli
Direct comparisons between 13C-MFA and FBA reveal significant differences in their ability to resolve intracellular fluxes. When both methods are applied to the same E. coli strains under identical growth conditions, 13C-MFA provides higher resolution flux maps that often contradict FBA predictions based on optimal growth assumptions [51] [12].
Table 1: Comparison of Key Metabolic Fluxes in E. coli Determined by 13C-MFA and FBA
| Metabolic Pathway/Reaction | 13C-MFA Flux Value | FBA Prediction | Discrepancy Notes |
|---|---|---|---|
| TCA Cycle Function | Non-cyclic, ~16% of glucose uptake | Typically complete cycle | FBA fails to predict incomplete TCA operation [12] |
| Pentose Phosphate Pathway Flux | Precisely resolvable | Often overestimated | Depends on tracer used [34] [75] |
| Glycolytic vs. Gluconeogenic Flux | Both directions quantifiable | Typically unidirectional | 13C-MFA can resolve parallel opposing fluxes [76] |
| Exchange Fluxes | Quantifiable with precision | Generally not resolved | COMPLETE-MFA greatly improves exchange flux resolution [34] |
| ATP Maintenance Costs | Directly calculable from flux maps | Requires prior assumption | MFA revealed 51% maintenance under anaerobiosis vs. 37% aerobically [12] |
A particularly revealing study by Chen et al. demonstrated that the TCA cycle operates non-cyclically in aerobically growing E. coli, with only about 16.1% of glucose uptake flux entering the non-cyclic TCA reactions—a finding that contradicts the complete TCA cycle typically predicted by FBA [12]. This fundamental discrepancy highlights how 13C-MFA can correct structural misconceptions in metabolic models.
Furthermore, 13C-MFA provides unique capabilities for quantifying exchange fluxes (forward and reverse reaction rates) through reversible reactions, which are notoriously difficult to resolve using FBA alone. The implementation of COMPLETE-MFA (complementary parallel labeling experiments) has significantly improved the precision of these exchange flux measurements, especially for central carbon metabolism in E. coli [34].
Experimental Protocols: Core Methodologies for 13C-MFA Validation
COMPLETE-MFA: Parallel Labeling Experiments
The most robust approach for 13C-MFA validation involves parallel labeling experiments, where multiple isotopic tracers are used simultaneously to generate a single, high-resolution flux map [34]. This methodology, termed COMPLETE-MFA, has been shown to significantly improve both flux precision and observability compared to single-tracer experiments.
The fundamental protocol involves:
- Growing E. coli cultures in parallel mini-bioreactors with different 13C-labeled glucose tracers
- Harvesting samples during mid-exponential growth phase for metabolic analysis
- Measuring mass isotopomer distributions of proteinogenic amino acids using GC-MS
- Integrating data from all parallel experiments into a unified flux estimation procedure
A landmark study analyzing 14 parallel labeling experiments in E. coli demonstrated that no single tracer optimally resolves all fluxes in the metabolic network [34]. Tracers that produced well-resolved fluxes in upper metabolism (glycolysis, pentose phosphate pathway) showed poor performance for fluxes in the lower metabolism (TCA cycle, anaplerotic reactions), and vice versa. The optimal tracer combination was found to be 75% [1-13C]glucose + 25% [U-13C]glucose for upper metabolism and [4,5,6-13C]glucose for lower metabolic fluxes [34].
Table 2: Essential Research Reagents for 13C-MFA Validation Studies
| Reagent/Category | Specific Examples | Function in 13C-MFA |
|---|---|---|
| 13C-Labeled Tracers | [1,2-13C]glucose, [1-13C]glucose, [U-13C]glucose, [4,5,6-13C]glucose | Create distinct isotopic labeling patterns for flux resolution [34] [77] |
| Analytical Instruments | GC-MS, LC-MS, NMR Spectrometers | Measure mass isotopomer distributions in metabolic intermediates [34] [12] |
| Strain Collections | Keio collection (E. coli BW25113 mutants) | Provide isogenic strains for validating model predictions of knockout effects [19] |
| Culture Systems | Controlled bioreactors, mini-bioreactor arrays | Maintain steady-state growth conditions required for flux determination [34] |
| Software Tools | COBRA Toolbox, cobrapy, EMU algorithms | Perform flux estimation, statistical analysis, and model validation [3] [76] |
Genome-Scale 13C-MFA Implementation
While traditional 13C-MFA focuses on central carbon metabolism, recent advances have enabled flux analysis at genome-scale [76]. This approach uses the same atomic mapping information but scales up to models containing hundreds of reactions and metabolites, allowing direct comparison with genome-scale FBA predictions.
The critical steps in genome-scale 13C-MFA include:
- Construction of a genome-scale mapping model with complete atom transition information for all reactions
- Application of flux constraints from experimental measurements including growth rates, substrate uptake, and product secretion
- Flux estimation using the EMU (Elementary Metabolite Units) framework or related algorithms
- Statistical evaluation using χ2-tests of goodness-of-fit and confidence interval analysis [3] [76]
This approach has revealed that expanding to genome-scale models results in wider flux confidence intervals for key reactions in central metabolism, reflecting the additional flexibility introduced by considering alternative metabolic routes [76]. For example, the glycolysis flux range doubled due to the possibility of active gluconeogenesis, and the transhydrogenase reaction flux became essentially unresolvable due to the presence of five alternative routes for NADPH/NADH interconversion in the genome-scale model.
Visualization of the 13C-MFA Validation Workflow
The following diagram illustrates the integrated experimental and computational workflow for validating genome-scale models using 13C-MFA:
13C-MFA Validation Workflow for Genome-Scale Models
Synergistic Applications: How 13C-MFA Reveals What FBA Cannot Predict
The integration of 13C-MFA with FBA has led to fundamental discoveries about E. coli metabolism that would not be possible using either method alone. Key insights include:
Metabolic Adaptation to Anaerobiosis
Comparative analysis of aerobic and anaerobic growth in E. coli revealed that the fraction of maintenance ATP consumption increases from 37.2% under aerobic conditions to 51.1% under anaerobiosis [12]. FBA helped explain this finding by revealing that increased ATP utilization is consumed by ATP synthase to secrete protons from fermentation—a critical insight into the bioenergetics of anaerobic growth.
Emergence of Metabolic Specialization in Colonies
When E. coli is grown on agar surfaces (as opposed to liquid culture), 13C-MFA revealed the emergence of two distinct metabolic subpopulations engaged in acetate cross-feeding [77]. Approximately 92% of cells metabolized glucose and secreted acetate, while 8% of cells consumed the secreted acetate without glucose uptake. This metabolic specialization, predicted theoretically by multi-scale FBA approaches, was experimentally validated using co-culture 13C-MFA methodology.
Resolution of Parallel Metabolic Pathways
Genome-scale 13C-MFA identified alternative metabolic routes that are typically unaccounted for in core metabolic models [76]. For instance, a bypass through arginine metabolism and multiple transhydrogenase routes for cofactor balancing were found to be active, explaining why flux confidence intervals expand when moving from core to genome-scale models.
Table 3: Metabolic Discoveries Enabled by 13C-MFA Validation of FBA Predictions
| Metabolic Phenomenon | FBA Prediction | 13C-MFA Validation | Biological Significance |
|---|---|---|---|
| TCA Cycle Operation | Complete cycle | Non-cyclic under aerobic conditions | Reveals suboptimal growth due to limited oxidative phosphorylation [12] |
| Acetate Cross-Feeding | Emergence possible in silico | Experimentally quantified subpopulations | Validates metabolic specialization in biofilms and colonies [77] |
| Transhydrogenase Flux | Single route | Multiple alternative pathways | Explains redundancy in cofactor balancing mechanisms [76] |
| Arginine Degradation | Often inactive | Non-zero flux identified | Uncovers alternative pathway for meeting biomass demands [76] |
The integration of 13C-MFA as a validation tool for genome-scale models represents a paradigm shift in metabolic engineering and systems biology. The complementary strengths of these approaches—FBA's ability to predict metabolic capacities and 13C-MFA's power to measure actual metabolic fluxes—create a powerful framework for understanding and engineering microbial metabolism [3] [12].
Future directions in this field include the development of more sophisticated model selection criteria beyond the traditional χ2-test of goodness-of-fit, increased incorporation of metabolite pool size information into flux estimation procedures, and the creation of standardized validation protocols that can be consistently applied across different laboratories and microbial systems [3]. As the field moves toward more complex microbial communities and multi-tissue systems, the principles established for E. coli will provide a foundation for understanding metabolism at even greater scales of biological complexity.
The synergy between 13C-MFA and FBA ultimately enhances confidence in constraint-based modeling as a whole and facilitates more widespread use of these methods in biotechnology applications. By grounding computational predictions in experimental measurements, researchers can more reliably engineer microbial systems for improved production of biofuels, pharmaceuticals, and biochemicals.
Conclusion
Flux Balance Analysis (FBA) and Metabolic Flux Analysis (MFA) are not competing but profoundly complementary techniques for elucidating E. coli metabolism. FBA excels in providing genome-scale predictions of metabolic capabilities and guiding strain design, while MFA delivers high-resolution, experimentally validated flux maps for core metabolism. The future of flux analysis lies in hybrid strategies that integrate the mechanistic foundations of FBA and MFA with machine learning, comprehensive model validation, and genome-scale experimental data. For biomedical and clinical research, these advanced, integrated approaches promise to accelerate the engineering of novel microbial cell factories for drug precursor synthesis and enhance our systems-level understanding of bacterial physiology in both industrial and pathogenic contexts.
References
- 1. Predicting internal cell fluxes at sub-optimal growth [https://bmcsystbiol.biomedcentral.com/articles/10.1186/s12918-015-0153-3]
- 2. Flux Balance Analysis | Virginia - iGEM 2025 [https://2025.igem.wiki/virginia/fba]
- 3. Model Validation and Selection in Metabolic Flux Analysis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10922127/]
- 4. Metabolic flux analysis of Escherichia coli knockouts [https://www.sciencedirect.com/science/article/abs/pii/S0958166914000378]
- 5. Prediction of Metabolic Flux Distribution by Flux Sampling [https://www.mdpi.com/2306-5354/10/6/636]
- 6. Enhanced flux prediction by integrating relative expression ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6532942/]
- 7. Characterizing cancer metabolism from bulk and single ... [https://www.nature.com/articles/s41467-023-40457-w]
- 8. Dynamic Metabolic Flux Analysis Demonstrated on Cultures ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2934775/]
- 9. Metabolic flux analysis: a comprehensive review on sample ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9449821/]
- 10. Model Validation and Selection in Metabolic Flux Analysis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10055486/]
- 11. Metabolic flux and flux balance analyses indicate the ... [https://www.sciencedirect.com/science/article/pii/S1096717625001211]
- 12. Synergy between 13 C-metabolic flux analysis and ... [https://www.sciencedirect.com/science/article/abs/pii/S1096717610001023]
- 13. A Method to Constrain Genome-Scale Models with 13 C ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1004363]
- 14. 13C metabolic flux analysis - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC9493264/]
- 15. Selection of Tracers for 13C-Metabolic Flux Analysis using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6474252/]
- 16. Evaluation of 13C isotopic tracers for metabolic flux analysis in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3026314/]
- 17. Optimal tracers for parallel labeling experiments and 13C ... [https://www.sciencedirect.com/science/article/abs/pii/S1096717616300428]
- 18. Rational design of 13C-labeling experiments for metabolic flux ... [https://bmcsystbiol.biomedcentral.com/articles/10.1186/1752-0509-6-43]
- 19. From Escherichia coli mutant 13 C labeling data to a core ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1007319]
- 20. Isotopically nonstationary 13C metabolic flux analysis in ... [https://www.sciencedirect.com/science/article/pii/S1096717621001932]
- 21. In vivo 2H/13C flux analysis in metabolism research [https://www.sciencedirect.com/science/article/abs/pii/S0958166921000653]
- 22. 13C Metabolic Flux Analysis [https://imsb.ethz.ch/research/zamboni/research/flux-analysis.html]
- 23. A guide to metabolic flux analysis in metabolic engineering [https://www.sciencedirect.com/science/article/abs/pii/S1096717620301683]
- 24. Guidelines for metabolic flux analysis in ... [https://www.biosynsis.com/blog/guidelines-for-metabolic-flux-analysis-in-metabolic-engineering-methods-tools-and-applications/]
- 25. RELATCH: relative optimality in metabolic networks explains ... [https://genomebiology.biomedcentral.com/articles/10.1186/gb-2012-13-9-r78]
- 26. Flux balance analysis [https://en.wikipedia.org/wiki/Flux_balance_analysis]
- 27. Metabolic flux balance analysis and the in silico ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC29061/]
- 28. Metabolic flux analysis of E. coli knockouts - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC5842030/]
- 29. Optimization-based framework with flux balance analysis (FBA ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1013635]
- 30. Systematic comparison of local approaches for isotopically ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10280729/]
- 31. Accurate prediction of gene deletion phenotypes with Flux ... [https://www.nature.com/articles/s41467-025-63436-9]
- 32. 13C metabolic flux analysis-guided metabolic engineering of ... [https://biotechnologyforbiofuels.biomedcentral.com/articles/10.1186/s13068-019-1372-4]
- 33. Rethinking 13C-metabolic flux analysis – The Bayesian ... [https://www.sciencedirect.com/science/article/pii/S1096717624000508]
- 34. Integrated 13C-metabolic flux analysis of 14 parallel labeling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5845449/]
- 35. The Design of FluxML: A Universal Modeling Language for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6543931/]
- 36. 13CFLUX - Third-generation high-performance engine for ... [https://arxiv.org/html/2509.23847]
- 37. A compact model of Escherichia coli core and biosynthetic ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1013564]
- 38. A genome-scale metabolic flux model of Escherichia coli K–12 derived from the EcoCyc database - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4086706/]
- 39. Modelling overflow metabolism in Escherichia coli with flux balance analysis incorporating differential proteomic efficiencies of energy pathways - PubMed [https://pubmed.ncbi.nlm.nih.gov/30630470/]
- 40. Analysis of optimality in natural and perturbed metabolic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC137552/]
- 41. Dynamic regulatory on/off minimization for biological systems ... [https://bmcsystbiol.biomedcentral.com/articles/10.1186/1752-0509-6-16]
- 42. Comparison of Optimization-Modelling Methods for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7734505/]
- 43. Flux balance analysis with or without molecular crowding ... [https://www.nature.com/articles/s41598-019-47935-6]
- 44. An objective function exploiting suboptimal solutions in ... [https://bmcsystbiol.biomedcentral.com/articles/10.1186/1752-0509-7-98]
- 45. An improved algorithm for flux variability analysis [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-05089-9]
- 46. From MFA to FBA: Defining linear constraints accounting ... [https://www.sciencedirect.com/science/article/abs/pii/S0959152417301312]
- 47. Maximizing multi-reaction dependencies provides more ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10538754/]
- 48. MINN: A metabolic-informed neural network for integrating ... [https://www.sciencedirect.com/science/article/pii/S2001037025003265]
- 49. Machine learning applied to enzyme turnover numbers ... [https://www.nature.com/articles/s41467-018-07652-6]
- 50. What is flux balance analysis? - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3108565/]
- 51. Synergy Between (13)C-metabolic Flux Analysis and ... [https://pubmed.ncbi.nlm.nih.gov/21129495/]
- 52. Integration of graph neural networks and genome-scale ... [https://www.nature.com/articles/s41540-024-00348-2]
- 53. ThermOptCobra: Thermodynamically optimal construction and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12329557/]
- 54. Maximizing multi-reaction dependencies provides more ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1011489]
- 55. Predicting internal cell fluxes at sub-optimal growth - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4397736/]
- 56. Flux Balance Analysis (FBA) [https://www.creative-proteomics.com/metabolic-flux/flux-balance-analysis-fba.html]
- 57. Metabolic flux analysis reaching genome wide coverage [https://www.sciencedirect.com/science/article/abs/pii/S2211339820300356]
- 58. Multiplex methods provide effective integration of multi-omic ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-016-0912-1]
- 59. Comparative multi-omics systems analysis of Escherichia coli ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3446290/]
- 60. Multi-omics Quantification of Species Variation ... [https://www.sciencedirect.com/science/article/pii/S2405471216302903]
- 61. The ETFL formulation allows multi-omics integration in ... [https://www.nature.com/articles/s41467-019-13818-7]
- 62. INTEGRATE: Model-based multi-omics data ... - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC8853556/]
- 63. Thermodynamic Constraints Improve Metabolic Networks [https://www.sciencedirect.com/science/article/pii/S0006349517306689]
- 64. Integration of graph neural networks and genome-scale ... [https://pubmed.ncbi.nlm.nih.gov/38448436/]
- 65. A neural-mechanistic hybrid approach improving the ... [https://www.nature.com/articles/s41467-023-40380-0]
- 66. Prediction of gene essentiality using machine learning and ... [https://www.sciencedirect.com/science/article/abs/pii/S240589632300006X]
- 67. BayFlux: A Bayesian method to quantify metabolic Fluxes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10664898/]
- 68. Bayesian metabolic flux analysis reveals intracellular flux ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6612884/]
- 69. Differential visual analysis of Flux Sampling based on ... [https://www.sciencedirect.com/science/article/abs/pii/S0097849322001583]
- 70. Model Validation and Selection in Metabolic Flux Analysis ... [https://arxiv.org/abs/2303.12651]
- 71. E. coli Keio Knockouts [https://horizondiscovery.com/en/non-mammalian-research-tools/products/e-coli-keio-knockouts]
- 72. Construction of Escherichia coli K-12 in-frame, single-gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1681482/]
- 73. Keio Strains [https://ecgrc.net/index.php/product-category/bacterial-cultures/keio-strains/]
- 74. Modelling overflow metabolism in Escherichia coli with flux ... [https://bmcsystbiol.biomedcentral.com/articles/10.1186/s12918-018-0677-4]
- 75. Investigation of useful carbon tracers for 13C-metabolic flux ... [https://www.sciencedirect.com/science/article/pii/S2214030116300189]
- 76. 13C metabolic flux analysis at a genome-scale [https://www.sciencedirect.com/science/article/abs/pii/S1096717615001032]
- 77. 13C metabolic flux analysis of E. coli grown on agar ... [https://www.sciencedirect.com/science/article/abs/pii/S1096717618301939]