Strategies for Heterologous Pathway Reconstruction in Yeast: From Foundational Concepts to Advanced Biomedical Applications
This article provides a comprehensive overview of heterologous pathway reconstruction in yeast, a cornerstone of modern metabolic engineering for drug development and sustainable biomanufacturing.
Strategies for Heterologous Pathway Reconstruction in Yeast: From Foundational Concepts to Advanced Biomedical Applications
Abstract
This article provides a comprehensive overview of heterologous pathway reconstruction in yeast, a cornerstone of modern metabolic engineering for drug development and sustainable biomanufacturing. It systematically explores the foundational principles of introducing foreign metabolic pathways into yeast hosts, details the cutting-edge methodological toolkit for pathway design and implementation, and addresses critical challenges in troubleshooting and optimization to achieve high-yield production. By presenting rigorous validation frameworks and comparative analyses of different yeast chassis, this resource equips researchers and scientists with the integrated knowledge needed to harness yeast cell factories for the efficient and scalable production of complex pharmaceuticals and high-value natural products.
Understanding the Blueprint: Principles and Host Selection for Yeast Pathway Engineering
Defining Heterologous Pathways and Their Value in Bioproduction
Heterologous expression refers to the expression of a gene or part of a gene in a host organism that does not naturally possess that genetic material, achieved through recombinant DNA technology [1]. In bioproduction, this typically involves transferring entire biosynthetic pathways—linked series of biochemical reactions—into microbial hosts to enable the production of valuable compounds that the host would not naturally synthesize [2]. This approach has evolved from simple single-gene expression to the complex introduction of multiple-gene clusters, spawning the field of metabolic engineering [2].
The fundamental value proposition of heterologous pathways lies in their ability to transform amenable host organisms into microbial cell factories for targeted compound production. This is particularly valuable for complex secondary metabolites—such as pharmaceuticals, fragrances, and flavors—that are difficult to obtain economically through chemical synthesis or extraction from their native biological sources [2] [3]. By transferring biosynthetic capabilities to optimized industrial hosts, researchers can achieve higher yields, better process control, and access to compounds from organisms that are uncultivable or slow-growing in their native state [3].
Key Host Organisms and Their Applications
Comparison of Major Production Hosts
The selection of an appropriate host organism is a critical determinant of success in heterologous pathway engineering. Different hosts offer distinct advantages and limitations based on their genetic background, metabolic capabilities, and physiological characteristics [2]. The table below summarizes the key hosts used in heterologous production:
Table 1: Key Host Organisms for Heterologous Pathway Expression
| Host Organism | Key Advantages | Major Limitations | Common Applications |
|---|---|---|---|
| Saccharomyces cerevisiae (Baker's Yeast) | GRAS status; Well-established genetic tools; Eukaryotic PTMs; Robust industrial performance [4] [5] | Hyperglycosylation; Tough cell wall; Low diversity of native secondary metabolites [2] | Pharmaceutical proteins; Plant terpenoids; Secondary metabolites [4] [6] |
| Komagataella phaffii (Pichia pastoris) | High cell density cultivation; Strong inducible promoters; Efficient secretion; Methylotrophic [5] [7] | Methanol requirement for some promoters; More limited genetic toolbox than S. cerevisiae [5] | Industrial enzymes; Pharmaceutical proteins; Antibody fragments [5] [7] |
| Escherichia coli | Rapid growth; Low-cost media; Extensive genetic knowledge [1] | Lack of eukaryotic PTMs; Intracellular protein aggregation; Endotoxin production [1] | Soluble prokaryotic proteins; Simple metabolic pathways [8] [1] |
| Filamentous Fungi (e.g., Aspergillus spp.) | High native secondary metabolite diversity; Efficient secretion [2] | Complex genetics; Native metabolic competition; Spore hazards [2] | Enzyme production; Fungal secondary metabolites [2] |
| Bacillus subtilis | Non-pathogenic; Protein secretion capability; No LPS production [1] | Extracellular proteases; Lower expression efficiency than E. coli [1] | Enzyme production; Industrial biotechnology [1] |
Yeast as a Preferred Eukaryotic Platform
Yeast systems, particularly S. cerevisiae and K. phaffii, have emerged as dominant platforms for heterologous production of eukaryotic proteins and complex natural products [8] [4]. These unicellular fungi represent an optimal compromise between bacterial simplicity and higher eukaryotic functionality, offering several distinct advantages:
- Eukaryotic Protein Processing: Yeast possess the necessary cellular machinery for proper protein folding, disulfide bond formation, and post-translational modifications essential for the functionality of many eukaryotic proteins [8] [4].
- Secretion Capability: Both major yeast hosts can secrete recombinant proteins into the extracellular space, significantly simplifying downstream purification processes compared to intracellular expression in bacteria [5].
- Genetic Tractability: Extensive molecular biology toolsets, including advanced CRISPR/Cas9 systems and synthetic biology parts, enable precise genetic manipulation [4] [6].
- Industrial Robustness: Yeast excel in high-density fermentation, tolerate various stress conditions, and can be cultivated on inexpensive media [4] [7].
The choice between S. cerevisiae and K. phaffii often depends on the specific application. S. cerevisiae is frequently preferred for metabolic pathway engineering and production of small molecules like terpenoids [6], while K. phaffii often excels in high-level protein production due to its strong inducible promoters and efficient secretion apparatus [5] [7].
Strategies for Heterologous Pathway Engineering
Host Engineering and Optimization
Successful heterologous pathway expression requires extensive optimization of the host organism to support the introduced genetic material and associated metabolic burden. Key host engineering strategies include:
- Central Metabolism Rewiring: Engineering core metabolic pathways to enhance precursor and cofactor supply. For terpenoid production in yeast, this involves modifying the mevalonate pathway to increase flux toward isoprenoid precursors (IPP, DMAPP, FPP) while downregulating competing pathways like sterol biosynthesis [6].
- Metabolic Burden Mitigation: Heterologous protein production consumes cellular resources, creating resource competition that can impair host fitness and reduce productivity [5]. Strategies to mitigate this burden include tuning expression levels, using genomic integration rather than plasmid-based expression, and engineering stress response pathways [5].
- Transport and Compartmentalization: Localizing heterologous pathways to specific subcellular compartments (e.g., peroxisomes, mitochondria) can concentrate substrates, isolate toxic intermediates, and exploit specialized cellular environments [9] [6].
Table 2: Quantitative Examples of Heterologous Production in Yeast
| Product Category | Specific Product | Host | Titer/Level | Production Scale | Citation |
|---|---|---|---|---|---|
| Medicinal Proteins | Antithrombin III | S. cerevisiae | 312 mg/L | Fed-batch, 5L bioreactor | [4] |
| Medicinal Proteins | Transferrin | S. cerevisiae | 2.33 g/L | Fed-batch, 10L bioreactor | [4] |
| Food Proteins | Brazzein | S. cerevisiae | 9 mg/L | Batch, shake flask | [4] |
| Industrial Enzymes | Lipase | S. cerevisiae | 11,000 U/L | Fed-batch, 5L bioreactor | [4] |
| Secondary Metabolites | Colletochlorins | S. cerevisiae | 35-fold increase vs. native producer | Not specified | [10] |
| Terpenoids | α-Santalene | S. cerevisiae | 164.7 mg/L | Not specified | [6] |
Genetic Tool Development for Pathway Assembly
Advanced genetic tools are essential for assembling and optimizing multi-gene heterologous pathways:
- Vector Systems: Yeast shuttle vectors that replicate in both E. coli and yeast enable convenient cloning and propagation [8]. These include:
- YEp (Yeast Episomal Plasmids): High-copy number vectors for strong expression
- YCp (Yeast Centromeric Plasmids): Low-copy, stable maintenance
- YIp (Yeast Integrating Plasmids): Chromosomal integration for genetic stability [8]
- Polycistronic Vectors: Recently developed systems enable coordinated expression of multiple genes from single constructs, significantly simplifying pathway assembly [10].
- CRISPR/Cas9 Genome Editing: The highly efficient CRISPR/Cas9 system has revolutionized yeast metabolic engineering by enabling precise genomic integration of pathway genes, multiplexed gene knockouts, and promoter engineering [4] [6]. This system is particularly powerful in S. cerevisiae due to its highly efficient homology-directed repair mechanism [6].
Experimental Protocol: Heterologous Expression of Fungal Secondary Metabolite Pathways inS. cerevisiae
Pathway Identification and Gene Isolation
- Bioinformatic Identification: Identify target biosynthetic gene clusters (BGCs) from fungal genomic data through comparative genomics and conserved domain analysis (e.g., presence of polyketide synthases, prenyltransferases, or tailoring enzymes) [10] [3].
- Gene Synthesis or Amplification: Based on identified sequences:
- Codon Optimization: Optimize heterologous gene sequences for S. cerevisiae codon usage to enhance translation efficiency [3].
Vector Assembly and Pathway Reconstruction
- Polycistronic Vector Assembly [10]:
- Select appropriate auto-inducible yeast promoters (e.g., PGK1, TEF1 for constitutive expression; GAL1, GAL10 for inducible systems)
- Employ transformation-assisted recombination (in vivo assembly) in E. coli for seamless, cost-effective cloning of large gene clusters
- Include different selection markers (e.g., URA3, LEU2) for versatile selection in auxotrophic yeast strains
- Modular Pathway Assembly: For complex pathways, assemble individual modules first, then combine using standardized parts and connectors [6].
- Vector Validation: Verify all constructs by restriction digest and Sanger sequencing before yeast transformation.
Yeast Transformation and Screening
- Strain Preparation: Use appropriate S. cerevisiae strain (e.g., BY4741, CEN.PK2) and cultivate in rich medium (YPD) to mid-log phase.
- Transformation: Employ lithium acetate/single-stranded carrier DNA/PEG method for plasmid transformation [8].
- Selection and Screening:
- Plate transformed yeast on appropriate selective dropout media
- Screen for successful transformants by colony PCR
- Verify pathway integration through analytical methods (e.g., HPLC, LC-MS) for expected metabolites [10]
Pathway Optimization and Production
- Fermentation Conditions: Optimize media composition, temperature, aeration, and induction parameters in shake flasks prior to bioreactor scale-up [4].
- Analytical Validation: Monitor pathway functionality and product formation using:
- LC-MS for metabolite identification and quantification
- RNA-seq to verify gene expression
- Protein electrophoresis for enzyme production confirmation [10]
- Iterative Engineering: Based on results, implement additional engineering strategies such as:
- Promoter swapping to balance expression levels
- Enzyme engineering to improve catalytic efficiency
- Co-factor regeneration to support redox reactions [6]
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagent Solutions for Yeast Heterologous Expression
| Reagent/Resource | Function | Examples/Specific Types |
|---|---|---|
| Shuttle Vectors | Enable gene expression in both E. coli and yeast | YEp, YCp, YIp vectors with selective markers (URA3, LEU2) [8] |
| CRISPR/Cas9 System | Precision genome editing | Cas9 nuclease, gRNA expression cassettes, repair templates [4] [6] |
| Promoter Systems | Transcriptional control of heterologous genes | Constitutive: PGK1, TEF1; Inducible: GAL1, GAL10, AOX1 (for K. phaffii) [2] [10] |
| Codon Optimization Tools | Enhance translation efficiency in heterologous host | Gene synthesis services with yeast-optimized codons [3] |
| Analytical Standards | Metabolite identification and quantification | Authentic chemical standards for LC-MS calibration [10] |
| Specialized Media | Selective growth and production conditions | Synthetic complete dropout media; Induction media with galactose or methanol [5] |
Visualizing Heterologous Pathway Engineering Workflows
Metabolic Engineering of Yeast for Terpenoid Production
Heterologous pathway reconstruction represents a powerful paradigm for microbial bioproduction of valuable compounds. Yeast systems, particularly S. cerevisiae and K. phaffii, have emerged as preferred eukaryotic platforms due to their unique combination of genetic tractability, eukaryotic processing capabilities, and industrial robustness. Successful implementation requires integrated strategies spanning host engineering, genetic tool development, and careful pathway design with iterative optimization. As synthetic biology tools continue to advance, particularly CRISPR-based genome editing and computational modeling approaches, the scope and efficiency of heterologous production will continue to expand, enabling more sustainable and economically viable manufacturing routes for high-value natural products and proteins.
Heterologous pathway reconstruction is a cornerstone of modern synthetic biology, enabling the production of valuable compounds in engineered microbial hosts. The yeast Saccharomyces cerevisiae is a particularly prominent chassis for this purpose, prized for its generally recognized as safe (GRAS) status, clear genetic background, and sophisticated eukaryotic structures that facilitate proper protein folding and essential post-translational modifications (PTMs) [11]. This protocol outlines the core principles and detailed methodologies for successfully reconstructing heterologous pathways in yeast, from initial gene isolation to long-term host maintenance. The process embodies a "Design-Build-Test-Learn" (DBTL) cycle, accelerated by advances in synthetic biology and metabolic engineering, allowing for the efficient production of a diverse range of molecules, from therapeutic proteins to complex natural products like naringenin and Asperosaponin VI [11] [12] [13].
Core Principles and Definitions
Heterologous Pathway Reconstruction refers to the process of introducing and optimizing genetic material from a donor organism into a host organism to confer the ability to produce a non-native compound. The ultimate goal is to achieve high-yield, sustainable production of the target molecule.
Key objectives for a successful process include:
- High Titer: The final concentration of the target compound in the fermentation broth.
- High Yield: The conversion efficiency of substrates into the desired product.
- High Productivity: The rate of product formation per unit volume per unit time. Optimizing these three metrics is mandatory to achieve a scalable and cost-effective process at an industrial scale [12].
Application Notes: A Step-by-Step Protocol
The following protocol provides a generalized workflow for heterologous pathway reconstruction in S. cerevisiae, integrating strategies from recent successful case studies.
Stage 1: Pathway Design and Gene Isolation
Objective: To design a functional biosynthetic pathway and isolate or design the corresponding genetic parts.
Step 1.1: Pathway Selection and Retrosynthesis
- Deconstruct the target molecule into its biosynthetic precursors.
- Identify all enzymatic steps required for the conversion from a central metabolic precursor in yeast.
- Research and select candidate genes for each enzymatic step from potential donor organisms. Consider enzyme kinetics, specificity, and compatibility with the yeast host. For example, in naringenin production, the pathway requires tyrosine ammonia-lyase (TAL), 4-coumarate-CoA ligase (4CL), chalcone synthase (CHS), and chalcone isomerase (CHI) [12].
Step 1.2: Codon Optimization and Gene Synthesis
- Perform in silico codon optimization of the heterologous gene sequences to match the codon usage bias of S. cerevisiae. This is a critical step to ensure high translation efficiency [11].
- Beyond simple codon preference, modern optimization should consider GC content, avoid base repeats, and eliminate sequences that might trigger unwanted regulation [11].
- Synthesize the optimized genes de novo.
Table 1: Key Considerations for Pathway Design
| Design Element | Consideration | Strategy |
|---|---|---|
| Codon Usage | Rare codons can drastically reduce translation efficiency [11]. | Full gene synthesis with host-optimized codons. |
| Enzyme Selection | Enzymes from different sources have varying kinetics and compatibility [12]. | Test orthologs from multiple organisms (e.g., TAL from Flavobacterium johnsoniae vs. other sources). |
| Promoter & Terminator | Controls transcriptional strength and mRNA stability [11]. | Use strong, inducible, or constitutive promoters (e.g., GPD, TEF) and optimized terminators. |
| Gene Copy Number | Influences enzyme expression levels [11]. | Utilize multi-copy plasmids or genomic integration at multiple loci. |
Stage 2: Host Engineering and Pathway Assembly
Objective: To build a robust yeast chassis and assemble the heterologous pathway.
Step 2.1: Host Strain Selection and Engineering
- Select a standard laboratory strain (e.g., CEN.PK, S288c) or an engineered derivative.
- Implement chassis engineering to enhance precursor supply. This often involves:
- Upregulating native pathways: Overexpressing key enzymes in central carbon metabolism (e.g., gluconeogenesis) or specific biosynthetic branches (e.g., the shikimate pathway for aromatic amino acids) [14].
- Deleting competing pathways: Knocking out genes that divert precursors away from the target product (e.g., deleting glycogen debranching enzyme GDB1 to reduce starch catabolism) [14].
- Improving cofactor supply: Engineering systems to regenerate essential cofactors like NADPH or ATP, though native metabolism is often sufficient [14].
Step 2.2: Vector Assembly and Transformation
- Assemble the expression cassettes for the heterologous genes into appropriate yeast vectors (e.g., episomal plasmids (YEp) for high copy number or integration plasmids (YIp) for stability) [11].
- Use advanced DNA assembly techniques (e.g., Gibson Assembly, Golden Gate Shuffling) to construct the pathway.
- Transform the assembled DNA into the engineered yeast host using standard methods (e.g., lithium acetate transformation).
Table 2: Common Genetic Tools for S. cerevisiae
| Tool | Type | Key Features | Best Use Case |
|---|---|---|---|
| YEp (Episomal Plasmid) | Plasmid | High copy number; uses 2µ origin; less stable without selection [11]. | Rapid testing of pathway variants; high-level expression. |
| YIp (Integration Plasmid) | Plasmid | Low copy; stable via chromosomal integration; requires homology [11]. | Creating stable, long-term production strains. |
| CRISPR/Cas9 | Genome Editing Tool | Enables precise gene knock-in, knockout, and mutation [11]. | Host chassis engineering; pathway integration. |
Stage 3: Cultivation and Bioprocess Optimization
Objective: To test the performance of the engineered strain and optimize the production process.
Step 3.1: Small-Scale Screening
- Inoculate transformed strains in deep-well plates or small shake flasks.
- Induce expression of the heterologous pathway under controlled conditions (temperature, inducer concentration).
- Use analytical methods (e.g., HPLC, LC-MS) to quantify the production of the target compound and key intermediates [13]. This helps identify potential rate-limiting steps in the pathway.
Step 3.2: Bioprocess Optimization in Bioreactors
- Scale up the best-performing strain to controlled bioreactors.
- Optimize key process parameters:
- Carbon Source: Test different carbon sources (e.g., glucose, glycerol, acetate) and concentrations. Using electrosynthetic acetate has been demonstrated for sustainable starch production [14].
- Feeding Strategy: Implement fed-batch processes to avoid substrate inhibition and maintain optimal metabolic activity. Fed-batch processes have been crucial for achieving high naringenin titers [12].
- Dissolved Oxygen: Critically important for aerobic processes; optimize aeration and agitation.
- pH and Temperature: Maintain at optimal levels for yeast growth and production.
Stage 4: Analysis and Learning for Iterative Engineering
Objective: To analyze strain performance and identify targets for the next DBTL cycle.
Step 4.1: Metabolite and Pathway Analysis
- Quantify the accumulation of intermediates to pinpoint enzymatic bottlenecks. For instance, in Asperosaponin VI production, tracing intermediates revealed downstream glycosylation and UDP-sugar supply as major limitations [13].
- Use transcriptomics or proteomics to analyze gene expression and protein levels of the heterologous pathway.
Step 4.2: Iterative Strain Engineering
- Based on the analysis, implement further engineering strategies:
- Enzyme Engineering: Improve enzyme kinetics or specificity through directed evolution.
- Fine-tuning Expression: Modulate the expression of bottleneck enzymes using promoters of different strengths or by adjusting gene copy number.
- Compartmentalization: Localize pathway enzymes in specific cellular organelles (e.g., peroxisomes) to improve flux and reduce toxicity [9].
- Cofactor Balancing: Fine-tune the expression of enzymes involved in cofactor regeneration.
- Based on the analysis, implement further engineering strategies:
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Heterologous Pathway Reconstruction in Yeast
| Reagent / Material | Function | Example |
|---|---|---|
| Codon-Optimized Genes | Ensures high translation efficiency in the host; the foundation of hyperexpression systems [11]. | gBlocks (Integrated DNA Technologies) or full gene synthesis services. |
| Yeast Shuttle Vectors | Plasmid-based systems for gene expression and maintenance in both E. coli (for cloning) and S. cerevisiae. | pRS series plasmids with different selectable markers and replication origins. |
| CRISPR/Cas9 System | A versatile genome editing tool for precise host genome modification [11]. | Plasmid expressing Cas9 and a guide RNA (gRNA) specific to the target locus. |
| Metabolic Pathway Enzymes | The heterologous enzymes that constitute the biosynthetic pathway of interest. | TAL, 4CL, CHS, CHI for naringenin production [12]. |
| Analytical Standards | Essential for calibrating instruments and quantifying target products and intermediates in complex broths. | Pure standards of the target molecule (e.g., Naringenin, Asperosaponin VI) [12] [13]. |
Workflow and Pathway Diagrams
The following diagrams, generated using Graphviz, illustrate the core experimental workflow and a generic metabolic pathway for heterologous production.
Within synthetic biology, the selection of an appropriate microbial chassis is paramount for the successful reconstruction of heterologous pathways. While Saccharomyces cerevisiae has long been the conventional model, non-conventional yeasts such as Komagataella phaffii (formerly Pichia pastoris) and Yarrowia lipolytica are emerging as powerful alternatives due to their unique and complementary metabolic capabilities [7] [15]. This application note provides a comparative analysis of these three yeast chassis, framing their distinct advantages within the context of heterologous pathway reconstruction for drug development and bio-manufacturing. We summarize key physiological and genetic characteristics, present standardized protocols for their engineering, and visualize core metabolic pathways to guide researchers in selecting and utilizing the optimal platform for their specific application.
Comparative Analysis of Yeast Chassis
The choice between S. cerevisiae, K. phaffii, and Y. lipolytica hinges on the nature of the target product and the process requirements. Below, we delineate their defining characteristics and optimal use cases.
Table 1: Key Characteristics of Yeast Chassis
| Feature | S. cerevisiae | K. phaffii (P. pastoris) | Y. lipolytica |
|---|---|---|---|
| Primary Application | Bioethanol, pharmaceuticals, model organism [16] [11] | High-yield protein production [7] [17] [15] | Lipids, oleochemicals, hydrophobic substrates [7] [15] |
| Key Strength | Extensive genetic toolbox, GRAS status, well-understood physiology [17] [11] | Strong, inducible promoters (e.g., pAOX1), high cell-density growth, efficient secretion [17] [18] | High flux through acetyl-CoA/Malonyl-CoA, innate lipid accumulation (>30% CDW) [7] [15] |
| Metabolic Mode | Crabtree-positive (mixed acid fermentation) [17] [19] | Crabtree-negative (respiratory) [17] [18] | Crabtree-negative (primarily respiratory) [17] |
| Exemplary Product | β-Farnesene, heterologous enzymes [16] [11] | Hepatitis B vaccine, human insulin, interferon [7] [15] | Carotenoids, omega-3 fatty acids, biofuels [7] [15] |
| Substrate Flexibility | Glucose, sucrose (engineered for xylose, glycerol) [11] [19] | Methanol, glycerol, sorbitol [7] [20] [18] | Fatty acids, waste oils, alkanes, glycerol, lignocellulosic hydrolysates [7] [15] |
| Genetic Tractability | High; CRISPR, in vivo assembly, vast parts library [7] [11] | Moderate; CRISPR, GoldenPiCS system [7] [17] | Moderate; CRISPR, Golden Gate system [7] [17] |
| Secretion Efficiency | Moderate [17] | High [17] [20] | High for native proteases and lipases [17] [20] |
Table 2: Quantitative Performance Comparison for Recombinant Protein Production (Model Protein: Candida antarctica Lipase B, CalB)
| Parameter | S. cerevisiae [17] | K. phaffii [20] | Y. lipolytica [20] |
|---|---|---|---|
| Maximal Biomass (gDCW/L) | ~5-10 (strain/variable dependent) | 4.8 | 10.6 |
| Specific Growth Rate (h⁻¹) | Variable | 0.27 | 0.31 |
| Time to Maximal Production (h) | Variable | ~24 | ~12 |
| Extracellular Lipase Activity | Baseline | 1X (Reference) | >5X |
Application Notes for Heterologous Pathway Reconstruction
Chassis Selection Guide
- For Proteins and Enzymes: K. phaffii is often superior for secreted, high-value proteins due to its strong inducible systems and high secretion capacity, simplifying downstream processing [17] [18]. S. cerevisiae remains a viable option for intracellular eukaryotic proteins, leveraging its extensive toolkit and GRAS status [11].
- For Lipid-Derived Chemicals: Y. lipolytica is the unequivocal choice. Its native metabolism is inherently geared toward acetyl-CoA and malonyl-CoA, the key precursors for fatty acids, terpenoids, and polyketides [7] [16]. Engineering efforts can further amplify this natural flux.
- For Rapid Bioprocess Development: S. cerevisiae offers the shortest design-build-test-learn cycle due to the availability of fully synthetic genomes (Sc2.0), advanced computational models, and the most mature CRISPR tools for multiplexed engineering [11].
- For Utilization of Waste Streams: Both K. phaffii (on methanol-rich waste streams) and Y. lipolytica (on lipid/fatty acid waste) provide sustainable, cost-effective bioprocessing options [7] [15]. Y. lipolytica can directly convert low-cost substrates like crude glycerol and waste oils into valuable products [7].
Critical Engineering Considerations
Successful pathway reconstruction extends beyond chassis selection. Key considerations include:
- Promoter Engineering: Utilize strong, tunable promoters for metabolic balancing. In K. phaffii, pAOX1 is a strong, methanol-inducible workhorse [20] [18]. In Y. lipolytica, hybrid promoters like pEYK1 offer strong induction with erythritol [20]. S. cerevisiae boasts a large library of constitutive and inducible promoters [11].
- Terminator Optimization: Employing strong terminators is crucial for enhancing mRNA stability and overall gene expression levels in all three yeasts [7].
- Secretion Engineering: For secreted products, the choice of signal peptide is critical. The α-mating factor (MF) is common in S. cerevisiae and K. phaffii, while the LIP2 signal is highly effective in Y. lipolytica [17] [20].
- Metabolic Burden Management: For complex pathways, dynamic regulation systems that separate growth from production can prevent metabolic burden and toxicity, enhancing final titers [21].
Experimental Protocols
Protocol: CRISPR-Cas9 Mediated Gene Knock-In inY. lipolytica
This protocol enables precise genomic integration of heterologous expression cassettes [7] [17].
I. Materials
- Y. lipolytica strain (e.g., PO1f)
- Plasmid expressing Cas9 and a customizable sgRNA (e.g., with a Golden Gate cloning site)
- Donor DNA fragment containing the gene of interest flanked by ~500 bp homology arms
- YPD media: 1% Yeast Extract, 2% Peptone, 2% Glucose
- Lithium Acetate (LiOAc) transformation kit
- Selective plates (e.g., YNB without uracil or hygromycin-containing YPD)
II. Procedure
- sgRNA Cloning: Design an sgRNA sequence targeting the desired genomic locus. Clone the annealed oligos into the Cas9/sgRNA plasmid using Golden Gate assembly [17].
- Donor DNA Preparation: Amplify the donor DNA (expression cassette + homology arms) via PCR. Purify the fragment.
- Transformation: a. Grow Y. lipolytica overnight in YPD to mid-exponential phase. b. Harvest cells and prepare competent cells using a LiOAc protocol. c. Co-transform 100-200 ng of the Cas9/sgRNA plasmid and 500 ng-1 µg of the purified donor DNA fragment. d. Plate onto appropriate selective media.
- Screening: a. After 2-3 days of growth at 28-30°C, pick colonies and perform colony PCR to verify correct integration. b. Streak verified positive clones to eliminate the Cas9/sgRNA plasmid.
Protocol: High-Density Fermentation for Protein Production inK. phaffii
This protocol outlines a two-stage process for high-level production of a recombinant protein [20] [18].
I. Materials
- K. phaffii strain (e.g., GS115 or X-33) with expression cassette integrated.
- Buffered Glycerol-Complex Medium (BMGY): 1% yeast extract, 2% peptone, 100 mM potassium phosphate pH 6.0, 1.34% YNB, 4 x 10⁻⁵% biotin, 1% glycerol.
- Buffered Methanol-Complex Medium (BMMY): Same as BMGY, but with 0.5% methanol instead of glycerol.
- Fermenter with dissolved oxygen (DO), pH, and temperature control.
- Methanol feed solution (100% w/v).
II. Procedure
- Biomass Accumulation (Batch Phase): a. Inoculate a shake flask with BMGY and grow for 16-20 hrs at 28-30°C until OD₆₀₀ reaches 2-10. b. Transfer the culture to a fermenter containing basal salts medium with an excess of glycerol. c. Grow while maintaining DO >20-30% via agitation/aeration, and pH at 5.0. Allow glycerol to be depleted (indicated by a sharp DO spike).
- Induction Phase (Fed-Batch Phase): a. Initiate a continuous feed of methanol (possibly mixed with glycerol or sorbitol to maintain cell vitality) [20]. b. The feed rate must be carefully controlled to prevent methanol accumulation (toxic) or starvation. c. Maintain induction for 60-100 hours, monitoring cell density and product titer. d. Harvest culture supernatant or cells for product purification.
Metabolic Pathways and Engineering Workflows
The following diagrams illustrate the core metabolic nodes targeted for reconstructing heterologous pathways in these yeast chassis.
Diagram 1: Core metabolic pathways for product synthesis. Dashed lines connect chassis to their exemplary product categories, highlighting their metabolic predispositions. Abbreviations: MVA, mevalonate; IPP, isopentenyl pyrophosphate; DMAPP, dimethylallyl pyrophosphate; ACC, acetyl-CoA carboxylase.
Diagram 2: Generalized engineering workflows. Workflows diverge based on chassis selection, with S. cerevisiae leveraging its superior in vivo assembly and non-conventional yeasts relying on precise CRISPR-mediated integration.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Yeast Metabolic Engineering
| Reagent / Tool | Function | Exemplar Use Case |
|---|---|---|
| CRISPR-Cas9 System | Enables precise gene knock-out, knock-in, and editing. | Integration of heterologous pathways into a defined genomic locus in Y. lipolytica or K. phaffii [7] [17]. |
| Golden Gate Cloning Kit | Modular, hierarchical assembly of multiple DNA parts into a single vector. | Construction of complex expression cassettes with multiple genes for K. phaffii (GoldenPiCS) or Y. lipolytica [17]. |
| Methanol-Inducible Promoter (pAOX1) | Strong, tightly regulated promoter for high-level expression. | Driving recombinant protein expression in K. phaffii; induction initiated upon glucose/glycerol depletion and methanol addition [20] [18]. |
| Erythritol-Inducible Promoter (pEYK1) | Strong, non-hydrophobic inducer-based promoter system. | Inducing gene expression in Y. lipolytica without the need for oils or fatty acids, simplifying process control [20]. |
| Protease-Deficient Strains | Host strains with knocked-out vacuolar proteases (e.g., pep4, prb1). | Minimizing degradation of secreted recombinant proteins in K. phaffii (e.g., strain SMD1163) and Y. lipolytica [17] [18]. |
| α-Mating Factor (MF) Signal Peptide | Directs secretion of recombinant proteins into the culture medium. | Used in S. cerevisiae and K. phaffii for efficient protein secretion [17] [20]. |
The Role of Computational and Retrosynthetic Algorithms in Pathway Prediction
The engineering of microbial cell factories, particularly the baker's yeast Saccharomyces cerevisiae, for the production of valuable chemicals represents a cornerstone of modern synthetic biology. A critical challenge in this field is the efficient design and reconstruction of heterologous metabolic pathways. Traditional methods for designing these pathways are often time-consuming and labor-intensive, sometimes requiring hundreds of person-years of effort for a single product [22]. The integration of computational and retrosynthetic algorithms has emerged as a transformative approach to accelerate this process. These methods leverage biological big data, sophisticated algorithms, and machine learning to predict viable biosynthetic routes, optimize pathway performance, and integrate heterologous pathways into host metabolism. This Application Note details protocols for employing these computational tools within the context of yeast research, providing a framework for researchers to streamline the development of yeast-based bioproduction platforms.
Fundamental Databases for Pathway Prediction
The effectiveness of any computational pathway prediction tool is contingent on the quality and scope of the underlying biological databases. These resources provide the essential compounds, reactions, and enzymatic data that algorithms use to construct plausible biosynthetic pathways [22]. The tables below categorize essential databases for pathway reconstruction.
Table 1: Essential Compound and Pathway Databases for Biosynthetic Pathway Design
| Data Category | Database Name | Description |
|---|---|---|
| Compound Information | PubChem [22] | Comprehensive information on chemical compounds, their structures, and biological activities. |
| ChEBI [22] | A curated database of small molecular entities focused on chemical biology. | |
| NPAtlas [22] | A curated repository of natural products with annotated structures and sources. | |
| Reaction/Pathway Information | KEGG [22] [23] | A comprehensive database integrating genomic, chemical, and systemic functional information. |
| MetaCyc [22] [23] | A database of metabolic pathways and enzymes from various organisms. | |
| Rhea [22] | A curated database of biochemical reactions with detailed reaction equations. | |
| Enzyme Information | BRENDA [22] | A comprehensive enzyme database providing functional and structural data. |
| UniProt [22] | A central resource for protein sequence and functional information. | |
| AlphaFold Protein Structure DB [22] | A database of highly accurate predicted protein structures. |
Algorithmic Approaches to Pathway Design
Computational methods for pathway design can be broadly classified into several categories, each with distinct strengths. Retrosynthesis algorithms work backwards from a target molecule to identify potential precursor molecules and enzymatic reactions, effectively decomposing the target into simpler, available building blocks [22]. Graph-based approaches model metabolism as a network of reactions (edges) and metabolites (nodes), using search algorithms to find connecting pathways [24]. In contrast, constraint-based methods, such as Stoichiometric Analysis, ensure that the proposed pathways are stoichiometrically feasible when integrated into a genome-scale metabolic model of the host organism (e.g., E. coli or S. cerevisiae) [24]. A powerful emerging trend is the hybrid approach, which combines the strengths of multiple methods. For instance, the SubNetX algorithm combines graph-search capabilities with constraint-based optimization to assemble balanced subnetworks that connect a target biochemical to the host's native metabolism via multiple precursors, enabling the production of complex molecules [24].
Furthermore, machine learning (ML) is playing an increasingly vital role. ML models can predict pathway yields, identify rate-limiting enzymes, and suggest optimal regulatory elements by learning from large biological datasets [25]. They are particularly useful for optimizing multistep pathways and can be integrated into the Design–Build–Test–Learn (DBTL) cycle to accelerate strain development [22] [25].
Application Notes & Protocols
Protocol 1: Computational Prediction of a Biosynthetic Pathway Using a Subnetwork Extraction Approach
This protocol describes the use of a tool like SubNetX to identify and evaluate heterologous pathways for a target molecule in S. cerevisiae [24].
Research Reagent Solutions
Table 2: Key Reagents for Computational Pathway Prediction and Validation
| Reagent / Resource | Function / Explanation |
|---|---|
| Biochemical Reaction Database (e.g., ARBRE, ATLASx) [24] | Provides the network of known and predicted balanced biochemical reactions from which pathways are extracted. |
| Genome-Scale Metabolic Model (GEM) | A computational representation of the host organism's metabolism (e.g., a yeast GEM) used to validate stoichiometric feasibility. |
| Mixed-Integer Linear Programming (MILP) Solver [24] | An optimization algorithm used to identify the minimal set of heterologous reactions (feasible pathway) from the extracted subnetwork. |
| Cheminformatics Tools | Used to calculate properties like Synthetic Accessibility (SA) to assess the complexity of target molecules [24]. |
Step-by-Step Procedure
Reaction Network Preparation: Define the input parameters.
- Select a database of elementally balanced biochemical reactions (e.g., ARBRE for aromatic compounds or ATLASx for a broader scope).
- Define the target compound (e.g., a pharmaceutical intermediate like scopolamine).
- Define the set of precursor metabolites native to S. cerevisiae (e.g., glucose, pyruvate, acetyl-CoA).
Graph Search for Linear Core Pathways: Execute a graph search to find all possible linear reaction paths from the defined precursor compounds to the target molecule.
Subnetwork Expansion and Extraction: The algorithm automatically expands the linear pathways into a balanced subnetwork. This critical step links essential cosubstrates and cofactors (e.g., ATP, NADPH) to the host's native metabolism, ensuring the pathway is not only connected but also thermodynamically and stoichiometrically feasible.
Host Integration: Integrate the extracted balanced subnetwork into a genome-scale metabolic model of S. cerevisiae. This models the pathway within the context of the entire cellular metabolic network.
Pathway Identification and Ranking:
- Use a MILP algorithm to find all minimal sets of reactions ("feasible pathways") within the subnetwork that enable production of the target.
- Rank these feasible pathways based on user-defined criteria such as:
- Theoretical Yield: Maximize the mass of product per mass of substrate.
- Pathway Length: Minimize the number of heterologous enzymatic steps.
- Enzyme Specificity: Prioritize reactions with known, highly specific enzymes.
- Thermodynamic Feasibility: Prefer pathways with favorable Gibbs free energy changes [24].
The following diagram illustrates the core computational workflow of this protocol:
Protocol 2: Experimental Validation and Optimization in Yeast
Once a pathway is predicted computationally, it must be built and tested in a yeast host. This protocol covers key steps for experimental implementation and optimization.
Research Reagent Solutions
Table 3: Key Reagents for Yeast Pathway Engineering
| Reagent / Resource | Function / Explanation |
|---|---|
| Constitutive Promoters (e.g., TDH3P, TEF1P) [26] | Strong, steady-state promoters used to drive the expression of heterologous enzymes. Performance must be tested under intended conditions. |
| Chaperone Overexpression Library [27] | A collection of yeast strains overexpressing cytosolic chaperones (e.g., YDJ1, SSA1) to improve the folding and activity of heterologous pathway enzymes. |
| CRISPR/Cas9 System for S. cerevisiae [26] | Enables precise genomic integration of heterologous expression cassettes. |
| Aerobic/Micro-aerobic Cultivation Systems [26] | Essential for testing pathway performance under different physiological conditions relevant to industrial scale-up. |
Step-by-Step Procedure
Promoter Selection and Vector Construction:
- Select strong constitutive promoters (e.g.,
TDH3P,TEF1P) and terminators (e.g.,DIT1T,CYC1) for constructing heterologous gene expression cassettes [26] [27]. - Critical Note: Promoter performance is unpredictable and highly dependent on the specific gene, protein, and cultivation conditions. It is essential to test several promoter-gene combinations empirically [26].
- Assemble expression cassettes for all heterologous genes in the pathway using yeast integration vectors or direct genomic integration tools like CRISPR/Cas9.
- Select strong constitutive promoters (e.g.,
Strain Engineering:
- Integrate the expression cassettes into well-characterized genomic loci (e.g.,
X-2,X-4,XII-5) in your S. cerevisiae host strain to ensure stable and comparable expression levels [27]. - If the pathway requires non-native cofactors or the metabolism of non-native sugars (e.g., xylose), engineer the host background accordingly [26].
- Integrate the expression cassettes into well-characterized genomic loci (e.g.,
Chaperone Co-expression Screening:
- To address potential issues with improper folding of heterologous enzymes, employ a chaperone overexpression library.
- Use a mating-based strategy to cross your engineered "query" strain (containing the heterologous pathway) with an arrayed library of strains overexpressing different cytosolic chaperones (e.g., HSP40, HSP70, HSP90) [27].
- Screen the resulting diploid strains for improved product titers. For the model compound aspulvinone E, the combined overexpression of chaperones
YDJ1andSSA1increased production by 84% [27].
Pathway Validation and Fermentation:
- Cultivate the engineered strains in relevant media, including defined media with the target carbon source (e.g., glucose, xylose, or lignocellulosic hydrolysates).
- Quantify pathway performance through enzymatic assays, measurement of intermediate metabolites, and final product titer, rate, and yield [26].
- Validate performance under both laboratory conditions and industrially relevant setups like consolidated bioprocessing (CBP) [26].
The experimental workflow for chassis engineering and validation is summarized below:
The integration of computational and retrosynthetic algorithms has fundamentally transformed the paradigm of heterologous pathway reconstruction in yeast. By leveraging vast biological databases, sophisticated pathway prediction tools like SubNetX, and machine learning for optimization, researchers can now move beyond simple linear pathways to design complex, balanced metabolic networks for the production of valuable and complex chemicals. The subsequent experimental protocols for promoter engineering, chaperone co-expression, and physiological validation provide a robust framework for translating these computational predictions into efficient yeast cell factories. This combined computational-experimental approach significantly accelerates the DBTL cycle, paving the way for more sustainable and efficient biomanufacturing processes.
The reconstruction of heterologous pathways in yeast represents a cornerstone of modern biotechnology, enabling the production of complex pharmaceuticals, industrial enzymes, and sustainable food proteins. Saccharomyces cerevisiae and other non-conventional yeasts have emerged as preferred chassis organisms due to their unique combination of eukaryotic processing capabilities and Generally Recognized as Safe (GRAS) status [11]. This designation, formalized by the U.S. Food and Drug Administration (FDA), signifies that these microorganisms are safe for use in pharmaceutical and food production, significantly streamlining regulatory approval pathways [28] [11]. The convergence of these attributes—eukaryotic machinery for proper protein processing and a validated safety profile—makes yeast systems indispensable for research and industrial applications requiring heterologous pathway engineering.
Yeasts offer distinctive advantages over both bacterial and mammalian expression systems. Unlike prokaryotic hosts such as E. coli, yeasts possess the subcellular machinery to perform eukaryotic post-translational modifications including glycosylation, disulfide bond formation, and proper protein folding, which are often essential for the biological activity of therapeutic proteins [11] [8]. Simultaneously, they avoid the technical complexities, high costs, and viral contamination risks associated with mammalian cell cultures while offering rapid growth on inexpensive media [29] [11]. Furthermore, yeast systems are highly amenable to genetic manipulation using a vast toolbox of molecular biology techniques, facilitating the precise engineering needed for heterologous pathway reconstruction [30].
Core Advantages of Yeast Expression Platforms
Eukaryotic Protein Processing Machinery
The capacity of yeast to correctly process and modify eukaryotic proteins is its most significant advantage for heterologous expression.
- Post-Translational Modifications: Yeast systems perform essential eukaryotic modifications such as proteolytic processing of signal peptides, protein folding facilitated by chaperones, formation of disulfide bonds, and initial N-linked glycosylation [11] [8]. These processes are crucial for the structural integrity, stability, and biological activity of complex proteins, including mammalian membrane receptors and secreted enzymes.
- Secretion and Purification: The eukaryotic secretory pathway in yeast allows for targeted secretion of heterologous proteins into the extracellular medium. This capability dramatically simplifies downstream purification processes, reducing costs and increasing yields for industrial-scale production [11]. Secretion also minimizes intracellular proteolytic degradation and facilitates the formation of disulfide bonds in the oxidizing environment of the endoplasmic reticulum.
- Membrane Protein Targeting: For structurally and functionally complex plant and mammalian membrane proteins—which constitute 20–30% of all eukaryotic genes—yeast provides the necessary machinery for proper integration into lipid bilayers and assembly into functional complexes [8]. This makes yeast an invaluable system for studying transporters, channels, and G-protein coupled receptors (GPCRs).
GRAS Status and Regulatory Acceptance
The GRAS status of S. cerevisiae and several non-conventional yeasts provides a significant regulatory and commercial advantage.
- Streamlined Product Approval: Products manufactured in GRAS-certified yeast hosts, such as many therapeutic proteins and food ingredients, benefit from more straightforward regulatory pathways. The FDA and European Medicines Agency (EMEA) have approved numerous recombinant protein products from yeast, with some processes even exempt from specific viral detection requirements based on risk assessment [11].
- Versatility Across Industries: The GRAS designation enables the use of the same yeast platform across diverse sectors, including pharmaceuticals, food, and cosmetics. Recent FDA updates for Q3 2025 confirm continued strong submission rates for yeast-related GRAS notices, particularly for microbial proteins and enzymes used in food applications [28].
- Industrial Robustness: Yeasts exhibit high tolerance to fermentation inhibitors, low pH, and industrial-scale processing conditions. Their general resilience lowers contamination risks and improves production consistency, making them ideal for large-scale manufacturing [29].
Table 1: Representative Therapeutic Proteins Produced in Yeast Systems
| Host Yeast | Therapeutic Protein | Reported Yield | Application |
|---|---|---|---|
| Pichia pastoris | Insulin | 3 g/L (insulin precursor) | Diabetes Treatment |
| S. cerevisiae | IFNα2b | 15 mg/L | Antiviral Therapy |
| Yarrowia lipolytica | IFNα2b | 425 mg/L | Antiviral Therapy |
| P. pastoris | Hepatitis B antigen | 7 g/L | Vaccine |
| H. polymorpha | HBV surface antigen | 250 mg/L | Vaccine |
| Kluyveromyces lactis | Human interferon β | Not specified | Antiviral Therapy |
| P. pastoris | Human serum albumin | 92.29 mg/L | Blood Volume Expander |
Engineering Strategies for Enhanced Protein Production
Genetic Toolbox for Pathway Engineering
Advanced genetic tools enable precise manipulation of yeast chassis strains to optimize heterologous pathway performance and protein yields.
Promoter Engineering: Robust, tunable promoters are critical for controlling heterologous gene expression. Advanced strategies include:
- Synthetic Promoter Libraries: Constructed via saturation mutagenesis of core promoter elements (e.g., PTDH3, PZEV), generating minimal promoters (20-30 bp) achieving up to 70% of native promoter strength [29].
- Inducible Systems: Methanol-inducible AOX1 promoter in P. pastoris enables high-level protein production (up to 22 g/L intracellular, 15 g/L extracellular). Engineered variants allow derepression upon glucose exhaustion, eliminating the need for methanol [29].
- Computational Design: Machine learning and convolutional neural networks (CNNs) predict promoter strength from sequence, enabling rational design of optimized synthetic promoters [29] [11].
CRISPR/Cas-Mediated Genome Editing: The CRISPR/Cas system has revolutionized yeast metabolic engineering by enabling rapid, multiplexed gene knockouts, knock-ins, and transcriptional regulation [30]. Applications include:
- Multiplexed Engineering: Simultaneous targeting of multiple genes to reconstruct complex pathways or eliminate competing reactions.
- Genome-Wide Screens: CRISPR-based libraries (e.g., CHAnGE method) enable high-throughput screening for improved phenotypes like stress tolerance [30].
- Transcriptional Control: Catalytically dead Cas9 (dCas9) fused to regulatory domains enables precise activation or repression of endogenous and heterologous genes [30].
Figure 1: Key Engineering Strategies for Optimizing Yeast Hosts. The diagram summarizes four major engineering approaches to enhance heterologous protein production and functionality in yeast systems.
Secretory Pathway and Glycosylation Engineering
Enhancing protein secretion and achieving human-compatible glycosylation are critical for producing functional biotherapeutics.
Secretory Pathway Optimization: Engineering the secretion machinery can dramatically increase yields of extracellular proteins.
- Signal Peptide Screening: Testing heterologous vs. native yeast signal peptides (e.g., from SUC2, MFα1) to identify optimal sequences for directing target proteins through the secretory pathway [11].
- Chaperone Co-expression: Overexpressing endoplasmic reticulum (ER) chaperones (e.g., BiP/Kar2p, PDI) to improve folding efficiency and prevent ER-associated degradation (ERAD) of complex proteins [11].
- Vesicular Trafficking Enhancement: Modulating genes involved in ER-to-Golgi transport and Golgi function to alleviate secretion bottlenecks [11].
Humanized Glycosylation Pathways: Native yeast glycosylation produces high-mannose structures potentially immunogenic in humans. Glycoengineering creates humanized yeast strains capable of synthesizing complex human N-glycans.
- Disruption of Yeast-Specific Glycosylation: Knockout of genes encoding α-1,6-mannosyltransferase (OCH1) and other enzymes responsible for hypermannosylation [29] [11].
- Introduction of Human Glycosylation Machinery: Heterologous expression of human glycosyltransferases (e.g., β-1,4-galactosyltransferase, sialyltransferases) to reconstruct human glycosylation pathways in yeast [11].
- Subcellular Relocalization: Targeting heterologous enzymes to specific compartments (e.g., Golgi) to ensure proper function within the glycosylation pathway [29].
Experimental Protocols and Workflows
Protocol: High-Throughput Yeast Strain Screening on Solid Media
This protocol enables efficient pre-screening of diverse yeast libraries under industrially relevant conditions [31].
Materials & Reagents:
- Yeast Library: Includes conventional (S. cerevisiae) and non-conventional yeasts (e.g., Kluyveromyces lactis, Yarrowia lipolytica).
- Solidified Industrial Media: Wort, Malt Extract, Synthetic Apple Juice with 2% agar.
- Automated Equipment: PIXL colony picker robot, ROTOR HDA replicator, PhenoBooth imaging system.
Procedure:
- Strain Revival: Revive yeast strains from -80°C storage on YPD agar plates at 30°C until colonies form.
- Array Generation: Use robotic platform to re-array strains in high-density formats (96, 384, or 1536 colonies per plate) onto YPD agar.
- Replication: Replicate source arrays onto solidified industrial media (wort, malt extract, apple juice) in triplicate using the ROTOR HDA.
- Incubation & Imaging: Incubate plates at relevant temperatures (e.g., 30°C) for 48-120 hours. Capture colony images automatically every ≈6 hours using the PhenoBooth.
- Data Analysis: Use imaging software to measure colony size (pixels) over time. Calculate fitness values based on maximum growth rate and biomass.
Applications: This method is ideal for initial hit generation from vast yeast libraries, identifying strains with superior growth under target fermentation conditions (e.g., beer, cider production) before moving to more resource-intensive liquid fermentation studies [31].
Protocol: Morphological Profiling for Target Identification
This protocol uses high-throughput microscopy and image analysis to predict intracellular targets of bioactive compounds in yeast [32].
Materials & Reagents:
- Drug-Hypersensitive Yeast Strain: Triple-deletion mutant (pdr1Δ pdr3Δ snq2Δ) with enhanced compound sensitivity.
- Yeast Mutant Library: ~1982 gene-deletion strains in the drug-hypersensitive background.
- Staining Solutions: Cell wall, actin, and nuclear DNA stains.
- HT Microscopy: Automated microscope with image capture capability.
- Image Analysis Software: CalMorph for morphological feature extraction.
Procedure:
- Chemical Treatment: Expose drug-hypersensitive yeast strain to dose gradients of target compounds in 96-well format.
- Staining and Imaging: Triple-stain treated cells for cell wall, actin, and DNA. Automatically capture high-resolution images using HT microscope.
- Morphological Feature Extraction: Process images with CalMorph to quantify 501 morphological traits (size, shape, intensity, spatial relationships).
- Data Integration & Modeling: Compare chemical-induced morphological profiles with those of gene-deletion mutants using a generalized linear model (GLM). Calculate correlation coefficients of Principal Component (PC) scores.
- Target Prediction: Identify gene deletions with morphological profiles most similar to chemical treatment, indicating potential functional targets.
Applications: Mechanism of action studies for novel bioactive compounds, antifungal drug discovery, and functional genomics research [32].
Figure 2: Integrated Workflow for Yeast Strain Screening and Development. The process begins with library preparation and progresses through automated screening, data analysis, and final validation to identify and characterize superior production strains.
The Scientist's Toolkit: Essential Research Reagents and Solutions
Table 2: Key Research Reagents for Yeast Heterologous Pathway Engineering
| Reagent / Tool Category | Specific Examples | Function & Application |
|---|---|---|
| Expression Vectors | YIp (integrating), YEp (episomal), YCp (centromeric) plasmids [8] | Shuttle vectors for gene expression in yeast and E. coli; differ in copy number and stability for various expression needs. |
| Selection Markers | URA3, LEU2, HIS3 [8] | Auxotrophic markers for selection of transformants on minimal media; essential for strain engineering and plasmid maintenance. |
| Promoter Systems | Constitutive: PGPD, PTEF1; Inducible: GAL1, AOX1 (P. pastoris) [29] [11] | Drive transcription of heterologous genes; inducible systems allow temporal control to mitigate toxicity during growth. |
| CRISPR Tools | Cas9 nucleases, sgRNA libraries, dCas9 fusion proteins [30] | Enable precise genome editing, knockout libraries, and transcriptional regulation for metabolic engineering. |
| Specialized Strains | Drug-hypersensitive (e.g., pdr1Δ pdr3Δ snq2Δ) [32]; Glyco-engineered strains [11] | Host backgrounds that enhance compound sensitivity or perform human-like protein glycosylation. |
| HTS Platforms | PIXL colony picker, ROTOR HDA replicator, PhenoBooth imager [31] [32] | Automated systems for high-density arraying, replication, and image-based screening of yeast libraries. |
The synergistic combination of eukaryotic processing machinery and GRAS status establishes yeast systems as powerful platforms for heterologous pathway reconstruction. These advantages translate directly into diverse real-world applications:
- Biopharmaceutical Production: Yeast systems produce commercially approved therapeutics including insulin, hepatitis vaccines, and virus-like particles, with the global therapeutic protein market representing approximately 25% of commercial pharmaceuticals [29] [11].
- Sustainable Food Ingredients: Precision fermentation in yeast generates sustainable proteins (e.g., Angel Yeast's AngeoPro with 80% protein content), dairy substitutes, and egg alternatives with significantly reduced environmental footprints compared to traditional agriculture [33] [34].
- Industrial Enzymes: Yeast-based production of enzymes (lipases, invertases, chymosin) for food processing, biofuels, and bioremediation represents a multi-billion dollar market growing at 4% annually [28] [11].
Future advancements will likely focus on enhancing yeast systems through more sophisticated engineering approaches. These include further humanization of glycosylation pathways, engineering of artificial organelles for compartmentalized biosynthesis, and the application of machine learning to predict optimal genetic configurations for heterologous pathway flux [29] [11]. As synthetic biology tools continue to evolve, particularly with the completion of the fully synthetic yeast genome (Sc2.0), the capabilities of yeast as programmable chassis for heterologous pathway reconstruction will expand further, solidifying their role as indispensable tools in both basic research and industrial biotechnology [11].
The Molecular Toolkit: Implementation and Application of Pathways in Yeast
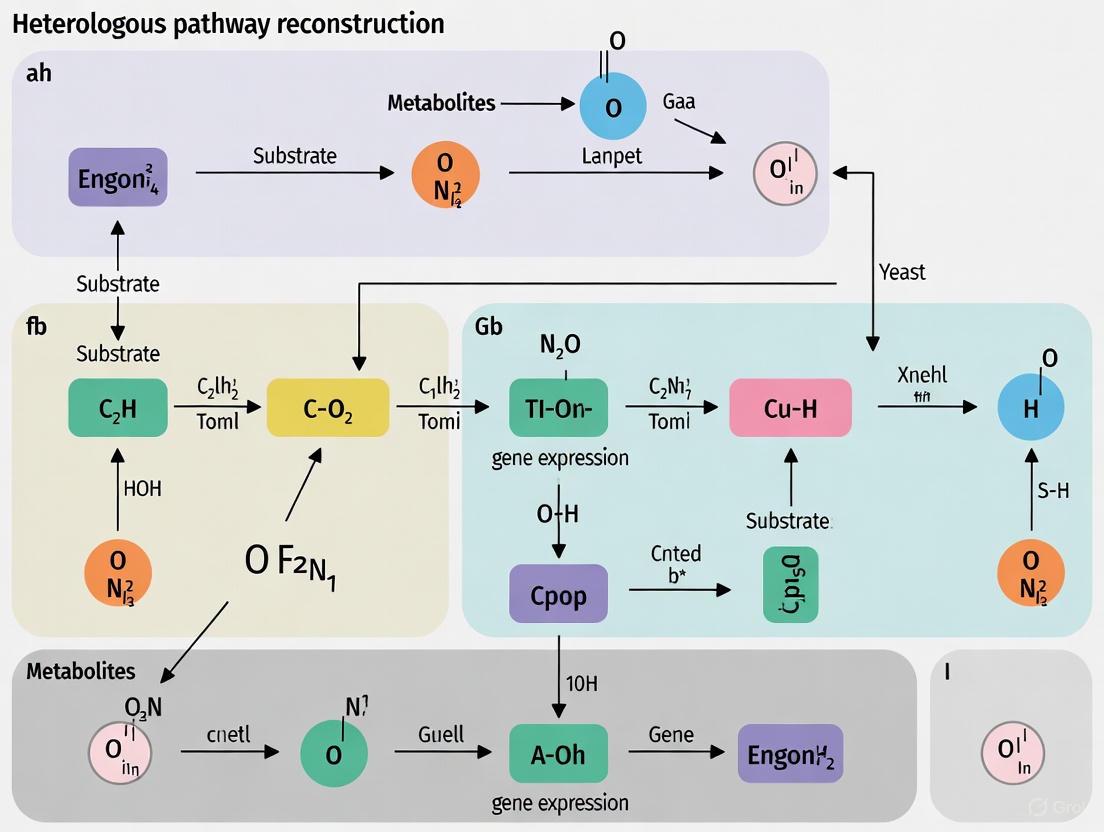
CRISPR-Cas9 and Advanced Genome-Editing Tools for Precision Engineering
The reconstruction of heterologous biosynthetic pathways in yeast represents a cornerstone of modern metabolic engineering and synthetic biology. CRISPR-Cas9 technology has revolutionized this field by enabling precise, efficient, and programmable manipulation of yeast genomes. This capability is crucial for inserting foreign genetic material and optimizing native metabolic networks to convert yeast into microbial cell factories for producing valuable chemicals, pharmaceuticals, and biofuels [35]. The budding yeast Saccharomyces cerevisiae is particularly valued for this work due to its efficient homology-directed repair (HDR) system, well-characterized genetics, and status as a generally recognized as safe (GRAS) organism [36] [37]. This protocol details the application of CRISPR-Cas9 and advanced editing tools specifically for heterologous pathway reconstruction in yeast, providing both foundational methods and cutting-edge approaches to address current challenges in precision genome engineering.
Foundational CRISPR-Cas9 Genome Editing Protocol
Mechanism and Core Components
The Type II CRISPR-Cas9 system from Streptococcus pyogenes functions as a RNA-guided DNA endonuclease. The system creates double-strand breaks (DSBs) 3 base pairs upstream of the protospacer adjacent motif (PAM: 5'-NGG-3') through the coordinated activity of two catalytic domains: the HNH domain cleaves the DNA strand complementary to the 20-nucleotide spacer sequence in the guide RNA (gRNA), while the RuvC-like domain cleaves the opposite strand [37]. In S. cerevisiae, these DSBs are predominantly repaired via homology-directed repair when a donor DNA template is provided, enabling precise integration of heterologous genes [37] [35].
Step-by-Step Experimental Protocol
gRNA Design and Expression Cassette Construction
- Target Identification: Select genomic integration sites that support high expression of heterologous genes. Common targets include intergenic regions, TEF loci, or rDNA regions.
- gRNA Design: Design 20-nucleotide spacer sequences targeting your selected site using the following criteria:
- Must be adjacent to a 5'-NGG PAM sequence on the genomic DNA
- Avoid targets with significant off-site matches (BLAST against yeast genome)
- For multiplex editing, design unique gRNAs for each target locus
- Expression Cassette Assembly:
Donor DNA Design and Construction
- Homology Arm Design: Flank your heterologous gene with 40-60 bp homology arms complementary to the genomic target site.
- Modular Assembly: For pathway reconstruction, design multiple donor DNAs with compatible overhangs for Golden Gate assembly or in vivo homologous recombination.
- Marker Selection: Include selection markers (e.g., antibiotic resistance, auxotrophic markers) for initial screening, preferably flanked by loxP or FRT sites for subsequent excision [36] [38].
Yeast Transformation and Screening
- Transformation:
- Co-transform yeast with: (1) Cas9 expression plasmid, (2) gRNA expression construct, and (3) donor DNA fragment(s)
- Use standard lithium acetate/single-stranded carrier DNA/PEG method
- Plate on appropriate selective media and incubate at 30°C for 2-3 days
- Screening:
- Patch colonies onto fresh selective plates
- Perform colony PCR with verification primers spanning integration junctions
- Confirm integration by sequencing and phenotypic assessment
Optimization Notes
- For non-conventional yeasts with predominant non-homologous end joining (NHEJ), delete KU70/KU80 genes to enhance HDR efficiency [39] [35]
- Use Cas9D147Y,P411T (iCas9) variants to improve editing efficiency in challenging strains [39]
- For Yarrowia lipolytica, employ the SCR1-tRNA promoter for gRNA expression, achieving up to 92.5% gene disruption efficiency [39]
Advanced Genome Editing Applications
Multiplex Editing for Pathway Reconstruction
Multiplex CRISPR-Cas9 editing enables simultaneous integration of multiple heterologous genes, dramatically accelerating reconstruction of complex metabolic pathways. The following workflow illustrates this process:
Protocol: Multiplex Gene Integration
gRNA Array Construction:
- Design 2-8 gRNAs targeting distinct genomic loci
- Assemble as a tandem array with tRNA spacers using Golden Gate assembly
- Clone into a high-copy plasmid under Pol III promoter
Donor DNA Preparation:
- Prepare linear donor DNAs for each heterologous gene with 40-60 bp homology arms
- Include different selectable markers for each integration or use recyclable marker systems
Transformation and Screening:
CRISPRi for Metabolic Flux Optimization
CRISPR interference (CRISPRi) using catalytically dead Cas9 (dCas9) enables precise metabolic flux control without permanent genetic alterations. The system can be dynamically regulated using engineered gRNA switches:
Protocol: Implementing CRISPRi with Switchable gRNAs
dCas9 Expression System:
- Express dCas9 fused to transcriptional repressor domains (e.g., Mxi1) under a constitutive promoter
- For yeast, codon-optimize dCas9 and include nuclear localization signals
Switchable gRNA Design:
- Engineer gRNAs with 5' or 3' extensions that block function until removed
- Incorporate ribozyme aptazymes that cleave in response to small molecules
- Implement toehold switches for mRNA-triggered activation [40]
Application for Pathway Optimization:
- Target dCas9-gRNA complexes to native genes that compete with heterologous pathway
- Implement dynamic control by linking gRNA activation to metabolic intermediates
- Use multi-input logic gates for sophisticated regulation of metabolic fluxes [40]
Quantitative Performance Data
Editing Efficiency Across Yeast Species
Table 1: CRISPR-Cas9 Performance Metrics in Different Yeast Hosts
| Yeast Species | Editing Type | Efficiency | Key Optimization Factors | Primary Applications |
|---|---|---|---|---|
| Saccharomyces cerevisiae | Gene disruption | 92.5% [39] | tRNA-sgRNA architecture | Multiplex pathway integration [37] |
| Gene integration | 82.7% correct edit rate [41] | MAGESTIC system with donor enrichment | Single nucleotide variants | |
| Yarrowia lipolytica | Gene disruption | 92.5% [39] | SCR1-tRNA promoter, KU70 deletion | Lipid metabolic engineering |
| Gene integration | Variable (NHEJ-dominated) | KU70 deletion, Rad52/Sae2 overexpression | Industrial chemical production | |
| Candida auris | Allele editing | 41.9% (plasmid-based) [38] | EPIC system with CpARS7 replicon | Functional genetics of pathogenicity |
| Integration-based editing | Unreliable [38] | Ectopic integration issues | - | |
| Multiple species | Base editing | ~20% (RNA editing) [35] | dCas13a-hADAR2d fusion | Transcript knockdown |
Structural Variant Risk Assessment
Table 2: Unintended Editing Outcomes and Mitigation Strategies
| Outcome Type | Frequency | Genomic Context | Prevention Strategy |
|---|---|---|---|
| Small indels (NHEJ) | 0.59% in S. cerevisiae [41] | All target sites | Enhance HDR with donor overexpression |
| Structural variants (large deletions) | 4.9% overall, up to 7% in high-coverage sites [41] | Repetitive regions, SV hotspots | Use SCORE prediction tool to identify risk regions [41] |
| Non-reciprocal translocations | 2.3% of edited clones [41] | Distal repetitive sequences | Avoid targets with significant homology elsewhere in genome |
| Off-target indels | Virtually nonexistent in S. cerevisiae [41] | Sites with 1-2 mismatches to gRNA | Design gRNAs with minimal off-target potential |
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Reagents for CRISPR Genome Editing in Yeast
| Reagent Category | Specific Examples | Function | Application Notes |
|---|---|---|---|
| Cas9 Variants | SpCas9, iCas9 (Cas9D147Y,P411T) [39] | DNA cleavage for DSB induction | iCas9 shows enhanced efficiency in Y. lipolytica |
| gRNA Expression Systems | SNR52 promoter, SCR1-tRNA, tRNA-sgRNA arrays [39] | Target specification and expression | SCR1-tRNA optimal for non-conventional yeasts |
| Donor DNA Templates | Linear dsDNA with homology arms, plasmid donors | HDR template for precise editing | 40-60 bp homology arms sufficient for S. cerevisiae |
| Selection Systems | Antibiotic resistance (Nourseothricin), auxotrophic markers (URA3, LEU2, HIS1) | Identification of successful transformants | Recyclable markers (loxP/FRT) enable iterative editing [36] [38] |
| Modulation Tools | Cre-loxP, FLP-FRT, serine integrases (φBT1, R4, BXB1, φC31) [36] | Marker recycling, sequence excision | Serine integrases offer higher efficiency than Cre-loxP |
| Advanced Editors | Base editors (Target-AID), prime editors (PE_Y18) [35] | Precise nucleotide changes without DSBs | Base editing successful for SPT15 evolution in S. cerevisiae |
The CRISPR-Cas9 toolkit for yeast precision engineering has evolved far beyond simple gene knockouts, now enabling sophisticated genome rewriting for heterologous pathway reconstruction. The protocols detailed here provide a foundation for implementing these technologies, from basic gene integration to advanced multiplexing and dynamic regulation. Future directions include the application of AI-designed editors like OpenCRISPR-1, which despite being 400 mutations away from natural Cas9, show comparable or improved activity and specificity [42], and the continued refinement of base and prime editing systems for nucleotide-precise modifications without double-strand breaks [35]. As these tools mature, they will further accelerate yeast metabolic engineering for sustainable bioproduction of pharmaceuticals, chemicals, and fuels.
Vector Systems and Strategies for Increasing Gene Copy Number
For metabolic engineers aiming to reconstruct heterologous pathways in yeast, achieving high-level expression of foreign genes is a fundamental challenge. The copy number of a gene within a cell is a primary determinant of transcriptional output and, consequently, the flux through engineered metabolic pathways [4]. While Saccharomyces cerevisiae remains a prominent host, non-conventional yeasts like Yarrowia lipolytica, Pichia pastoris (Komagataella phaffii), and Kluyveromyces marxianus are increasingly valued for their robust physiology, ability to utilize low-cost carbon sources, and innate high-capacity metabolisms [7]. This application note, framed within the context of heterologous pathway reconstruction, details current vector systems and methodologies for enhancing gene copy number. It provides actionable protocols and a structured analysis of key parameters to guide researchers and scientists in drug development and industrial biotechnology.
Strategic Approaches to Enhance Gene Copy Number
Several strategies can be employed to increase the dosage of a target gene in yeast. The choice of strategy depends on the specific host, the desired stability of expression, and the experimental timeline. The table below summarizes the core strategic approaches.
Table 1: Core Strategies for Increasing Gene Copy Number in Yeast
| Strategy | Underlying Principle | Key Features | Ideal Use Case |
|---|---|---|---|
| High-Copy Plasmid Vectors | Engineering the plasmid's origin of replication (ORI) to increase its copy number per cell [43]. | - Rapid testing and transient expression.- Can be burdensome to the host.- Potential instability without selective pressure. | Pathway prototyping and initial gene function validation. |
| Genomic Integration (Multi-Copy) | Targeted or random insertion of multiple gene copies into the host genome. | - Enhanced genetic stability without antibiotic selection.- Requires efficient DNA delivery and integration tools.- Copy number can be variable. | Creating stable production strains for long-term fermentation. |
| Directed Evolution of ORIs | Using high-throughput growth-coupled selection to identify mutations in the origin of replication that lead to higher plasmid copy number [43]. | - Can significantly boost copy number and transformation efficiency.- Provides a deployable framework for diverse hosts.- Is an advanced molecular biology technique. | Optimizing binary vectors for Agrobacterium-mediated transformation or specific yeast hosts. |
| Adaptive Laboratory Evolution (ALE) | Non-GMO method involving iterative growth and selection under a selective pressure that rewards a desired phenotype [44]. | - Can rewire complex fitness-related phenotypes.- Does not require prior knowledge of genetic basis.- Can be time-consuming. | Improving complex, polygenic traits like overall pathway performance and host fitness. |
The following diagram illustrates the strategic decision-making workflow for selecting and implementing these approaches to optimize heterologous pathway expression.
Detailed Experimental Protocols
Protocol: Engineering High-Copy Number Plasmid Vectors
This protocol outlines a method for increasing plasmid copy number by mutating the replication initiator protein, RepA, based on a directed evolution pipeline successfully used to improve Agrobacterium-mediated transformation [43].
Principle
Plasmid copy number is regulated by the Rep protein and its interaction with the origin of vegetative replication (oriV). Mutations that weaken RepA dimerization on the oriV can reduce replication inhibition, leading to a higher final plasmid copy number [43].
Materials
- Plasmid Backbone: Contains the target Origin of Replication (ORI: e.g., pVS1, RK2, pSa, BBR1) and a bacterial selectable marker.
- Error-Prone PCR (epPCR) Kit: To randomly mutagenize the repA open reading frame (ORF).
- Host Strain: E. coli for library construction and Agrobacterium tumefaciens C58C1 (or target yeast) for selection.
- Growth Media: LB with appropriate antibiotics. Use a range of antibiotic concentrations for selection.
Procedure
- Amplify and Mutagenize: Design primers to amplify the entire repA ORF. Perform error-prone PCR to create a library of mutagenized repA sequences.
- Library Construction: Clone the mutagenized repA pool back into the plasmid backbone, replacing the wild-type sequence.
- Transformation and Pooling: Transform the library into the host strain (e.g., A. tumefaciens C58C1) and pool ~100,000 colonies to create the mutant library.
- Growth-Coupled Selection: Grow the pooled library under wild-type-lethal conditions, typically a high concentration of an antibiotic to which resistance is encoded on the plasmid. Higher-copy-number plasmids confer greater antibiotic resistance [43].
- Enrichment and Sequencing: Isolate the plasmid population from surviving cells. Sequence the repA gene using Illumina MiSeq to identify significantly enriched mutations.
- Validation: Clone identified mutant repA sequences into fresh vectors and quantitatively measure the resulting plasmid copy number (e.g., by qPCR) and transformation efficiency in the target host.
Protocol: Multi-Copy Genomic Integration via rDNA Targeting
This protocol describes a method for integrating multiple copies of an expression cassette into the highly repetitive ribosomal DNA (rDNA) locus of the yeast genome, a well-established strategy for achieving stable, high-copy expression.
Principle
The rDNA region is present in hundreds of tandem repeats in the yeast genome. An integration vector containing a segment of the rDNA sequence can undergo homologous recombination into this locus. Selection for a marker on the vector, followed by counter-selection, can lead to the amplification of the integrated cassette, resulting in strains with dozens of copies [4].
Materials
- Integration Vector: Contains a yeast selectable marker (e.g., URA3), the target heterologous gene, and a ~1-2 kb fragment of the yeast 25S or 18S rDNA sequence.
- Yeast Strain: An auxotrophic strain (e.g., ura3-) suitable for selection.
- Transformation Reagents: PEG/LiAc method or electroporation.
- Selection Media: Synthetic Defined (SD) media lacking uracil and SD media containing 5-Fluoroorotic Acid (5-FOA).
Procedure
- Linearize Vector: Digest the integration vector within the rDNA fragment to stimulate homologous recombination.
- Yeast Transformation: Transform the linearized vector into the host yeast strain and plate onto SD -Ura plates to select for successful integration events.
- Initial Clone Selection: Pick and culture several transformants.
- Counter-Selection for Amplification: Plate the cultures onto SD plates containing 5-FOA. 5-FOA is toxic to cells expressing the URA3 gene. Surviving colonies often have the URA3 marker "looped out" via homologous recombination between rDNA repeats, which can sometimes lead to the amplification of the remaining, non-excised integrated cassette.
- Copy Number Verification: Screen 5-FOA resistant colonies for the presence and copy number of the heterologous gene using quantitative PCR (qPCR) or Southern blotting.
Table 2: Troubleshooting Guide for Multi-Copy Integration
| Problem | Potential Cause | Suggested Solution |
|---|---|---|
| Low transformation efficiency | Inefficient DNA delivery or linearization. | Optimize transformation protocol; verify complete vector linearization by gel electrophoresis. |
| No growth on 5-FOA plates | Insufficient amplification or incorrect URA3 function. | Ensure the host strain is a ura3- mutant; try increasing the number of cells plated on 5-FOA. |
| Low heterologous gene expression despite high copy number | Transcriptional silencing or metabolic burden. | Use strong, constitutive promoters from the host; consider insulators or integrating into a transcriptionally active genomic locus. |
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents and tools critical for implementing gene copy number enhancement strategies in yeast.
Table 3: Key Research Reagent Solutions for Gene Copy Number Engineering
| Reagent / Tool | Function | Example & Notes |
|---|---|---|
| Broad-Host-Range ORIs | Enables plasmid replication in diverse hosts, including Agrobacterium and non-conventional yeasts. | pVS1, RK2, pSa, BBR1 [43]. Mutations in their RepA proteins can be engineered for higher copy number. |
| CRISPR/Cas9 System | Enables precise genome editing for targeted multi-copy integration. | CRISPR-GPT, an LLM agent system, can assist in designing gRNAs and experiment planning for knockout and activation [45]. |
| Error-Prone PCR Kit | Introduces random mutations into a target DNA sequence for directed evolution. | Commercial kits from suppliers like Jena Bioscience or NEB. Critical for creating repA mutant libraries [43]. |
| Type IIS Restriction Enzymes | Facilitates advanced DNA assembly methods like Golden Gate Assembly. | BsaI, BsmBI. Allows seamless, scarless, and simultaneous assembly of multiple DNA fragments, useful for building complex multi-gene pathways [46]. |
| Synthetic Promoters & Terminators | Provides precise control over gene expression levels in engineered pathways. | Engineered parts for fine-tuned control in non-conventional yeasts like Y. lipolytica and P. pastoris [7]. |
| Fluorescent Reporters (e.g., GFP) | Allows rapid, quantitative screening of transformation efficiency and expression levels. | Used in transient expression assays in systems like Nicotiana benthamiana to rapidly screen for high-performing ORI variants [43]. |
The strategic enhancement of gene copy number is a cornerstone of successful heterologous pathway reconstruction in yeast. The choice between high-copy plasmids, stable genomic integrations, and evolved systems depends on the balance required between speed, stability, and final titers. As synthetic biology advances, the integration of these methods with AI-assisted design tools like CRISPR-GPT [45] and the expanding genetic toolbox for non-conventional yeasts [7] will continue to push the boundaries of yeast metabolic engineering, enabling more efficient and sustainable biomanufacturing of drugs and chemicals.
Promoter and Terminator Engineering for Controlled Gene Expression
Within the context of heterologous pathway reconstruction in yeast, controlling gene expression is a cornerstone of synthetic biology and metabolic engineering. Achieving optimal product yields, particularly for high-value compounds such as therapeutics, requires precise balancing of the expression levels of every gene in a biosynthetic pathway to maximize flux while avoiding the buildup of toxic intermediates or undue cellular burden [47]. While promoters have traditionally been the primary tool for this regulation, it is now well-established that terminator sequences are equally critical genetic elements. Terminators not only ensure proper transcriptional termination but also profoundly influence mRNA stability and abundance, thereby exerting significant post-transcriptional control over final protein levels [48] [49] [50]. The emerging paradigm is that the combinatorial pairing of promoters and terminators provides a powerful, modular strategy for fine-tuning gene expression. This application note details practical methodologies and recent advances in promoter and terminator engineering, providing validated protocols for researchers aiming to reconstruct and optimize heterologous pathways in yeast for drug development and other applications.
Quantitative Analysis of Regulatory Elements
The strength of promoters and terminators is quantifiable, enabling rational design. The data below summarize performance ranges for these genetic parts across various yeast species, providing a reference for selection.
Table 1: Promoter Strength Characterization in Yeasts
| Yeast Species | Promoter Name | Expression Strength/Characteristics | Regulation/Induction |
|---|---|---|---|
| Saccharomyces cerevisiae | Synthetic iSynP (110 bp) | >100-fold induction | DAPG-inducible [51] |
| Komagataella phaffii | 94-bp minimal iSynP | 1730-fold induction, 2x stronger than constitutive KpGAPDH | DAPG-inducible [51] |
| Ogataea polymorpha | pMOX, pCAT | Comparable high strength on methanol; pCAT reaches peak expression >24h earlier | Methanol-induced/repressed on glucose [52] |
Table 2: Terminator Strength and Characterization
| Yeast Species | Terminator Name | Relative Effect/Fold-Range | Key Mechanism/Note |
|---|---|---|---|
| Saccharomyces cerevisiae | High-capacity (e.g., DIT1t) | Up to 11x higher than CYC1t; 6.5-fold transcript level difference [50] | Increased mRNA half-life is a major cause [50] |
| Komagataella phaffii | Catalog of 72 terminators | 17-fold tunable range [49] | Effect is independent of the upstream promoter and ORF [49] |
| Ogataea polymorpha | MOX terminator | ~50% higher expression than next strongest; 6-fold range across 15 terminators [52] | Stabilizes mRNA, increasing transcript level [52] |
Application Notes & Experimental Protocols
Protocol: Designing a Strong, Low-Leakage Inducible Promoter
This protocol describes the construction of tightly regulated, synthetic inducible promoters (iSynPs) in yeast, based on a study that achieved >1000-fold induction [51].
Key Reagents:
- Insulator DNA: A >1 kbp sequence from a genomic region with no enhancer activity (e.g., K. phaffii ARG4).
- Core Promoter: A short sequence containing a TATA box and transcription start site (e.g., 53-bp KpAOX1 core).
- Operator Repeats: Multiple copies of the bacterial operator sequence (e.g., phlO, tetO) for the desired inducer.
- Synthetic Transcription Activator (sTA): A plasmid expressing a fusion protein that binds the operator only in the presence of its inducer.
Procedure:
- Insulator Insertion: Clone a >1 kbp insulator sequence directly upstream of the planned iSynP location to shield it from cryptic transcriptional activation from upstream genomic sequences [51].
- Operator-TATA Fusion: Directly fuse the bacterial operator repeats (e.g., 2-4 copies) upstream of the TATA-box sequence of the core promoter. Minimize the spacer between the operator and the TATA box to ≤40 bp for maximal induction [51].
- Operator Screening/Mutation: If leakiness persists, randomize the sequence immediately upstream of the operator and screen for clones with reduced basal expression, as this region may contain cryptic enhancer elements [51].
- sTA Co-expression: Integrate the expression cassette into a yeast strain that constitutively expresses the corresponding sTA.
- Validation: Measure reporter protein (e.g., GFP) levels in the absence and presence of the inducer to calculate the fold-induction. A successful design should show negligible leakiness and induction of >1000-fold [51].
Protocol: Implementing the GEMbLeR System for Combinatorial Optimization
This protocol utilizes the GEMbLeR (Gene Expression Modification by LoxPsym-Cre Recombination) system for rapid, in vivo generation of diverse expression levels for multiple pathway genes [47].
Key Reagents:
- GEM-blocks: Arrays of diverse upstream promoter elements (UPEs) or terminator sequences, each flanked by orthogonal LoxPsym sites.
- Cre Recombinase: A plasmid with an inducible promoter controlling Cre recombinase expression.
- Orthogonal LoxPsym Sites: Use different, non-cross-reacting LoxPsym sequences for the 5' and 3' GEMs of each gene to prevent inter-module recombination [47].
Procedure:
- Strain Construction: Replace the native promoter and terminator of each target pathway gene with a 5' GEM (UPE array) and 3' GEM (terminator array), respectively.
- Library Generation: Introduce the Cre recombinase plasmid into the engineered strain and induce its expression. Cre-mediated recombination will shuffle the GEM-blocks, creating a vast library of strains, each with unique promoter and terminator combinations for every gene [47].
- Screening/Selection: Screen the resulting library for the desired phenotype (e.g., high production of a compound like astaxanthin). A single round of GEMbLeR has been shown to double production titers [47].
- Genotype Verification: Sequence the genomic loci of the GEMs in the best-performing strains to identify the optimal promoter and terminator combinations.
Protocol: Engineering a Terminator-Promoter Bifunctional Element
This protocol outlines the rational design of a single DNA part that functions as both a terminator for an upstream gene and a promoter for a downstream gene, simplifying pathway assembly [53].
Key Reagents:
- Terminator Elements: Efficiency element (e.g., TATATA), positioning element (e.g., AATAAA), and poly(A) site (e.g., TTTCAAA).
- Core Promoter Elements: TATA box (e.g., TATATAA), BRE, Inr, MTE, and DPE.
- Upstream Activating Sequence (UAS): A sequence containing transcription factor binding sites to enhance promoter strength.
Procedure:
- Design and Synthesize:
- Design the terminator segment by linking the efficiency element, positioning element, and poly(A) site with short linkers [53].
- Design the core promoter segment by assembling the BRE, TATA box, Inr, MTE, and DPE with appropriate spacers. Note that shortening spacer sequences can reduce promoter strength [53].
- Fuse the UAS upstream of the core promoter.
- Assemble Bifunctional Element: Fuse the 3' end of the synthetic terminator (including the poly(A) site) to the 5' end of the synthetic promoter (upstream of the UAS) to create the final bifunctional element [53].
- Characterization:
- Terminator Strength: Place the element between a constitutive promoter-driven reporter gene and a downstream gene. Measure read-through transcription to assess termination efficiency. Strength can be regulated by the efficiency element [53].
- Promoter Strength: Use the element to drive expression of a reporter gene (e.g., GFP). Measure output to assess promoter activity, which can be regulated by the UAS and spacer sequences [53].
- Pathway Application: Use the characterized bifunctional elements in a heterologous pathway (e.g., lycopene biosynthesis) to improve assembly efficiency and product yield [53].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Key Reagents for Promoter and Terminator Engineering
| Reagent / Tool Name | Function / Key Feature | Example Application |
|---|---|---|
| Orthogonal LoxPsym Sites [47] | Enable independent, parallel recombination at multiple genomic loci without cross-talk. | Multiplexed gene expression tuning via the GEMbLeR system. |
| Synthetic Transcription Activators (sTAs) [51] | Engineered proteins that activate transcription from a minimal core promoter only upon binding a specific small molecule inducer (e.g., DAPG, Dox). | Creating custom, high-induction, low-leakage expression systems. |
| Insulator Sequences [51] | Genomic DNA fragments (>1 kbp) that prevent cryptic transcriptional activation of synthetic promoters from upstream regions. | Eliminating basal expression (leakiness) in inducible promoter systems. |
| Bifunctional Element [53] | A single DNA sequence that combines promoter and terminator functions, streamlining pathway assembly. | Simplifying the construction of multi-gene pathways in yeast. |
| Terminator Catalog [49] | A pre-characterized library of terminator sequences with a known range of activities in a specific host (e.g., 17-fold range in K. phaffii). | Fine-tuning protein expression levels via terminator exchange. |
Visual Guide: Engineering Workflows and Element Structures
Bifunctional Element Architecture
Diagram 1: Bifunctional element structure for simplified pathway assembly.
GEMbLeR Combinatorial Shuffling Workflow
Diagram 2: GEMbLeR system workflow for generating expression diversity.
Application Notes
Heterologous pathway reconstruction in yeast has established Saccharomyces cerevisiae as a premier platform for the sustainable production of valuable natural products. This approach bypasses the limitations of plant extraction and chemical synthesis, enabling robust microbial synthesis of complex molecules. The following case studies exemplify strategies for reconstructing and optimizing pathways for alkaloids, terpenoids, and xylose catabolism, highlighting methodologies that enhance precursor supply, manage cofactor balances, and improve pathway flux through systematic engineering.
Case Study 1: Reconstruction of Alkaloid Biosynthetic Pathways
Monoterpenoid Indole Alkaloid (MIA) Biosynthesis
Experimental Objective: To reconstruct the biosynthetic pathway for the vinblastine precursors catharanthine and tabersonine in S. cerevisiae [54].
Key Achievements:
- Successfully expressed the challenging flavoenzyme O-acetylstemmadenine oxidase by modifying its signal peptide, enabling functional expression in yeast [54].
- Integrated ~18 kb of MIA biosynthetic gene cassettes as single copies into four genomic loci using CRISPR-Cas9 [54].
- Demonstrated pathway promiscuity by producing fluorinated and hydroxylated catharanthine and tabersonine derivatives from unnatural substrates [54].
Table 1: Key Enzymes for Monoterpenoid Indole Alkaloid Production in Yeast
| Enzyme Function | Engineering Strategy | Key Outcome |
|---|---|---|
| O-acetylstemmadenine oxidase | Signal peptide modification with yeast CPY signal | Enabled functional expression in yeast |
| Multiple MIA pathway enzymes | Simultaneous integration into 4 genomic loci via CRISPR-Cas9 | Biosynthesis of vinblastine precursors from secologanin and tryptamine |
Chelerythrine Biosynthesis
Experimental Objective: To reconstruct the complete biosynthesis pathway for the benzophenanthridine alkaloid chelerythrine from (S)-reticuline in S. cerevisiae [55].
Protocol:
- Strain Construction: Use S. cerevisiae W303-1A as background strain
- Pathway Assembly: Integrate seven plant-derived enzymes (McoBBE, TfSMT, AmTDC, EcTNMT, PsMSH, EcP6H, and PsCPR) using CRISPR-Cas9 genome editing
- Strain Optimization:
- Integrate multiple copies of rate-limiting genes (TfSMT, AmTDC, EcTNMT, PsMSH, EcP6H, PsCPR, INO2, and AtATR1)
- Engineer heme and NADPH supply
- Enhance product trafficking via heterologous expression of MtABCG10 transporter
- Fermentation: Conduct pH-based fed-batch fermentation in a 0.5 L bioreactor with optimized media and inoculum concentrations
Results: Achieved chelerythrine titers of 12.61 mg/L, representing a 37,000-fold increase over first-generation strains [55].
Dihydrosanguinarine Biosynthesis
Experimental Objective: To reconstitute a 10-gene pathway for dihydrosanguinarine synthesis from (R,S)-norlaudanosoline in S. cerevisiae [56].
Protocol:
- Pathway Division: Separate nine enzymatic reactions into three blocks of three sequential enzymes
- Vector Design: Clone each block into separate plasmids with a fourth plasmid containing cytochrome P450 reductase from P. somniferum (PsCPR)
- Functional Expression: Verify enzyme functionality by supplementing cultures with pathway intermediates
- Product Detection: Analyze end products via LC-MS
Key Findings: Achieved dihydrosanguinarine synthesis with 20% yield of reticuline from norlaudanosoline, along with detection of unnatural side products N-methylscoulerine and N-methylcheilanthifoline [56].
Case Study 2: Reconstruction of Terpenoid Biosynthetic Pathways
Engineering Terpenoid Production via the Mevalonate Pathway
Experimental Objective: To enhance terpenoid production by optimizing the native mevalonate (MVA) pathway in yeast [57].
Protocol:
- Precursor Enhancement:
- Overexpress HMG-CoA reductase (HMGR), the pathway's rate-limiting enzyme
- Consider N-terminal truncation of HMGR to prevent self-degradation (though native HMGR may be superior in some yeasts like Yarrowia lipolytica)
- Overexpress IPP isomerase (IDI) to improve flux distribution
- Pathway Amplification: Overexpress multiple MVA pathway genes (ERG10, ERG13, ERG12, ERG8, and ERG19)
- Validation: Measure terpenoid production improvements (e.g., 251-fold, 29-fold, and 72-fold enhancement in α-bisabolene, β-bisabolene and γ-bisabolene production, respectively, with full pathway overexpression) [57]
Implementing the Isopentenol Utilization (IU) Pathway
Experimental Objective: To replace the native MVA pathway with a more efficient isopentenol utilization pathway for terpenoid precursor synthesis [58].
Protocol:
- MVA Pathway Inactivation: Knock out ERG13 (encoding HMG-CoA synthase) to disable the native MVA pathway
- IU Pathway Integration: Introduce choline kinase from S. cerevisiae (ScCKI1) and isopentenyl phosphate kinase from A. thaliana (AtIPK)
- Substrate Utilization: Supplement with prenol (isopentenol) as substrate for the IU pathway
- Adaptive Evolution: Use growth-coupled selection to enhance ATP supply and IU pathway efficiency
Results: The IU pathway-dependent strain showed 152.95% increased squalene accumulation compared to MVA pathway-dependent strains, demonstrating superior efficiency for complex terpenoid synthesis [58].
Table 2: Comparison of Terpenoid Synthesis Pathways in Yeast
| Parameter | Mevalonate (MVA) Pathway | Isopentenol Utilization (IU) Pathway |
|---|---|---|
| Steps | Multiple enzymatic reactions | Two-step phosphorylation |
| Cofactor Requirements | ATP, NADPH | ATP only |
| Theoretical Yield | Lower | Higher |
| Engineering Complexity | High (multiple gene regulation) | Lower (fewer enzymes) |
| Substrate | Endogenous acetyl-CoA | Exogenous isopentenol |
Case Study 3: Reconstruction of Xylose Catabolic Pathways
Engineering Xylose Metabolism in Non-Native Yeasts
Experimental Objective: To enable xylose utilization in S. cerevisiae for production of natural products from lignocellulosic biomass [59].
Protocol:
- Pathway Selection: Choose from three established xylose catabolic pathways:
- XR-XDH Pathway: Xylose reductase (XR) and xylitol dehydrogenase (XDH) from native xylose-utilizing yeasts
- XI Pathway: Xylose isomerase from bacteria or fungi
- Weimberg Pathway: Bacterial pathway converting xylose to α-ketoglutarate
- Gene Expression: Express selected pathway genes in S. cerevisiae
- Cofactor Engineering: Address redox imbalance issues through enzyme engineering or regulatory modifications
- Transport Engineering: Express specific xylose transporters to overcome uptake limitations
Key Findings: The XR-XDH pathway is most commonly implemented, though it creates redox imbalances that require additional engineering to resolve [59] [60].
Utilizing Native Xylose-Fermenting Yeasts
Experimental Objective: To leverage naturally xylose-fermenting yeasts like Scheffersomyces stipitis for natural product synthesis [59].
Protocol:
- Strain Selection: Use S. stipitis for its native XR-XDH pathway and high xylose uptake capability
- Genetic Tools: Implement codon optimization (as S. stipitis uses CUG to encode serine)
- Pathway Engineering: Introduce heterologous natural product pathways
- Fermentation Optimization: Control oxygen levels to regulate metabolic flux between respiration and fermentation
Advantages: S. stipitis has the highest native capability for xylose fermentation among known microbes, with xylose uptake rates one order of magnitude higher than engineered S. cerevisiae [59].
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents for Heterologous Pathway Reconstruction in Yeast
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Genome Editing Tools | CRISPR-Cas9 system, p414-TEF1p-Cas9 | Precise genomic integration of pathway genes |
| Vector Systems | pRS416, episomal plasmids | Heterologous gene expression |
| Pathway Enzymes | McoBBE, TfSMT, EcTNMT, PsMSH (for alkaloids); ScCKI1, AtIPK (for IU pathway) | Catalyzing specific biosynthetic steps |
| Signal Peptides | Yeast CPY signal peptide | Improving plant glycoprotein expression in yeast |
| Transporters | MtABCG10, PRM10L156Q mutation | Enhancing product trafficking and substrate uptake |
| Cofactor Systems | Cytochrome P450 reductase (PsCPR), AtATR1 | Supporting P450 enzyme function and redox balance |
| Chassis Strains | S. cerevisiae W303-1A, S. stipitis CBS 6054 | Host organisms for pathway reconstruction |
Pathway Diagrams
Diagram 1: Alkaloid Biosynthetic Pathway Reconstruction
Diagram 2: Terpenoid Synthesis Pathway Comparison
Diagram 3: Xylose Catabolic Pathway Engineering
The reconstruction of heterologous pathways in yeast represents a cornerstone of modern metabolic engineering, enabling the production of high-value pharmaceuticals, biofuels, and chemicals. However, conventional cytoplasmic expression of biosynthetic pathways often encounters significant limitations, including suboptimal local enzyme concentrations, competition with native metabolism, and cytotoxicity of pathway intermediates or products [61]. Spatial reconfiguration through compartmentalization has emerged as a powerful strategy to overcome these challenges by harnessing yeast's native organelles as specialized microreactors with unique physicochemical environments [62].
Organelles such as mitochondria, peroxisomes, and the endoplasmic reticulum offer distinct advantages for pathway engineering, including sequestered metabolite pools, favorable cofactor availability, and reduced interference with competing metabolic reactions [61] [63]. This application note details experimental protocols and case studies for implementing compartmentalization strategies, with a specific focus on mitochondrial targeting to enhance pathway efficiency and product yield in Saccharomyces cerevisiae.
Case Study: Compartmentalizing the Isobutanol Pathway in Mitochondria
Experimental Rationale and Design
The biosynthesis of isobutanol in yeast involves a branched-chain amino acid pathway that is naturally split between mitochondria and cytoplasm. The upstream pathway (pyruvate to α-ketoisovalerate) is naturally mitochondrial, while the downstream Ehrlich pathway (α-ketoisovalerate to isobutanol) is typically cytoplasmic [61]. This natural division creates inherent inefficiencies due to the required transport of intermediates across mitochondrial membranes, where they become vulnerable to competing pathways.
To address this bottleneck, Avalos et al. engineered a complete mitochondrial isobutanol pathway by targeting all downstream enzymes to the mitochondrial matrix, creating a unified biosynthetic compartment [61]. This spatial reconfiguration resulted in a 260% increase in isobutanol production compared to strains expressing the same pathway cytoplasmically, which showed only marginal improvement (10%) over controls [61].
Key Findings and Quantitative Outcomes
Table 1: Comparative Isobutanol Production from Cytoplasmic vs. Mitochondrial Compartmentalization
| Strain Configuration | Isobutanol Titer (mg/L) | Fold Increase vs. Control | Relative Improvement |
|---|---|---|---|
| Control (Empty Plasmid) | 28 ± 2 | 1x | Baseline |
| Upstream ILV Genes Only | 136 ± 23 | ~5x | Reference |
| Complete Cytoplasmic Pathway | 151 ± 34 | ~5.4x | 10% vs. ILV Only |
| Complete Mitochondrial Pathway | 486 ± 36 | ~18x | 260% vs. ILV Only |
Table 2: Benefits of Mitochondrial Compartmentalization for Metabolic Engineering
| Advantage | Mechanism | Impact on Production |
|---|---|---|
| Increased Local Enzyme Concentration | Reduced volume of mitochondrial matrix concentrates enzymes and substrates | Higher reaction rates and pathway efficiency |
| Enhanced Intermediate Availability | Sequestration of α-ketoisovalerate within mitochondria | Reduced diversion to competing pathways |
| Optimal Cofactor Environment | Higher reducing potential and unique pH in mitochondria | Improved activity of iron-sulfur cluster enzymes |
| Toxicity Mitigation | Isolation of cytotoxic intermediates from cytoplasm | Improved host cell viability and productivity |
Protocol: Engineering Mitochondrial Compartmentalization for Metabolic Pathways
Vector Design and Assembly for Mitochondrial Targeting
Principle: Construction of standardized vector systems enables parallel assembly of pathways targeted to different subcellular compartments while maintaining identical enzyme expression levels and characteristics.
Materials:
- pJLA Vector Series: Standardized plasmid system for S. cerevisiae [61]
- CoxIV Mitochondrial Localization Signal (MLS): N-terminal targeting sequence from yeast cytochrome c oxidase subunit IV [61]
- Yeast Codon-Optimized Genes: α-ketoacid decarboxylase (Kivd, ARO10, KID1) and alcohol dehydrogenase (ADH7, FucO, AdhARE1) [61]
- Strong Constitutive Promoters: TDH3, PGK1, TEF1 for driving high-level expression [61]
Procedure:
- MLS Fusion Construct Design:
- Amplify coding sequences of target enzymes without native localization signals
- Fuse N-terminally with CoxIV MLS (25-35 amino acid presequence) via flexible glycine-serine linkers
- Maintain identical peptide sequence except for single N-terminal glutamine in mitochondrial variants [61]
Multigene Pathway Assembly:
- Clone upstream pathway genes (ILV2, ILV3, ILV5) under control of TDH3, PGK1, and TEF1 promoters respectively
- Assemble downstream pathway components (α-KDC and ADH) with identical promoter strength in both cytoplasmic and mitochondrial configurations
- Utilize standardized restriction sites or Gibson assembly for combinatorial construction [61]
Vector Verification:
- Confirm reading frame preservation across fusion junctions by sequencing
- Verify promoter-gene combinations and transcriptional termination
- Validate plasmid stability in E. coli and copy number determination
Strain Engineering and Transformation
Materials:
- S. cerevisiae strain background (e.g., BY4741, CEN.PK113-7D)
- Standard yeast transformation reagents (LiAc/PEG method)
- Selective media plates lacking appropriate auxotrophic markers
Procedure:
- Strain Preparation:
- Grow recipient yeast strain in YPD to mid-log phase (OD600 = 0.5-0.8)
- Harvest cells and render competent using LiAc treatment
Transformation and Selection:
- Transform with 500 ng-1 μg of verified plasmid DNA using high-efficiency LiAc/PEG method
- Plate on appropriate selective medium and incubate at 30°C for 2-3 days
- Pick multiple transformants (minimum 5-10) to account for positional effects
Strain Validation:
- Confirm plasmid retention by colony PCR
- Verify protein expression by Western blotting with HA or Myc tags
- Assess growth phenotypes in selective media
Subcellular Localization and Enzyme Activity Validation
Principle: Confirmation of proper mitochondrial targeting and functionality of relocated enzymes is essential before production characterization.
Materials:
- Mitochondrial isolation kit (commercial)
- Antibodies against mitochondrial markers (porin, citrate synthase)
- Protease protection assay reagents (proteinase K, Triton X-100)
- GC-MS system for isobutanol quantification
Procedure:
- Mitochondrial Isolation and Fractionation:
- Culture engineered strains to mid-log phase in appropriate selective media
- Harvest cells and disrupt using enzymatic digestion or mechanical disruption
- Purify mitochondria using differential centrifugation and density gradient separation
- Collect cytosolic and mitochondrial fractions for analysis [61]
Localization Validation:
- Perform Western blotting on subcellular fractions using anti-HA (for tagged enzymes) and organelle markers
- Conduct protease protection assays to confirm intramitochondrial localization
- Visualize localization using fluorescence microscopy with GFP-tagged constructs
Enzyme Activity Assays:
- Measure α-ketoacid decarboxylase activity by following α-ketoisovalerate-dependent NADH oxidation
- Determine alcohol dehydrogenase activity by ethanol-dependent NAD+ reduction
- Compare specific activities in mitochondrial vs. cytoplasmic fractions
Fermentation and Product Analysis
Materials:
- High-cell density fermentation media (minimal or complete)
- Anaerobic chambers or sealed fermentation systems
- GC-MS with headspace autosampler
Procedure:
- Fermentation Conditions:
- Inoculate pre-culture in selective medium and grow overnight
- Dilute to OD600 = 0.1 in fresh medium for low-cell density fermentations
- Use OD600 = 5.0 for high-cell density fermentations in minimal medium
- Conduct fermentations for 24 hours at 30°C with moderate shaking (200 rpm) [61]
Product Quantification:
- Collect samples at 0, 6, 12, 18, and 24 hours
- Analyze isobutanol content by GC-MS with appropriate internal standards
- Measure optical density to correlate production with biomass
- Calculate specific productivity (mg/L/OD600) and total titer [61]
Data Analysis:
- Compare production kinetics between mitochondrial and cytoplasmic strains
- Calculate fold-improvements relative to control strains
- Perform statistical analysis (Student's t-test) on biological replicates (n ≥ 3)
The Scientist's Toolkit: Essential Research Reagents
Table 3: Key Research Reagents for Compartmentalization Engineering
| Reagent/Category | Specific Examples | Function/Application |
|---|---|---|
| Targeting Signals | CoxIV MLS (yeast), PEX5 (peroxisomal), Pex15 (peroxisomal membrane) | Directing enzymes to specific organelles [61] [64] |
| Vector Systems | pJLA series, 2μ-based high-copy plasmids | Standardized pathway assembly and expression [61] |
| Promoter Systems | TDH3, PGK1, TEF1 (constitutive); hybrid carbon-responsive promoters | Controlling temporal and strength of gene expression [61] [64] |
| Assembly Tools | Gibson Assembly, Golden Gate, Cre-loxP | Combinatorial construction of multigene pathways |
| Validation Tools | Anti-HA/Myc antibodies, organelle-specific dyes, subcellular fractionation kits | Confirming proper localization and function |
Pathway Engineering and Compartmentalization Workflow
Figure 1: DBTL Cycle for Compartmentalization Engineering illustrating the iterative workflow for designing, building, testing, and learning from organelle-targeted pathway engineering experiments.
Comparative Organelle Characteristics for Pathway Engineering
Figure 2: Organelle Properties and Applications highlighting the unique metabolic features of different yeast organelles and their suitability for specific biosynthetic pathways.
Spatial reconfiguration through compartmentalization represents a paradigm shift in heterologous pathway engineering in yeast. The strategic targeting of biosynthetic pathways to organelles leverages natural microenvironments and substrate channeling to overcome limitations of cytoplasmic expression. The mitochondrial compartmentalization of the isobutanol pathway demonstrates the profound impact of this approach, with nearly 3-fold higher production compared to conventional cytoplasmic expression [61].
Future developments in compartmentalization engineering will likely focus on multi-organelle coordination, where different pathway modules are targeted to their optimal subcellular environments. Additionally, emerging techniques for expanding organelle size and storage capacity [62] [63], combined with dynamic regulatory systems [64], will further enhance the potential of spatial reconfiguration strategies. As our understanding of organelle biology and trafficking mechanisms deepens, so too will our ability to engineer yeast as efficient cell factories for increasingly complex natural products and pharmaceuticals.
Enhancing Performance: Strategies for Troubleshooting and Flux Optimization
Identifying and Overcoming Metabolic Bottlenecks and Cofactor Imbalances
The reconstruction of heterologous pathways in microbial cell factories like Saccharomyces cerevisiae is a cornerstone of modern biotechnology, enabling the sustainable production of high-value compounds. However, the productivity of these engineered pathways is often hampered by metabolic bottlenecks and cofactor imbalances, which restrict metabolic flux and limit yields. Identifying and overcoming these limitations is critical for developing economically viable bioprocesses. This Application Note details standardized protocols for diagnosing flux restrictions and optimizing cofactor metabolism, framed within the context of heterologous alkaloid biosynthesis in yeast. The methodologies presented herein, which leverage recent advances in metabolic engineering and synthetic biology, provide a systematic framework for enhancing the production of functional products in microbial systems.
Key Concepts and Quantitative Benchmarks
Metabolic bottlenecks are enzymatic steps within a pathway that significantly limit the overall flux towards a desired product due to low enzyme activity, insufficient cofactor supply, or poor enzyme stability. Cofactor imbalances occur when heterologous pathways disrupt the delicate balance of intracellular redox carriers (e.g., NADPH/NADP⁺) or energy currencies (e.g., ATP), leading to suboptimal performance and potential cellular toxicity. The impact of addressing these issues is profound, as demonstrated in the reconstruction of the chelerythrine pathway, where a combinatorial engineering strategy achieved a nearly 900-fold increase in production, culminating in a titer of 12.61 mg/L in a bioreactor [55].
Table 1: Key Enzymes and Cofactors in Heterologous Alkaloid Biosynthesis
| Enzyme/Component | Function in Pathway | Origin | Key Cofactor/Regulator |
|---|---|---|---|
| Berberine Bridge Enzyme (BBE) | Catalyzes the first oxidation and C-C bond formation from (S)-reticuline [55] | Macleaya cordata [55] | |
| Scoulerine-9-O-methyltransferase (SMT) | Methylates (S)-scoulerine [55] | Thalictrum flavum [55] | |
| Tetrahydroprotoberberine cis-N-methyltransferase (TNMT) | Methylates (S)-canadine [55] | Eschscholzia californica [55] | |
| Protopine 6-hydroxylase (P6H) | Oxidizes allocryptopine [55] | Eschscholzia californica [55] | Cytochrome P450, NADPH [55] |
| Cytochrome P450 Reductase (CPR) | Supports P450 enzyme function (e.g., P6H) [55] | Papaver somniferum [55] | NADPH |
| IN O2 | Transcriptional regulator | Saccharomyces cerevisiae [55] | |
| ABC Transporter (MtABCG10) | Enhances product trafficking and potentially relieves feedback inhibition [55] | Macleaya cordata [55] | ATP |
Experimental Protocols
Protocol 1: Diagnostic Metabolite Extraction and Analysis from Yeast
This protocol is optimized for the rapid quenching of metabolism and efficient extraction of intracellular metabolites from yeast, facilitating the accurate identification of accumulated intermediates that indicate metabolic bottlenecks [65].
Materials
- Strain: Recombinant S. cerevisiae strain with heterologous pathway.
- Quenching Solution: 60% (v/v) Aqueous Methanol, containing 4 mM N-Ethylmaleimide (NEM), pre-chilled to -70 °C in a dry-ice/ethanol bath. NEM is toxic; use appropriate personal protective equipment. [65]
- Extraction Solvent: Chlorform:Methanol:Internal Standard mixture (e.g., 1:3:0.001 ratio).
- Equipment: Centrifuge capable of 20,000 × g at -9 °C, bead-beater with zirconia/silica beads (0.5 mm diameter), lyophilizer.
Procedure
Sampling and Quenching:
- From a growing culture, rapidly withdraw a 0.5 mL sample using a pre-chilled syringe.
- Immediately expel the sample into 0.5 mL of pre-chilled Quenching Solution. The entire process from sampling to quenching must be completed within 10 seconds to minimize metabolic changes [65].
- Vortex mix briefly and centrifuge at 20,000 × g, -9 °C for 5 minutes.
- Carefully separate the supernatant (containing extracellular metabolites) and the cell pellet.
Cell Disruption and Metabolite Extraction:
- To the cell pellet, add 1 mL of ice-cold Extraction Solvent and a volume of zirconia/silica beads equivalent to the pellet.
- Lyse the cells by bead-beating for 3 minutes at 4 °C.
- Centrifuge the lysate at 20,000 × g, -9 °C for 10 minutes to pellet cell debris.
- Transfer the clear supernatant to a new tube.
Sample Preparation for Analysis:
- Lyophilize both the extracellular supernatant and the intracellular metabolite extract.
- Store the dried metabolites at -80 °C until analysis.
- For analysis by Capillary Electrophoresis-Mass Spectrometry (CE-MS) or Liquid Chromatography-Mass Spectrometry (LC-MS), reconstitute the dried samples in an appropriate, MS-compatible buffer [65] [66].
Protocol 2: Combinatorial Engineering for Bottleneck Relief and Cofactor Balancing
This protocol outlines a CRISPR-Cas9-based strategy for the multi-locus integration of rate-limiting genes and cofactor-regulating enzymes to overcome metabolic bottlenecks, as demonstrated for chelerythrine production [55].
Materials
- Plasmids: pRS416-based expression vectors or integrative cassettes for target genes [55].
- gRNA Plasmids: Plasmids expressing guide RNAs (gRNAs) targeting specific, neutral genomic integration sites (e.g., intergenic regions). gRNAs can be designed using online tools like CRISPRdirect [55].
- Cas9 Plasmid: p414-TEF1p-Cas9 for expressing the Cas9 nuclease in yeast [55].
- Yeast Strain: S. cerevisiae W303-1A or another suitable background strain.
- Media: Synthetic Complete (SC) media lacking appropriate amino acids for selection.
Procedure
Identification of Rate-Limiting Steps:
- Analyze diagnostic metabolite data from Protocol 1. The accumulation of a specific pathway intermediate immediately upstream of an enzymatic step strongly indicates a bottleneck at that step.
- For P450-dependent steps, confirm cofactor limitation by analyzing NADPH/NADP⁺ ratios or by co-expressing a cognate reductase.
Strain Construction via CRISPR-Cas9:
- Design Integration Cassettes: For each rate-limiting gene (e.g., TfSMT, EcTNMT, PsMSH, EcP6H) and cofactor-regulating gene (e.g., AtATR1, INO2), assemble an expression cassette containing a strong promoter, the codon-optimized gene, and a terminator.
- Co-transform: Co-transform the yeast strain with:
- The p414-TEF1p-Cas9 plasmid.
- gRNA plasmids targeting the desired genomic loci.
- The donor DNA fragments (integration cassettes) containing homology arms to the targeted sites. A prominent study used this approach to integrate up to eight key genes [55].
- Plate the transformation mixture on appropriate selective media.
Validation and Screening:
- Screen for successful integrants using colony PCR and verify by sequencing.
- Cultivate validated strains in SC medium supplemented with pathway precursors (e.g., 100 µM (S)-reticuline).
- Quantify the final product titers using LC-MS/MS to identify the highest-producing engineered strain.
Diagram 1: A workflow for diagnosing and overcoming metabolic bottlenecks in yeast.
Results and Data Analysis
The systematic application of these protocols enables dramatic improvements in pathway performance. The data generated from diagnostic metabolite analysis should be quantified on a per-cell basis (e.g., attomol/cell) where possible, and the effectiveness of engineering strategies is quantified by final product titers.
Table 2: Impact of Combinatorial Engineering on Chelerythrine Production in S. cerevisiae [55]
| Engineering Strategy | Key Genetic Modifications | Reported Titer | Fold Improvement |
|---|---|---|---|
| First-Generation Strain | Heterologous expression of 7 plant-derived enzymes [55] | 0.34 µg/L | (Baseline) |
| Combinatorial Engineering | Multi-copy integration of TfSMT, EcTNMT, PsMSH, EcP6H, PsCPR, INO2, AtATR1 [55] | ~300 µg/L | ~900-fold |
| Integrated Bioprocess | Combined metabolic engineering with product trafficking (MtABCG10) and fed-batch fermentation [55] | 12.61 mg/L | >37,000-fold |
The relocation of metabolic pathways to subcellular compartments, such as mitochondria or peroxisomes, can further enhance productivity by concentrating substrates and enzymes, isolating toxic intermediates, and leveraging localized cofactor pools [67].
Diagram 2: Strategic solutions for overcoming metabolic bottlenecks.
The Scientist's Toolkit
Table 3: Essential Research Reagents and Solutions
| Reagent / Tool | Function / Application | Example / Source |
|---|---|---|
| CRISPR-Cas9 System | Precision genome editing for multi-locus gene integration [55] | p414-TEF1p-Cas9 plasmid; gRNA plasmids [55] |
| Codon-Optimized Genes | Enhances heterologous expression of plant or bacterial enzymes in yeast. | Synthetic genes for McoBBE, TfSMT, EcTNMT, etc. [55] |
| N-Ethylmaleimide (NEM) | Thiol-protecting agent for accurate quantification of redox metabolites during extraction [65] | Quenching Solution (4 mM in methanol) [65] |
| Cytochrome P450 Reductase (CPR) | Essential partner for NADPH-dependent activity of P450 enzymes (e.g., P6H, MSH) [55] | Papaver somniferum PsCPR [55] |
| ABC Transporter | Facilitates product secretion, alleviating potential feedback inhibition and cytotoxicity [55] | Macleaya cordata MtABCG10 [55] |
| Metabolomics Standards | Internal standards for quantitative LC-MS/CE-MS analysis of intracellular metabolites. | Stable isotope-labeled amino acids, organic acids, cofactors |
The systematic identification and removal of metabolic bottlenecks and cofactor imbalances are indispensable for the efficient operation of heterologous pathways in yeast. The integrated use of advanced diagnostic metabolomics, combinatorial genetic engineering, and compartmentalization strategies provides a powerful and generalizable framework. As demonstrated by the extraordinary 37,000-fold improvement in chelerythrine titer, this rigorous approach enables the transformation of rudimentary pathway reconstructions into highly productive microbial cell factories, paving the way for the sustainable bioproduction of complex natural products and pharmaceuticals.
Balancing Pathway Flux through Combinatorial Promoter and Terminator Shuffling
Within the field of microbial metabolic engineering, heterologous pathway reconstruction in yeast has become a cornerstone for the sustainable production of high-value compounds, ranging from pharmaceuticals to biofuels [47]. A critical challenge in this process is achieving optimal product titers, as the imbalanced expression of heterologous pathway genes often leads to suboptimal flux, accumulation of toxic intermediates, and reduced cell fitness [47]. Consequently, balancing the expression levels of every gene in a biosynthetic pathway is paramount.
Traditional methods for optimizing gene expression heavily rely on iterative, trial-and-error approaches, which are often time-consuming and labor-intensive [47]. To address these limitations, combinatorial optimization strategies have emerged, enabling the simultaneous exploration of a vast landscape of gene expression levels. This application note focuses on one such powerful method: the combinatorial shuffling of promoters and terminators to balance pathway flux in yeast. We will detail the principle, provide a protocol for its implementation, and highlight key reagent solutions, framing the discussion within the broader objective of efficient heterologous pathway reconstruction for drug development and industrial biotechnology.
Key Technologies and Performance
Two primary in vivo methods for combinatorial promoter and terminator shuffling have been recently developed: GEMbLeR and PULSE. Both leverage the Cre-LoxPsym recombination system to generate diversity in gene expression.
Table 1: Comparison of Key Pathway Optimization Tools in Yeast
| Tool Name | Core Technology | Key Components Shuffled | Reported Performance | Key Application Example |
|---|---|---|---|---|
| GEMbLeR [47] | Cre-LoxPsym Recombination | Promoter and Terminator modules | Doubled astaxanthin production titers; Pathway gene expression ranged over 120-fold. | Astaxanthin biosynthetic pathway |
| PULSE [68] | Cre-LoxPsym Recombination & FACS-screened UAS | Upstream Activating Sequences (UAS) in hybrid promoters | 8-fold increase in β-carotene production; Improved growth on high xylose. | β-carotene pathway, Xylose utilization pathway |
The GEMbLeR (Gene Expression Modification by LoxPsym-Cre Recombination) approach involves designing arrays of upstream promoter elements (UPEs) and terminator sequences, each flanked by orthogonal LoxPsym sites [47]. When Cre recombinase is induced, these modules are independently shuffled through deletion, inversion, and duplication events. This process generates a vast library of strain variants, each possessing a unique expression profile for the targeted genes. When applied to a six-gene astaxanthin pathway, a single round of GEMbLeR successfully doubled production titers [47].
The PULSE (Promoter engineering by shuffling Upstream activating sequences via LoxPsym Supported Evolution) tool utilizes a similar LoxPsym-based recombination mechanism but focuses on shuffling Upstream Activating Sequence (UAS) elements that have been pre-screened for activity via FACS [68]. This workflow allows for the creation of synthetic hybrid promoters that can exceed the strength of native strong promoters. Its application has led to an eight-fold increase in β-carotene production [68].
Figure 1: Generalized Workflow for LoxPsym-Mediated Expression Shuffling. The process begins with a platform strain harboring Gene Expression Modifier (GEM) arrays integrated at target gene loci. Induction of Cre recombinase triggers shuffling of the modules, creating a library of strains with diverse expression profiles for subsequent screening.
Detailed Experimental Protocol
This protocol outlines the steps for implementing the GEMbLeR approach to optimize a heterologous biosynthetic pathway in S. cerevisiae.
Strain Construction and Library Generation
Materials:
- Yeast strain with integrated heterologous pathway of interest.
- Plasmids or DNA fragments for constructing 5' GEM (UPE array) and 3' GEM (terminator array) modules.
- A Cre recombinase expression system (typically an inducible plasmid).
Procedure:
- Design GEM Modules: For each gene in the pathway to be optimized, design a 5' GEM module and a 3' GEM module. Each module should consist of a series of distinct genetic parts (e.g., 6 different UPEs for the 5' GEM, 6 different terminators for the 3' GEM), each flanked by orthogonal LoxPsym sites to prevent cross-recombination between modules [47].
- Replace Native Regulatory Elements: For each target gene, replace its native promoter and terminator with the designed 5' and 3' GEM modules, respectively. This can be achieved through standard yeast genetic techniques like homologous recombination. This creates your "Platform Strain."
- Introduce Cre Recombinase: Transform the platform strain with a plasmid carrying the Cre recombinase gene under the control of an inducible promoter (e.g., GAL1).
- Induce Recombination: Inoculate the transformed strain into an appropriate induction medium to express Cre recombinase. Incubate for a sufficient time to allow for recombination events. This step generates a complex library of variants where the order, orientation, and copy number of the UPEs and terminators have been shuffled.
Library Screening and Validation
Materials:
- Selective plates or medium for library outgrowth.
- Equipment for high-throughput screening (e.g., FACS, microplate readers, HPLC).
Procedure:
- Library Outgrowth: After induction, plate the cell library on solid medium or inoculate into liquid medium to allow outgrowth of the individual variants.
- Primary Screening: Employ a high-throughput screening method to identify top producers. For pathways producing colored compounds (e.g., carotenoids like astaxanthin or β-carotene), visual screening or FACS can be highly effective [47] [68]. For other products, use a method linked to the product, such as a growth-coupled assay or a rapid, small-scale extraction followed by analytical chemistry (e.g., HPLC).
- Secondary Screening and Validation: Isolate the top-performing clones from the primary screen. Inoculate them in small-scale liquid cultures to quantify production yields more accurately (e.g., using HPLC-MS). Ensure genetic stability by passaging the strains and re-measuring production.
- Characterization (Optional): For lead isolates, sequence the shuffled promoter and terminator regions to correlate the specific regulatory element combination with the high-production phenotype.
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for Combinatorial Shuffling
| Reagent / Tool | Function and Key Features | Application Note |
|---|---|---|
| Orthogonal LoxPsym Sites [47] | Mutated LoxP sequences that recombine only with their identical partner, not with other LoxPsym variants. | Enables independent shuffling of multiple GEM modules for different genes within the same pathway without cross-talk. |
| 5' GEM Module [47] | An array of diverse Upstream Promoter Elements (UPEs) flanked by LoxPsym sites. | Provides the variation for transcriptional initiation rates. Strategic placement of LoxPsym sites (e.g., upstream of TATA box) is critical to avoid inhibiting translation [47]. |
| 3' GEM Module [47] | An array of diverse terminator sequences flanked by LoxPsym sites. | Influences mRNA stability and 3' end formation, contributing to variation in steady-state mRNA levels. |
| Inducible Cre Recombinase | A system to control the timing of the shuffling event, preventing premature recombination during strain construction. | Typically supplied on a plasmid with an inducible promoter (e.g., Galactose-inducible GAL1). Allows library generation on demand. |
| FACS (Fluorescence-Activated Cell Sorting) [68] | A high-throughput method to screen large variant libraries when production is linked to a fluorescent reporter or intrinsic fluorescence. | Instrumentation is critical for screening PULSE libraries and can be adapted for other pathways by creating transcriptional fusions to fluorescent proteins. |
Combinatorial promoter and terminator shuffling represents a significant advancement over traditional, sequential metabolic engineering strategies. Tools like GEMbLeR and PULSE leverage synthetic biology to perform multiplexed, in vivo optimization, allowing researchers to rapidly navigate the vast combinatorial space of gene expression levels. The ability to generate libraries where pathway gene expression varies over more than two orders of magnitude from a single experiment drastically accelerates the strain development cycle [47]. By integrating these powerful combinatorial methods into the heterologous pathway reconstruction workflow, scientists and drug development professionals can more efficiently engineer robust microbial cell factories, thereby shortening the development timeline for novel therapeutics and industrially relevant compounds.
Codon Optimization and Managing Translational Efficiency
Codon optimization is an essential technique in synthetic biology and heterologous pathway reconstruction in yeast, enhancing recombinant protein expression by fine-tuning genetic sequences to match the translational machinery and codon usage preferences of the host organism, Saccharomyces cerevisiae [69] [70]. The degeneracy of the genetic code allows multiple synonymous codons to encode the same amino acid, and different organisms exhibit distinct preferences for certain codons—a phenomenon known as codon usage bias [69]. This bias significantly affects translation rates and accuracy, ultimately impacting the yield of recombinant proteins [69]. For researchers engineering yeast to produce valuable biopharmaceuticals or industrial enzymes, codon optimization represents a critical step in overcoming the challenge of achieving high expression levels for genes originating from non-yeast systems [70]. By aligning the codon sequence of a heterologous gene with the preferred codon usage of yeast, scientists can significantly enhance translational efficiency and protein yield, thereby maximizing the performance of yeast as a microbial cell factory [69] [70].
Fundamental Principles of Codon Optimization
The Genetic Code and Codon Usage Bias
The standard genetic code consists of 64 codons, with 61 coding for amino acids and 3 serving as stop signals [71]. Most amino acids are encoded by multiple codons; for example, leucine can be encoded by six different codons (UUA, UUG, CUU, CUC, CUA, CUG) [71]. However, organisms do not use these synonymous codons with equal frequency. In S. cerevisiae, this codon usage bias reflects the relative abundance of specific transfer RNA (tRNA) molecules, which act as adapters during translation [72]. Codons that are complementary to abundant tRNAs (optimal codons) are typically translated more rapidly and accurately than those read by scarce tRNAs (non-optimal codons) [72]. This concept, termed codon optimality, is a major determinant of both translation efficiency and mRNA stability [72]. When a ribosome encounters a non-optimal codon, it may pause transiently due to limited tRNA availability. These pauses can lead to co-translational protein misfolding or recruitment of mRNA degradation complexes, thereby reducing protein yield [72].
Key Parameters for Optimization
Effective codon optimization extends beyond merely matching codon frequencies. Several interdependent sequence features must be considered to achieve maximal protein expression:
- Codon Adaptation Index (CAI): This metric quantifies the similarity between the codon usage of a target gene and the preferred codon usage of highly expressed genes in the host organism. A CAI value closer to 1.0 indicates better adaptation and is generally correlated with higher protein expression levels [69].
- GC Content: The percentage of guanine and cytosine nucleotides in a sequence can influence mRNA stability and secondary structure. In yeast, moderately balanced GC content is often targeted to avoid extremes that might impair transcriptional or translational efficiency [69].
- mRNA Secondary Structure: Stable secondary structures, particularly in the 5' coding region, can hinder ribosome scanning and elongation. The stability of these structures is often evaluated by calculating the minimum free energy (MFE), where more negative values indicate stronger, more stable folding that can inhibit translation [73] [69].
- Codon Context/Codon-Pair Bias: This refers to the non-random occurrence of specific pairs of adjacent codons. Optimizing for host-preferred codon pairs can enhance translational accuracy and speed by improving ribosome kinetics [69].
Codon Optimization Tools and Strategies
Comparison of Optimization Tools
Various computational tools have been developed to implement codon optimization, each employing different algorithms and prioritizing different parameters. A comparative analysis of widely used tools reveals significant variability in their design principles and outputs [69].
Table 1: Comparative Analysis of Codon Optimization Tools and Strategies
| Tool / Strategy | Primary Optimization Method | Key Parameters | Notable Features |
|---|---|---|---|
| Traditional Rule-Based Tools (e.g., JCat, OPTIMIZER) [69] | Matching host organism's codon usage frequency. | CAI, Individual Codon Usage (ICU) | Straightforward, but may overlook mRNA structure and other regulatory elements. |
| Multi-Parameter Tools (e.g., GeneOptimizer, ATGme) [69] | Iterative optimization considering multiple, sometimes conflicting, parameters. | CAI, GC content, mRNA secondary structure, restriction sites | Provides a balanced approach, aiming to harmonize various sequence features. |
| Deep Learning Frameworks (e.g., RiboDecode) [73] | Deep learning trained on large-scale biological data (e.g., ribosome profiling). | Translation level prediction, MFE, cellular context | Data-driven; can explore vast sequence space and generate novel, highly efficient sequences. |
| tRNA-Based Enhancement [72] | Supplementing with exogenous tRNAs to match codon demand. | tRNA abundance, decoding efficiency | A complementary strategy to sequence optimization, useful for genes with unavoidable non-optimal codons. |
Advanced Data-Driven Approaches
Emerging deep learning frameworks like RiboDecode represent a paradigm shift from rule-based to data-driven, context-aware optimization [73]. This tool integrates a translation prediction model trained on over 320 paired ribosome profiling (Ribo-seq) and RNA sequencing (RNA-seq) datasets from human tissues and cell lines, allowing it to learn complex relationships between codon sequences and their translation levels directly from experimental data [73]. Furthermore, RiboDecode incorporates a dedicated MFE prediction model and a codon optimizer that uses gradient ascent to explore a vast sequence space, generating synonymous sequences that maximize a fitness score combining both translation efficiency and stability [73]. This approach has demonstrated robust performance across different mRNA formats, including unmodified, m1Ψ-modified, and circular mRNAs, making it highly relevant for advanced therapeutic applications [73].
Experimental Protocol for Codon Optimization in Yeast
This protocol provides a step-by-step methodology for optimizing and validating codon usage for a heterologous gene intended for expression in S. cerevisiae.
Computational Design and Sequence Optimization
- Gene Sequence Acquisition: Obtain the amino acid sequence of the target protein. If starting from a DNA sequence, translate it to confirm the correct amino acid sequence.
- Host Codon Usage Analysis: Retrieve the codon usage table for Saccharomyces cerevisiae from a reliable database such as the Kazusa DNA Research Institute database.
- Tool Selection and Optimization:
- Select one or more optimization tools from Table 1 (e.g., OPTIMIZER, ATGme, or RiboDecode if available).
- Input the target amino acid sequence and select S. cerevisiae as the host organism.
- Set parameters to optimize for CAI, GC content (aim for a balanced percentage), and reduce stable mRNA secondary structures, especially near the start codon.
- Generate 3-5 candidate optimized DNA sequences.
- In silico Analysis:
- Calculate the CAI, GC content, and predict MFE for each candidate sequence and the wild-type sequence using tools like RNAfold [69].
- Check for the inadvertimental introduction of cryptic regulatory elements (e.g., splice sites, polyadenylation signals).
- Select the top candidate sequence for synthesis.
Gene Synthesis and Cloning
- DNA Synthesis: Commission the synthesis of the optimized gene sequence from a commercial provider.
- Vector Construction:
- Clone the synthesized gene into a suitable yeast expression vector (e.g., a plasmid with a strong, inducible promoter like GAL1, and a selectable marker like URA3).
- Use standard cloning techniques such as Gibson assembly or restriction enzyme digestion and ligation.
- Transform the constructed plasmid into E. coli for amplification and verify the plasmid sequence by Sanger sequencing.
Functional Validation in Yeast
- Yeast Transformation: Transform the verified plasmid into an appropriate S. cerevisiae strain (e.g., BY4741) using the lithium acetate/single-stranded carrier DNA/polyethylene glycol (LiAc/SS-DNA/PEG) method.
- Culture and Induction: Inoculate positive transformants into selective medium and grow to mid-log phase. Induce protein expression by adding the relevant inducer (e.g., galactose for the GAL1 promoter).
- Protein Expression Analysis:
- Harvesting: Collect cells by centrifugation at various time points post-induction.
- Cell Lysis: Lyse cells using glass bead beating or enzymatic lysis in an appropriate lysis buffer.
- SDS-PAGE and Western Blotting: Separate total proteins by SDS-PAGE and perform Western blotting with an antibody specific to the target protein to confirm identity and estimate expression levels relative to a control (e.g., yeast expressing the wild-type gene).
- Assessment of Translational Efficiency:
- Quantitative PCR (qPCR): Extract total mRNA from induced cultures and perform qPCR with primers for the target gene and a housekeeping gene (e.g., ACT1). This normalizes for transcript abundance.
- Calculate Protein per mRNA: Combine Western blot densitometry and qPCR data to estimate the protein produced per unit of mRNA, a direct measure of translational efficiency.
The experimental workflow for this protocol is summarized in the diagram below.
Table 2: Essential Research Reagents for Codon Optimization and Validation in Yeast
| Reagent / Resource | Function / Application | Example / Notes |
|---|---|---|
| Codon Optimization Tools | Computational design of optimized gene sequences. | OPTIMIZER [69], ATGme [69], RiboDecode [73]. |
| Codon Usage Table | Reference for the preferred codons of the host organism. | S. cerevisiae table from Kazusa DB [74]. |
| Yeast Expression Vector | Plasmid for gene expression in yeast; contains promoter and marker. | pYES2 (GAL1 promoter, URA3 marker). |
| S. cerevisiae Strain | Host organism for heterologous protein expression. | BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0). |
| Yeast Transformation Kit | Introducing plasmid DNA into yeast cells. | LiAc/SS-DNA/PEG method kit. |
| Antibody for Target Protein | Detecting and quantifying recombinant protein expression. | Primary antibody for Western blot. |
| qPCR Reagents | Quantifying mRNA transcript levels of the target gene. | SYBR Green mix, primers for target and reference gene (e.g., ACT1). |
Supplementary tRNA Enhancement Strategy
A complementary strategy to codon optimization involves directly modulating the host's tRNA pool. Research has shown that overexpressing specific tRNAs that correspond to abundant, non-optimal codons in a target gene can enhance protein expression [72]. For instance, co-expressing tRNA^Ala^AGC-2-1 alongside a target mRNA rich in its cognate codons led to a 4.7-fold increase in protein production in human cells [72]. Furthermore, chemically synthesizing tRNAs with site-specific modifications in the anticodon loop (to enhance decoding efficacy) and the TΨC loop (to improve stability and interaction with elongation factors) can yield tRNAs with ~4-fold higher decoding efficacy than unmodified tRNAs [72]. While more common in mammalian systems, this strategy represents a powerful tool for yeast metabolic engineers facing expression bottlenecks with genes containing codons that are rare in yeast.
The relationship between codon choice, tRNA availability, and translational efficiency is illustrated below.
Rewiring Central Metabolism to Enhance Precursor and Cofactor Supply
Within the framework of heterologous pathway reconstruction in yeast, the efficient production of target compounds is often constrained by the limited supply of essential metabolic precursors and cofactors. Native metabolism in Saccharomyces cerevisiae is intrinsically optimized for growth and ethanol production, not for the high-yield synthesis of non-native chemicals [75]. Rewiring central carbon metabolism is therefore a fundamental strategy to overcome these limitations, enhancing the flux toward key building blocks like erythrose-4-phosphate (E4P), acetyl-CoA, and redox cofactors required for the success of engineered pathways. This application note details key engineering strategies and provides actionable protocols to implement these solutions in a research setting.
Quantitative Analysis of Metabolic Rewiring Outcomes
Table 1: Key Performance Metrics from Metabolic Rewiring Strategies
| Engineering Strategy | Target Molecule | Maximum Titer Achieved | Yield on Glucose | Key Genetic Modifications |
|---|---|---|---|---|
| E4P & AAA Pathway Enhancement | ( p )-Coumaric acid | 12.5 g L⁻¹ | 154.9 mg g⁻¹ | Phosphoketolase pathway for E4P; Feedback-insensitive Aro4, Aro7; Overexpression of Aro1, Aro2, Aro3, Pha2 [76] |
| Cytosolic Acetyl-CoA Generation | Various products (in Pdc⁻ strain) | N/A | N/A | Heterologous pathways: Pfl, A-Ald, Pdhcyto, Po/Pta, Pk/Pta [75] |
| Growth-Product Coupling (in silico) | 29 diverse products | N/A | N/A | Model-predicted knockout strategies (e.g., SDH3, SER3, SER33; ICL1, KGD1, PYC1) [77] |
Detailed Experimental Protocols
Protocol: Enhancing the Aromatic Amino Acid (AAA) Biosynthesis Pathway
This protocol outlines the steps to rewire central carbon metabolism to increase the supply of erythrose-4-phosphate (E4P) and enhance flux into the aromatic amino acid biosynthesis pathway, based on the work for high-level production of ( p )-coumaric acid [76].
Materials
- Strain Background: Saccharomyces cerevisiae BY4741 or similar laboratory strain.
- Engineering Tools: CRISPR-Cas9 system for yeast [78] or traditional homologous recombination with recyclable markers.
- Plasmids: Vectors for heterologous gene expression (e.g., pRS series). For high-throughput pathway assembly, consider the YeastFab system using Golden Gate assembly [79].
- Key Genetic Elements:
- ARO4^{K229L} and ARO7^{G141S} alleles (feedback-insensitive mutants).
- xfpk gene (phosphoketolase) from B. subtilis or other sources.
- Genes for overexpression: ARO1, ARO2, ARO3, PHA2.
- Strong, constitutive promoters (e.g., TEF1, PGK1) for gene overexpression.
Procedure
- Relieve Feedback Inhibition: Genomically integrate the feedback-insensitive alleles of ARO4^{K229L} and ARO7^{G141S} using CRISPR-Cas9 mediated gene replacement [76] [78].
- Introduce Phosphoketolase Pathway: Assemble an expression cassette for the heterologous xfpk gene. Integrate this cassette into a genomic locus to create a new flux route from fructose-6-phosphate and xylose-5-phosphate to E4P [76].
- Overexpress Downstream Pathway Enzymes: Systematically overexpress genes encoding enzymes of the AAA pathway. A significant boost in flux is achieved by co-overexpressing ARO1, ARO2, ARO3, and PHA2 [76]. Assemble these expression cassettes and integrate them into the yeast genome.
- Optimize Promoter Strength: Replace the native promoters of key genes at critical nodes between glycolysis and the AAA pathway (e.g., TKL1) with promoters of varying strengths to fine-tune carbon distribution [76].
- Fermentation and Validation:
- Inoculate the engineered strain in a suitable synthetic defined (SD) medium with 2% glucose.
- Grow cultures at 30°C with shaking. Monitor growth (OD600) and, for ( p )-coumaric acid production, quantify the titer using HPLC [76].
- Compare the performance of the final engineered strain against the base strain to validate the enhancement in precursor supply.
Protocol: Rewiring Redox Metabolism in Pyruvate Decarboxylase-Deficient (Pdc⁻) Strains
This protocol describes strategies to enable growth and production in strains where ethanol synthesis is abolished, focusing on restoring redox balance and cytosolic acetyl-CoA supply [75].
Materials
- Strain Background: Pdc⁻ strain (e.g., pdc1Δ pdc5Δ pdc6Δ).
- Engineering Tools: CRISPR-Cas9 system [78].
- Key Genetic Elements:
- Heterologous acetyl-CoA pathways (e.g., pfl, A-ALD, PDH_{cyto}, or the pk/pta operon).
- Mutations in glucose-sensing regulators (e.g., mth1-Δ or MTH1 mutants).
Procedure
- Circumvent C2-Auxotrophy: Introduce a heterologous pathway for cytosolic acetyl-CoA synthesis. For example, clone and express the phosphoketolase (xfpk)-phosphotransacetylase (pta) operon to generate acetyl-CoA from sugar phosphates [76] [75].
- Address Glucose Derepression: Delete or mutate MTH1 to reduce the expression of high-affinity hexose transporters (Hxt), thereby limiting glucose uptake and alleviating the Crabtree effect [75].
- Adaptive Laboratory Evolution (ALE):
- Grow the engineered Pdc⁻ mth1-Δ strain in serial batch cultures or chemostats with glucose as the sole carbon source.
- Monitor for the emergence of faster-growing mutants. Isolate and sequence evolved clones to identify causative mutations (e.g., in YAK1) [75].
- Provide Alternative Redox Sinks: Introduce pathways that consume NADH and produce a valuable or benign compound, helping to regenerate NAD⁺ and maintain redox balance [75] [77].
Pathway and Workflow Visualizations
Metabolic Rewiring of the AAA Biosynthesis Pathway
Workflow for Computational Pathway Expansion and Derivatization
This diagram outlines a computational workflow for identifying and producing novel derivatives of heterologous pathways, as applied to the noscapine pathway [80].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Research Reagents for Metabolic Rewiring in Yeast
| Reagent / Tool Name | Function / Application | Specific Example / Note |
|---|---|---|
| CRISPR-Cas9 System | Enables efficient, multiplexed gene editing, knockout, and integration. | High-efficiency gRNA plasmids for targeted integration; Used for introducing point mutations (e.g., ARO4^{K229L}) and deleting multiple genes (e.g., PDC genes) [78]. |
| YeastFab Assembly | Standardized, high-throughput assembly of genetic parts for pathway construction. | Golden Gate-based method for modular assembly of Promoters, ORFs, and Terminators (PRO, ORF, TER); Ideal for combinatorial testing of pathway expression levels [79]. |
| Phosphoketolase (XFPK) | Diverts glycolytic flux to enhance E4P or acetyl-CoA supply. | Heterologous enzyme from B. subtilis; Creates a non-native pathway from F6P/X5P to E4P and acetyl-phosphate [76] [75]. |
| Feedback-Insensitive Mutants | Relieves allosteric regulation to increase flux into desired pathways. | ARO4^{K229L} (DAHP synthase) and ARO7^{G141S} (chorismate mutase) are key for AAA pathway [76]. |
| Alternative Acetyl-CoA Pathways | Generates cytosolic acetyl-CoA in Pdc⁻ strains, overcoming C2-auxotrophy. | Pathways include Pyruvate-formate lyase (Pfl), Acetylating acetaldehyde dehydrogenase (A-Ald), or cytosolic pyruvate dehydrogenase (Pdhcyto) [75]. |
| Synthetic Promoter Libraries | Fine-tunes gene expression strength to optimize metabolic flux. | Used to replace native promoters of key nodes (e.g., TKL1) to balance carbon distribution between glycolysis and target pathways [76] [11]. |
| Polycistronic Vectors | Allows coordinated expression of multiple genes from a single transcript. | Useful for compact expression of entire biosynthetic gene clusters (BGCs) from fungal or plant pathways in yeast [10]. |
Adaptive Laboratory Evolution (ALE) for Improved Strain Robustness and Titers
Within the framework of heterologous pathway reconstruction in yeast, achieving high product titers and robust industrial performance is a central challenge. Rational metabolic engineering often encounters unforeseen physiological bottlenecks, such as metabolic imbalances, stress induced by pathway intermediates, or suboptimal flux through introduced pathways [81]. Adaptive Laboratory Evolution (ALE) serves as a powerful complementary strategy to address these complex, multigenic traits by harnessing the power of natural selection under defined selective pressures [82]. This approach allows for the selection of beneficial mutations that enhance overall host fitness, which often concomitantly improves stress tolerance, substrate utilization, and ultimately, the production of target compounds [83] [84]. This Application Note provides detailed protocols and methodologies for implementing ALE to develop superior yeast chassis for synthetic biology applications.
ALE Experimental Design and Workflow
A well-designed ALE experiment involves serial passaging of microbial populations over numerous generations under a specific selective pressure, such as the presence of a toxic intermediate or product, or the utilization of a non-preferred carbon source [82].
Core Protocol: Continuous Transfer Culture
This foundational method is ideal for most ALE experiments in yeast [82].
- Objective: To evolve yeast populations with enhanced tolerance to lactic acid and improved production titers.
- Principle: Repeatedly transferring a growing microbial culture to fresh medium maintains constant selective pressure, enabling the accumulation of beneficial mutations.
Methodology:
- Inoculum Preparation: Start with a genetically diverse population of the engineered yeast strain (e.g., Kluyveromyces marxianus with a lactic acid production pathway [83]).
- Culture Conditions: Use a defined medium with the target carbon source (e.g., glucose or xylose). To drive evolution towards lactic acid tolerance, supplement the medium with progressively increasing concentrations of lactic acid, starting from a sub-inhibitory level (e.g., 20 g/L).
- Serial Passaging:
- Grow the culture in flasks with appropriate aeration.
- Monitor growth (OD600) to determine the transition into the early stationary phase.
- At each passage, inoculate fresh medium with a small volume of the prior culture (typically 1-10% transfer volume). A lower transfer volume increases selection pressure but may reduce genetic diversity [82].
- The transfer interval should be dynamically adjusted based on the growth rate, typically occurring every 24-72 hours for yeasts.
- Evolution Duration: Continue the serial passaging for a sufficient number of generations (often 200-400 generations) to allow for significant phenotypic improvement. Periodically archive frozen stocks (at -80°C in 15-25% glycerol) of the evolving population.
- Endpoint Analysis: After evolution, isolate single clones from the final population and characterize them for target traits (e.g., lactic acid titer, yield, and growth rate under stress).
The following workflow diagrams the complete ALE process from setup to mutant characterization.
Protocol for Automated Evolution in Bioreactors
For greater control and scalability, ALE can be conducted in automated bioreactor systems like turbidostats or chemostats [82].
- Objective: To maintain a constant, optimal growth rate under high-density fermentation conditions.
- Principle: A turbidostat maintains a constant cell density by continuously adding fresh medium based on turbidity measurements, ensuring evolution under rapid, nutrient-rich growth. A chemostat maintains a constant dilution rate, favoring evolution under nutrient-limited conditions and specific metabolic fluxes.
Methodology:
- System Setup: Initialize the bioreactor with the engineered yeast strain and a defined medium.
- Parameter Control:
- For a turbidostat, set the OD600 set-point. The system will automatically add fresh medium when this density is exceeded.
- For a chemostat, set a fixed dilution rate (D) that is less than the maximum growth rate (μmax) of the strain.
- Evolution: Allow the culture to evolve for a predetermined number of generations or volume changes. The continuous culture in a turbidostat can achieve a very high number of generations in a short time.
- Sampling: Periodically sample the effluent or the culture vessel to monitor phenotypic changes and archive populations for later analysis.
Quantitative Performance of ALE-Evolved Yeasts
ALE has been successfully applied to enhance various traits in yeasts relevant to heterologous pathway reconstruction. The table below summarizes key performance metrics from documented cases.
Table 1: Performance Metrics of ALE-Evolved Yeast Strains
| Yeast Species | Target Trait / Product | Selective Pressure | Key Outcomes | Reference |
|---|---|---|---|---|
| Kluyveromyces marxianus | Lactic Acid Production | General fitness in production medium | Titer: 120 g/LYield: 0.81 g/g18% increase in production; Enhanced xylose fermentation | [83] |
| Saccharomyces cerevisiae | Aroma Compound (2-Phenylethanol) | Not Specified | Increased production of the rose-scented compound 2-PE to meet market demand | [84] |
| Saccharomyces cerevisiae | Ethanol Tolerance | High ethanol concentrations | Achieved >1 order of magnitude improvement in tolerance within ~80 generations | [82] |
| Escherichia coli (as reference) | Autotrophic Growth | CO₂ as sole carbon source | Successfully evolved to use the Calvin cycle for growth on CO₂ | [82] |
The relationship between ALE, core cellular processes, and the resulting industrially relevant phenotypes can be visualized as a signaling network.
The Scientist's Toolkit: Key Research Reagent Solutions
Successful implementation of ALE requires specific laboratory materials and reagents. The following table details essential items and their functions.
Table 2: Essential Research Reagents and Materials for ALE
| Item | Function / Application in ALE |
|---|---|
| CRISPR/Cas9 System | Precision genome editing for pathway reconstruction prior to ALE and for reverse engineering of identified mutations post-ALE [81]. |
| Defined Medium Components | For preparing selective growth media with specific carbon sources (e.g., glucose, xylose) and incorporating stress-inducing agents like lactic acid or other inhibitors [83] [82]. |
| Cryopreservation Reagents (Glycerol) | For archiving intermediate and final evolved populations and isolates at -80°C to maintain a frozen fossil record of the evolution experiment [82]. |
| Next-Generation Sequencing (NGS) | For whole-genome sequencing of evolved clones to identify causal mutations, a critical step for linking genotype to phenotype [83] [82]. |
| Automated Bioreactor (Turbidostat/Chemostat) | For running controlled, high-throughput, and long-term evolution experiments with minimal manual intervention [82]. |
| Plasmids for Heterologous Expression | Vectors (YIp, YCp, YEp) for integrating or maintaining heterologous pathways, such as lactate dehydrogenase (LDH) for lactic acid production [83] [11]. |
Measuring Success: Validation, Analytical Methods, and Chassis Comparison
The reconstruction of heterologous pathways in yeast is a cornerstone of modern industrial biotechnology, enabling the production of valuable chemicals and pharmaceuticals. However, the simple introduction of foreign genes into a host like Saccharomyces cerevisiae is often insufficient for achieving economically viable product yields [2]. Success hinges on the application of quantitative frameworks that allow researchers to calculate pathway yield and understand the stoichiometric limits imposed by yeast metabolism. These frameworks are essential for predicting the theoretical maximum yield of a target compound, identifying rate-limiting steps, and guiding rational strain optimization [85]. This document provides application notes and detailed protocols for employing these critical quantitative tools within the context of heterologous pathway reconstruction in yeast, providing researchers and drug development professionals with methodologies to enhance the efficiency and output of their engineered strains.
Theoretical Foundations: From Stoichiometry to Thermodynamics
A robust quantitative analysis of a heterologous pathway begins with a thorough understanding of the theoretical constraints that govern microbial growth and product formation.
Stoichiometric Limits of Biomass and Product Yield
The conversion of a carbon source into biomass or a target product is bound by fundamental stoichiometric limits. The maximum possible biomass yield is determined by two primary factors: the carbon lost as CO₂ during anabolic processes and the carbon substrate required for generating essential reducing equivalents, specifically NADPH [85]. This can be conceptualized as a mass balance problem where the carbon from the substrate is partitioned between biomass, products, CO₂, and cellular energy.
The biomass yield on ATP (YATP) is a key parameter in these calculations, representing the grams of dry biomass produced per mole of ATP consumed. This value is not constant; it depends strongly on the nature of the carbon source and the macromolecular composition of the cell, particularly its protein content [85]. Furthermore, the efficiency of energy-generating systems, quantified by the P/O ratio (the number moles of ATP synthesized per atom of oxygen reduced in the mitochondrial respiratory chain), has a profound impact on the overall cellular yield. A higher P/O ratio signifies more efficient energy generation and thus a higher potential biomass yield [85].
Incorporating Thermodynamic Constraints
While stoichiometric models define what is possible, thermodynamic analysis reveals what is feasible. The direction and flux of biochemical reactions are constrained by their change in Gibbs free energy (ΔG). System-Level Thermodynamic Analysis of Elementary Flux Modes (EFMs) has emerged as a powerful method to integrate quantitative metabolite concentration data with network stoichiometry to rule out thermodynamically infeasible flux distributions [86].
The principle is that any thermodynamically feasible flux distribution in a metabolic network can be described as a non-negative linear combination of thermodynamically feasible EFMs [86]. By calculating all EFMs for a yeast metabolic network and then classifying them as thermodynamically feasible or infeasible based on experimental metabolome data, researchers can significantly narrow the solution space of possible metabolic operations. This approach has provided system-level insights, for example, demonstrating how compartmental concentrations of NADH and NAD+ prevent certain intercompartmental redox shuttles from operating under typical glucose batch conditions [86].
Table 1: Key Quantitative Parameters in Yeast Metabolic Models
| Parameter | Description | Impact on Yield Calculation |
|---|---|---|
| YATP (g biomass/mol ATP) | Biomass yield on ATP | Determines the ATP cost of building biomass; varies with carbon source and cell composition [85]. |
| P/O Ratio | ATP yield from oxidative phosphorylation | Defines efficiency of energy metabolism; lower ratio means more substrate needed for same biomass [85]. |
| Maintenance Energy (mATP) | ATP used for non-growth functions | Accounts for energy used for cell motility, osmotic stress response, and protein turnover [85]. |
| Cofactor Coupling (NADPH/ATP demand) | Stoichiometric demand for redox cofactors | Limits maximum carbon assimilation; dictates carbon source needed for anabolism [85]. |
Quantitative Frameworks in Practice: Protocols and Applications
Protocol: In Silico Stoichiometric Modeling for Yield Prediction
This protocol outlines the steps for using a genome-scale metabolic model (GEM) to calculate the theoretical maximum yield of a target compound from a specified substrate.
Principle: GEMs are computational representations of an organism's metabolism. By applying constraints (e.g., substrate uptake rate, reaction reversibility), one can use Flux Balance Analysis (FBA) to identify a flux distribution that maximizes for a specific objective, such as product formation.
Materials:
- Software: CobraPy, COBRA Toolbox for MATLAB, or similar environment.
- Model: A curated genome-scale metabolic model for Saccharomyces cerevisiae (e.g., Yeast8, iMM904).
- Computing Environment: A standard desktop computer is sufficient for most models.
Procedure:
- Model Import and Curation: Import the chosen GEM into your software environment. Verify that the heterologous pathway for your target compound (e.g., itaconic acid, 3-methyl-1-butanol) is accurately represented in the model, including cofactor balances (NADH, NADPH, ATP) [87].
- Define Constraints: Set the constraints that reflect your experimental conditions.
- Set the glucose uptake rate to a realistic value (e.g., -10 mmol/gDW/h).
- Set the oxygen uptake rate for aerobic or anaerobic conditions.
- Define the non-growth associated ATP maintenance (NGAM) requirement.
- If known, constrain the fluxes of deleted or non-functional genes to zero.
- Set the Objective Function: Define the reaction representing the export of your target metabolite as the objective function to be maximized.
- Run Flux Balance Analysis: Execute the FBA simulation. The output will provide a flux value for every reaction in the network, including the maximum theoretical production rate of your target compound.
- Calculate Maximum Yield: The maximum yield (YP/S) in gram product per gram substrate is calculated from the simulated production flux and substrate uptake flux, adjusted for their molecular weights.
Application Note: This in silico approach was used to predict gene deletion targets that could reduce acetate byproduct formation in a strain engineered for 3-methyl-1-butanol co-production with ethanol, leading to a 4.4-fold increase in 3MB yield [87].
Protocol: Thermodynamic Analysis of Elementary Flux Modes
This protocol describes a method for refining flux predictions by incorporating metabolite concentration data to eliminate thermodynamically infeasible pathways.
Principle: This analysis uses quantitative metabolomics data to calculate the Gibbs free energy change (ΔG) for reactions within each Elementary Flux Mode (EFM). EFMs with reactions operating in a thermodynamically infeasible direction (positive ΔG for a reaction carrying flux) are discarded [86].
Materials:
- Stoichiometric Model: A medium- to large-scale metabolic network of S. cerevisiae.
- Software: EFMTool or similar for EFM calculation; software for NET analysis.
- Metabolome Data: Quantitative intracellular metabolite concentration data for key central metabolites, ideally compartmentalized (cytosol, mitochondrion), obtained from LC-MS/MS or GC-MS.
Procedure:
- Calculate Elementary Flux Modes: Compute all EFMs for the metabolic network under defined environmental conditions (e.g., glucose batch growth) [86].
- Integrate Metabolite Data: Input the experimentally determined, compartment-specific metabolite concentrations into the thermodynamic model.
- Classify EFMs: For each EFM, calculate the ΔG for every reaction carrying a non-zero flux. An EFM is classified as thermodynamically feasible only if all its reactions operate with a negative ΔG under the given metabolite concentrations.
- Analyze Results: The resulting set of feasible EFMs defines the restricted solution space. Compare feasible and infeasible EFMs to identify system-level thermodynamic bottlenecks, such as infeasible intercompartmental redox shuttles [86].
Application Note: Applying this method to a model of yeast central metabolism with 71 million EFMs allowed researchers to classify 54% as thermodynamically infeasible based on metabolome data, providing critical insight into the impossibility of certain metabolic cycles under standard conditions [86].
Diagram 1: Workflow for thermodynamic analysis of Elementary Flux Modes.
Case Study: Calculating Yield in an Engineered Itaconic Acid Pathway
The production of itaconic acid in S. cerevisiae provides a clear example of yield optimization through quantitative analysis.
Background: Itaconic acid is a platform chemical with applications in polymer and resin production. While naturally produced by fungi like Ustilago maydis, its pathway was introduced into the industrially robust host S. cerevisiae [88].
Quantitative Analysis and Outcome:
- Pathway Identification: Researchers systematically compared different fungal itaconic acid pathways and identified the Ustilago route, including its mitochondrial transport mechanism, as the most efficient in yeast [88].
- Yield Calculation: The yield was quantified as the titer (g/L) in the fermentation broth. Initial shake flask cultivations in pH-buffered Verduyn media produced 1.3 g/L [88].
- Identifying Limitations: In silico and experimental analyses identified native yeast transporters (Aqr1p, Dtr1p, Qdr3p) that promiscuously exported itaconic acid but were suboptimal. Replacing the main promiscuous transporter (Dtr1p) with the specialized Ustilago transporter (Itp1) significantly increased biosynthesis [88].
- Maximizing Yield via Process Control: The final step involved transferring the production to a controlled 3.8 L bioreactor. This allowed for optimized fed-batch fermentation, controlling parameters like dissolved oxygen and nutrient feed, which pushed the final titer to 4.7 g/L—the highest reported in S. cerevisiae at the time of the study [88]. This demonstrates how yield is a function of both genetic design and process stoichiometry/kinetics.
Table 2: Comparative Yields from Engineered Yeast Pathways
| Target Product | Host | Key Engineering Strategy | Reported Yield / Titer | Stoichiometric / Thermodynamic Consideration |
|---|---|---|---|---|
| Itaconic Acid [88] | S. cerevisiae | Expression of Ustilago pathway & specialized transporter Itp1 | 4.7 g/L (Bioreactor) | Optimized transport efficiency to overcome export bottleneck. |
| 3-Methyl-1-Butanol (3MB) [87] | S. cerevisiae | Alleviation of valine/leucine feedback inhibition; byproduct reduction. | 1.5 mg/g sugars (4.4-fold increase) | Redirected carbon flux from byproducts (e.g., acetate) to target product. |
| Ethylene Glycol [89] | S. cerevisiae | Dahms oxidative pathway for xylose; iron metabolism engineering. | 1.5 g/L | Engineered Fe-S cluster supply to activate bottleneck enzyme XylD. |
| Abscisic Acid (ABA) [90] | S. cerevisiae | Balanced expression of P450 enzymes (BcABA1, BcABA2); CPR overexpression. | 4.1-fold increase from baseline | Addressed limiting heterologous enzymes and cofactor (NADPH) supply for P450s. |
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Reagents for Quantitative Analysis of Yeast Pathways
| Research Reagent / Tool | Function / Application | Example Use in Context |
|---|---|---|
| Genome-Scale Metabolic Model (GEM) | In silico prediction of maximum yields and identification of gene knockout targets. | Yeast8 model used to predict deletions that reduce acetate in 3MB-producing strains [87]. |
| CRISPR-Cas9 System | Precision genome editing for gene knock-outs, knock-ins, and promoter swaps. | Used to integrate heterologous genes (e.g., bcaba cluster) and mutate regulatory sites (e.g., LEU4) [87] [90]. |
| LC-MS / GC-MS Platforms | Quantitative metabolomics for measuring intracellular metabolite concentrations. | Provides essential data for thermodynamic flux analysis and calculating Gibbs free energy [86]. |
| Cytochrome P450 Reductase (CPR) | Supplies reducing equivalents (NADPH) to heterologous cytochrome P450 enzymes. | Overexpression (native or heterologous) increased titers in abscisic acid production [90]. |
| Inducible / Constitutive Promoters | Fine-tuning the expression levels of heterologous pathway genes. | PAOX1 in P. pastoris; used to balance expression of BcABA1 and BcABA2 to reduce bottlenecks [2] [90]. |
| Fed-Batch Bioreactor | Provides controlled environmental conditions (pH, O₂, nutrient feed) for maximizing yield. | Enabled a ~3.6x increase in itaconic acid titer compared to shake flasks [88]. |
Diagram 2: Metabolic node competition between native ethanol production and a heterologous pathway.
The reconstruction of heterologous biosynthetic pathways in yeast, such as Saccharomyces cerevisiae, is a cornerstone of modern metabolic engineering for producing high-value pharmaceuticals, nutraceuticals, and natural products [91] [37]. However, the successful engineering of these complex pathways requires more than just the integration of foreign genes; it demands rigorous validation to ensure that the intended enzymes are expressed and functional. Multi-omics integration, specifically the combined application of transcriptomics and proteomics, provides a powerful framework for this validation. By comparing the transcriptional output (transcriptomics) with the actual protein abundance (proteomics), researchers can confirm the correct expression of heterologous pathways, identify potential bottlenecks, and diagnose unexpected physiological responses in the host organism [92] [93]. This protocol details the application of these techniques within the context of yeast metabolic engineering, providing a structured approach to data acquisition, analysis, and interpretation to assure the validity of engineering outcomes [92].
Key Research Reagent Solutions
The following table catalogs essential reagents and materials critical for the execution of transcriptomic and proteomic analyses in yeast.
Table 1: Essential Research Reagents for Multi-omics Validation in Yeast
| Reagent/Material | Function in Multi-omics Workflow |
|---|---|
| CRISPR/Cas9 System [91] [37] | Enables multiplex genomic integration of heterologous biosynthetic pathway genes into the yeast chromosome. |
| Promoter/Terminator Library [91] | Provides a set of well-characterized genetic parts for fine-tuned, stable expression of integrated heterologous genes. |
| Liquid Chromatography (LC) System [92] | Separates complex mixtures of peptides or proteins prior to mass spectrometric analysis. |
| High-Resolution Mass Spectrometer (HRMS) [92] | Precisely measures the mass-to-charge ratio of ions to identify and quantify peptides and proteins. |
| Calibration Mixtures [92] | Standard solutions used to calibrate the mass spectrometer, ensuring high mass accuracy for reliable identification. |
| Quality Control (QC) Samples [92] | Simple or complex peptide/protein samples used to monitor and ensure the stability and performance of the LC-HRMS instrumentation. |
Experimental Protocols
Genome Engineering for Heterologous Pathway Reconstruction
A critical first step is the stable integration of the heterologous pathway into the yeast genome. CRISPR/Cas9-mediated multiplex editing is the preferred method for its efficiency and precision [91] [37].
- gRNA Design and Synthesis: Design single-guide RNAs (gRNAs) with 20-nt spacer sequences complementary to the genomic loci targeted for integration. The target site must be immediately followed by a 5'-NGG-3' Protospacer Adjacent Motif (PAM) [37].
- Donor DNA Preparation: For each heterologous gene, prepare a linear donor DNA fragment containing the gene of interest flanked by a selected promoter and terminator from the library. This "gene expression cassette" must also include 25-50 bp homology arms on each end, corresponding to the sequences immediately upstream and downstream of the Cas9-induced double-strand break (DSB) or to adjacent cassettes for in-locus assembly [91].
- Yeast Transformation: Co-transform the recipient yeast strain with:
- A plasmid expressing the Cas9 enzyme and the designed gRNAs.
- The pooled linear donor DNA fragments for all pathway genes.
- Selection and Validation: Plate the transformed yeast on selective media. Isolate colonies and validate correct genomic integration via colony PCR and DNA sequencing.
Transcriptomic Analysis (RNA-seq)
This protocol outlines the steps for using RNA sequencing to profile gene expression in the engineered yeast strain.
- Sample Harvest: Culture the engineered yeast strain and an appropriate control strain under defined conditions. Harvest cells at the desired growth phase (e.g., mid-logarithmic or stationary phase) by rapid centrifugation. Immediately flash-freeze the cell pellet in liquid nitrogen [93].
- RNA Extraction and QC: Extract total RNA using a hot phenol method or a commercial kit. Assess RNA integrity and purity using an instrument such as a Bioanalyzer; an RNA Integrity Number (RIN) > 8.0 is typically required.
- Library Preparation and Sequencing: Deplete ribosomal RNA and prepare sequencing libraries using a standardized kit. Perform paired-end sequencing (e.g., 150 bp) on an Illumina platform to a depth of at least 20 million reads per sample.
- Bioinformatic Analysis:
- Quality Control: Use FastQC to assess raw read quality.
- Alignment: Map cleaned reads to the S. cerevisiae reference genome and a custom reference containing the heterologous pathway sequences using a splice-aware aligner like HISAT2 or STAR.
- Quantification: Count reads mapping to each gene using featureCounts.
- Differential Expression: Identify significantly differentially expressed genes between engineered and control strains using packages such as DESeq2 or edgeR. Focus on the expression of integrated heterologous genes and any perturbed native pathways.
Proteomic Analysis (LC-HRMS)
Proteomics validates the presence of the proteins encoded by the integrated genes. Liquid Chromatography-High-Resolution Mass Spectrometry (LC-HRMS) is the gold standard [92].
- Sample Preparation: Lyse the yeast cell pellets mechanically or chemically. Digest the extracted proteins into peptides using a sequence-grade protease like trypsin. Desalt the resulting peptides.
- Liquid Chromatography: Separate the complex peptide mixture using a reverse-phase UHPLC system with a C18 column and a water-acetonitrile gradient [92].
- High-Resolution Mass Spectrometry: Analyze the eluting peptides using a high-resolution mass spectrometer (e.g., Orbitrap) coupled to the LC system. Operate in data-dependent acquisition (DDA) mode, cycling between a full MS1 scan and subsequent MS2 fragmentation scans of the most intense ions [92].
- Data Processing and Protein Identification:
- Database Search: Search the raw MS/MS spectra against a protein database containing the yeast proteome and the sequences of the heterologous enzymes using search engines such as MaxQuant or FragPipe.
- Quality Control: Adhere to a standardized protocol to ensure validity [92]. This includes:
- Quantification: Use label-free (e.g., MS1 intensity) or isobaric labeling (e.g., TMT) methods to quantify protein abundance.
Data Integration and Validation
The core of the validation process lies in the integrated analysis of transcriptomic and proteomic datasets.
- Data Correlation: Perform a correlation analysis between the transcript levels (from RNA-seq) and the protein abundance (from LC-HRMS) for the heterologous pathway genes. Strong correlation confirms that transcriptional engineering has successfully translated to the protein level.
- Identification of Discordance: Investigate cases where high transcript levels do not result in correspondingly high protein levels. This discordance can indicate post-transcriptional regulation, protein instability, or inefficient translation due to codon usage.
- Contextual Analysis with Phenotypic Data: Integrate the multi-omics data with measured product titers and growth phenotyping. For example, if the desired product is not detected, low abundance of a key pathway enzyme (proteomics) despite high mRNA levels (transcriptomics) would point to a post-translational bottleneck. Conversely, if the proteomics confirms the presence of all pathway enzymes but the product titer is low, the issue may lie with enzyme activity, cofactor availability, or metabolic flux constraints.
Table 2: Interpreting Multi-omics Data for Pathway Validation
| Transcriptomic Data | Proteomic Data | Product Titer | Potential Interpretation |
|---|---|---|---|
| High | High | High | Successful pathway reconstruction and function. |
| High | Low | Low | Bottleneck: Post-transcriptional regulation, protein degradation, or translation inefficiency. |
| Low | Low | Low | Bottleneck: Weak promoter, failed integration, or genetic instability. |
| High | High | Low | Bottleneck: Possible issue with enzyme activity, substrate/cofactor availability, or metabolic flux. |
Case Study: Investigating Host Response to Cannabinoid Production
An integrated omics approach was used to study the physiological response of S. cerevisiae to cannabidiol (CBD), a heterologous product, revealing critical insights for bioengineers [93].
- Transcriptomics: Revealed the significant overexpression of genes including PDR5, an ABC transporter, indicating a pleiotropic drug resistance (PDR) response was activated to efflux CBD [93].
- Proteomics: Can be applied to validate the increased abundance of the Pdr5p transporter protein and other stress-related proteins.
- Metabolomics: Further analysis identified lipidomic remodeling, including changes in fatty acids and glycerophospholipids, suggesting the yeast was adapting its membrane composition in response to CBD [93].
- Integrated Conclusion: The multi-omics data revealed that the engineered yeast was not only producing the target compound but also actively expending energy to export it and remodel its cellular structures. This explained the observed growth perturbations and highlighted the need to engineer host tolerance alongside pathway reconstruction for improved yields [93].
Quality Control and Data Presentation
Ensuring Analytical Validity
For proteomics, adherence to a standardized protocol based on the principles of ISO/IEC 17025:2017 is recommended to ensure the validity of results [92]. Key steps include:
- Mass Accuracy: Regular calibration of the HRMS with suitable calibrants [92].
- Chromatographic Stability: Monitoring retention time, peak shape, and width using quality control samples before and during analyses [92].
- Replication: Performing technical and biological replicates to assess variability.
Principles for Clear Data Presentation
When presenting quantitative multi-omics data in tables, adhere to the following design principles to aid comprehension [94]:
- Alignment: Left-flush align text headers; right-flush align numerical data and their headers to facilitate comparison [94].
- Precision: Use a consistent and appropriate level of decimal precision for all numbers in a column [94].
- Fonts: Use a tabular font where each character has equal width, ensuring numerical place values align vertically [94].
- Significance: Clearly highlight statistical significance (e.g., with asterisks and a footnote).
Within metabolic engineering, the metrics of Titer, Rate, and Yield (TRY) are paramount for evaluating the performance and economic viability of a microbial fermentation process [95]. For researchers focused on heterologous pathway reconstruction in yeast, selecting an appropriate chassis organism is a critical decision that directly influences these key outcomes. This Application Note provides a comparative analysis of TRY metrics across two primary yeast species used in industrial biotechnology: the conventional workhorse, Saccharomyces cerevisiae, and the non-conventional yeast, Brettanomyces bruxellensis. We frame this comparison within the context of pathway engineering for the production of biofuels and high-value natural products, providing structured data, experimental protocols, and visualizations to guide research planning.
TRY Performance Metrics in Yeast
Definition of TRY Metrics and Their Impact on Technoeconomics
In microbial biotechnology, TRY metrics are used to conduct technoeconomic analysis prior to scaling a fermentation process to commercial levels [95]. Each metric impacts the production cost differently:
- Titer: The concentration of the product achieved in the fermentation broth, typically reported in g/L or mg/L. A high titer reduces the volume that needs to be processed during downstream purification.
- Rate: The speed of product formation, often measured as volumetric (g/L/h) or specific (g/g cell/h) productivity. A high rate increases the output of a bioreactor of a given size over time.
- Yield: The efficiency of converting the substrate (e.g., sugar) into the product (g product/g substrate or mol/mol). A high yield minimizes raw material costs and waste generation [95].
A comprehensive technoeconomic analysis must consider all three parameters, as they can involve trade-offs. For instance, maximizing yield might come at the expense of a slower production rate.
Comparative TRY Performance Across Species and Products
The choice of yeast species imposes distinct physiological boundaries that directly influence TRY performance. The table below summarizes representative data for different products and metabolic pathways.
Table 1: Comparative TRY Metrics for Saccharomyces cerevisiae and Brettanomyces bruxellensis
| Yeast Species | Product | Substrate | Titer | Rate | Yield | Key Context |
|---|---|---|---|---|---|---|
| S. cerevisiae | Ethanol | Glucose (CSL Media) | >40 g/L [96] | >0.7 g/L/h [96] | >0.42 g/g [96] | Co-fermentation, high performance |
| Chelerythrine | Glucose | 12.61 mg/L [55] | Information Missing | Information Missing | Engineered strain, bioreactor | |
| Ethanol (2G) | Lignocellulosic Hydrolysate | Robust Performance [97] | Varies by strain/condition [97] | Varies by strain/condition [97] | High inhibitor tolerance | |
| B. bruxellensis | Ethanol | Glucose | Lower than S. cerevisiae [98] | Significantly lower than S. cerevisiae [98] | Information Missing | 1G ethanol, lower productivity |
| Ethanol | D-Xylose | Low [99] | Low [99] | Low [99] | 2G ethanol, inefficient fermentation |
Performance and Robustness Trade-offs
Beyond raw performance under ideal conditions, robustness—the ability of a strain to maintain stable performance under perturbations—is critical for industrial applications. A study simulating lignocellulosic hydrolysate conditions for 24 S. cerevisiae strains revealed a key trade-off: a negative correlation between the performance and robustness of ethanol yield, biomass yield, and cell dry weight [97]. This means strains optimized for maximum yield in a single condition may be more susceptible to process variability. Notably, the Ethanol Red strain was identified as a candidate with both high performance and robustness in the tested perturbation space [97].
Application Notes for Heterologous Pathway Engineering
Case Study: Reconstruction of the Chelerythrine Pathway inS. cerevisiae
The biosynthesis of the alkaloid chelerythrine in yeast serves as an exemplary case of multi-factorial metabolic engineering to enhance TRY metrics [55].
Experimental Workflow Overview: The process involved initial pathway reconstruction from (S)-reticuline to chelerythrine, followed by iterative systems-level optimization to overcome limitations, finally leading to high-titer production in a controlled bioreactor.
Key Optimization Strategies:
- Combinatorial Pathway Engineering: The initial low titer of 0.34 µg/L in the first-generation strain (Z4) was dramatically improved by integrating multiple-copy genes for rate-limiting enzymes, including TfSMT, AmTDC, EcTNMT, PsMSH, EcP6H, and PsCPR [55].
- Cofactor Engineering: The supply of essential cofactors (heme and NADPH) for P450 enzymes was tailored to enhance metabolic flux [55].
- Product Trafficking: A heterologous transporter (MtABCG10) was expressed to enhance the secretion of chelerythrine, potentially reducing feedback inhibition [55].
- Bioreactor Process Control: A pH-based fed-batch fermentation strategy was employed, which optimized the cultivation conditions to achieve the final reported titer of 12.61 mg/L, representing an increase of over 37,000-fold from the initial strain [55].
The Scientist's Toolkit: Essential Research Reagents
Successful pathway reconstruction and optimization rely on a suite of key reagents and tools.
Table 2: Key Research Reagent Solutions for Yeast Metabolic Engineering
| Reagent / Tool | Function / Application | Example Use Case |
|---|---|---|
| CRISPR-Cas9 System | Precision genome editing for gene knock-ins, knock-outs, and multiplexed engineering. | Integration of heterologous expression cassettes into specific genomic loci [55]. |
| Codon-Optimized Genes | Heterologous genes optimized for the yeast's tRNA pool to ensure high expression levels. | Expression of plant-derived enzymes (e.g., McoBBE, TfSMT, EcP6H) in S. cerevisiae [55]. |
| Promoter/Terminator Libraries | Fine-tuning of gene expression levels by using regulatory parts of varying strengths. | Balancing metabolic flux in multi-enzyme pathways to minimize intermediate accumulation [100]. |
| Plasmid Vectors & Shuttle Systems | Stable maintenance and expression of heterologous genes (e.g., pRS416 series). | Cloning and expression of genes in E. coli prior to transformation into yeast [55]. |
| Synthetic Complete (SC) Media | Defined cultivation media allowing for selection and maintenance of engineered strains. | Cultivating auxotrophic strains and selecting for markers like URA3 and TRP1 [55]. |
Protocols
Protocol: Cultivation and TRY Analysis in Microtiter Plates for Robustness Screening
This protocol is adapted from a high-throughput methodology designed to quantify phenotype performance and robustness across multiple strains and perturbation conditions [97].
Part I: Experimental Setup and Cultivation
- Strain Preparation: Aliquot 24 S. cerevisiae strains, including laboratory, industrial (e.g., Ethanol Red, PE-2), and environmental isolates.
- Perturbation Space Preparation: Prepare a base chemically defined medium (e.g., Delft medium). Dispense this medium into a 96-well microtiter plate. Add a single perturbation component to each column/row to simulate industrial conditions. Perturbations should include:
- Acids: Acetic, formic, levulinic, lactic acid.
- Aldehydes: Furfural, 5-hydroxymethylfurfural (HMF), vanillin.
- Pentoses: Xylose, arabinose.
- Other: NaCl, ethanol [97].
- Inoculation and Cultivation: Use an automated pipetting system to inoculate each well to an initial OD600 of ~0.05. Seal the plates with a breathable membrane and incubate in a plate reader at 30°C with continuous shaking. Monitor OD600 every 15 minutes for 48-72 hours.
Part II: Data Analysis and Robustness Quantification
- Phenotype Calculation: For each cultivation well, calculate the following phenotypes from the growth curve data:
- Maximum Specific Growth Rate (μmax): Calculated from the exponential phase of growth.
- Lag Phase (λ): Duration before the onset of exponential growth.
- Final Cell Dry Weight (CDW): Estimated from final OD600 using a predetermined correlation.
- Biomass Yield (Yx/s): g CDW per g substrate consumed.
- Ethanol Yield (Yp/s): g ethanol per g substrate consumed (requires off-line ethanol assay) [97].
- Performance and Robustness Scoring:
- Calculate the performance of a given phenotype for a strain as its average value across all perturbations.
- Calculate the robustness using a previously published metric that quantifies the stability of a phenotype across perturbations, resulting in a dimensionless number where zero represents complete robustness [97].
- Trade-off Analysis: Perform Spearman correlation tests between the performance and robustness of each phenotype across the strain library to identify potential trade-offs.
Protocol: Fed-Batch Fermentation for High-Titer Production in a Bioreactor
This protocol outlines the key steps for scaling up a engineered yeast strain from shake flasks to a controlled bioreactor to maximize titer, as demonstrated for chelerythrine production [55].
Seed Train Preparation:
- Inoculate a single colony of the engineered S. cerevisiae strain into 10 mL of SC medium lacking appropriate auxotrophic amino acids. Incubate overnight at 30°C with shaking at 250 rpm.
- Use this culture to inoculate a 250 mL baffled flask containing 50 mL of YPD or SC medium. Grow for ~16 hours until the late exponential phase (OD600 ~6-10). This is the seed culture for the bioreactor.
Bioreactor Setup and Fermentation:
- Use a 0.5 L or larger bioreactor with working volume of 0.3 L. Equip with controls for temperature, pH, dissolved oxygen (DO), and agitation.
- Prepare the initial batch medium containing defined salts, vitamins, trace elements, and the carbon source (e.g., 20 g/L glucose).
- Sterilize the bioreactor and medium in situ or by autoclaving.
- Calibrate the pH and DO probes. Set initial fermentation parameters to 30°C, pH 5.5 (controlled with addition of base, e.g., 2 M KOH, and acid, e.g., 1 M HCL), and agitation at 300-500 rpm. Sparge with air at 0.5-1 vvm to maintain DO above 20-30%.
- Inoculate the bioreactor with the seed culture to an initial OD600 of 0.5-1.0.
Fed-Batch Operation:
- Once the initial batch of glucose is depleted (indicated by a sharp rise in DO), initiate the feed.
- Use a concentrated feed solution (e.g., 500 g/L glucose) and connect it to a peristaltic pump. Employ a pre-determined feeding profile (exponential, constant, or pH-based) to maintain a controlled, low growth rate and avoid overflow metabolism.
- For pH-stat feeding, the addition of base to control pH can trigger the feed pump, supplying carbon source as it is consumed [55].
- Continue the fermentation for 3-10 days, sampling periodically for analysis of cell density, substrate consumption, and product formation.
The data and protocols presented herein highlight fundamental differences in the metabolic engineering landscape for different yeast species. S. cerevisiae remains the premier chassis for heterologous pathway reconstruction, as evidenced by its well-developed genetic tools [100], high TRY metrics for ethanol [96], and successful production of complex molecules like chelerythrine [55]. A critical insight for researchers is the demonstrated trade-off between performance and robustness; strain selection and engineering must therefore be guided by the specific demands of the industrial process, prioritizing robust performance under variable conditions when necessary [97].
In contrast, B. bruxellensis presents a case of unfulfilled potential. While its native metabolism offers intriguing traits like the ability to assimilate pentoses, its inefficient conversion of D-xylose to ethanol and lower productivity on hexoses currently limit its application [98] [99]. Its future as a platform for second-generation ethanol or other products is contingent upon the development of effective genetic engineering tools to overcome its metabolic bottlenecks.
In conclusion, the strategic selection of a yeast host, followed by systematic pathway engineering and process optimization as detailed in this note, is essential for achieving the TRY metrics required for commercially viable bioprocesses. The continued development of both conventional and non-conventional yeasts will expand the toolbox available to scientists and drug development professionals for the sustainable production of biofuels, chemicals, and pharmaceuticals.
Computational Workflows for Predicting and Validating Novel Pathway Derivatives
The reconstruction of heterologous biosynthetic pathways in engineered microbial hosts, such as yeast, is a fundamental strategy in synthetic biology for the sustainable production of high-value plant natural products (PNPs) and their derivatives [80]. These compounds, which include many modern medicines, are often difficult to obtain in sufficient quantities from their native plant sources due to low yield, laborious extraction, and environmental variability [80]. While microbial production can address these concerns, a significant challenge remains in efficiently expanding beyond native pathway products to create novel derivatives with potentially improved pharmaceutical properties.
Computational workflows are now pivotal in systematically addressing this challenge. They enable researchers to explore the biochemical space around a known pathway, prioritize high-value targets, and identify enzyme candidates capable of producing these targets, thereby accelerating the design of new microbial cell factories [80] [101]. This Application Note details a proven computational and experimental protocol for predicting and validating novel pathway derivatives, framed within the context of heterologous pathway reconstruction in yeast. The described workflow is based on a published study that successfully led to the de novo biosynthesis of benzylisoquinoline alkaloid (BIA) derivatives in Saccharomyces cerevisiae [80].
Computational Pathway Expansion and Target Prioritization
This phase involves the in-silico generation of potential derivatives from a core pathway and the subsequent selection of the most promising candidates for experimental pursuit.
Cheminformatic Network Expansion
The process begins with the computational expansion of a defined heterologous pathway using generalized enzymatic reaction rules that simulate known biochemical transformations [80].
- Tool Example: The Biochemical Network Integrated Computational Explorer (BNICE.ch) is one such tool that can be applied for this purpose [80].
- Procedure:
- Input: Define the set of initial metabolites from the heterologous pathway of interest. For example, in a noscapine pathway reconstruction, this would include the 17 intermediates from (S)-norcoclaurine to noscapine [80].
- Reaction Rule Application: Apply a comprehensive set of enzymatic reaction rules (e.g., oxidation, reduction, methylation, acetylation) to every functional group on every pathway intermediate.
- Iterative Expansion: Repeat this process for multiple generations (e.g., four generations) from each starting compound, generating a network of both known and novel compounds accessible via one or more predicted enzymatic steps [80].
- Network Curation: Filter the resulting network to retain chemically relevant structures. For instance, in the case of BIAs, the network can be trimmed to compounds containing the core 1-benzylisoquinoline scaffold [80].
Table 1: Key Cheminformatic Tools for Pathway Expansion and Enzyme Prediction
| Tool Name | Primary Function | Application in Workflow |
|---|---|---|
| BNICE.ch [80] | Generation of novel biochemical reactions and pathways | Expands a core set of pathway metabolites into a network of potential derivatives. |
| BridgIT [80] | Prediction of enzymes for novel reactions | Identifies candidate enzymes likely to catalyze a predicted transformation. |
| RetroPath2.0 [80] | Retrobiosynthetic pathway design | Determines bioproduction pathways from a target back to host metabolism. |
| Selenzyme [80] | Enzyme sequence selection for synthetic biology | Selects enzyme sequences to catalyze a given reaction. |
| Model SEED [102] | High-throughput generation of genome-scale metabolic models | Aids in metabolic network reconstruction, an essential first step. |
Ranking and Selection of Target Compounds
The expanded network can contain thousands of compounds, necessitating a robust ranking system to identify the most promising targets for experimental validation [80].
- Ranking Criteria:
- Popularity/Interest: Rank compounds by the sum of scientific citations and patents to prioritize those with known biomedical relevance or commercial potential [80].
- Pharmaceutical Potential: Filter for molecules with documented or potential therapeutic activity.
- Biosynthetic Feasibility: Apply critical filters to ensure experimental tractability:
- Thermodynamic Feasibility: The predicted pathway to the target should be thermodynamically favorable.
- Proximity to Pathway: Prioritize targets that are only one enzymatic transformation away from an existing pathway intermediate.
- Enzyme Availability: Enzymes must be available (natively or through engineering) that can perform the desired transformation [80].
Table 2: Example Ranking of High-Priority Compounds Derived from a Noscapine Pathway [80]
| Compound Name | Total Annotations (Citations + Patents) | Therapeutic Activity | Biosynthetic Step from Pathway Intermediate |
|---|---|---|---|
| Papaverine | 22,918 | Vasodilator | Multiple Steps |
| Bicuculline | 16,118 | GABA~A~ receptor antagonist; research chemical | Multiple Steps |
| Berberine | 12,154 | Antibacterial, antidiabetic | Multiple Steps |
| (S)-Tetrahydropalmatine (THP) | Not Specified | Analgesic, Anxiolytic | Single O-methylation step from (S)-tetrahydrocolumbamine |
Application Note: In the foundational study, applying these criteria identified (S)-tetrahydropalmatine (THP)—a known analgesic and anxiolytic—as a high-priority, feasible target. It was one enzymatic step (O-methylation) away from the noscapine pathway intermediate (S)-tetrahydrocolumbamine, and enzyme candidates for this transformation were predicted to be available [80].
Diagram 1: Computational workflow for predicting novel pathway derivatives.
Experimental Validation in a Yeast Chassis
Following the computational predictions, the proposed pathways require experimental validation in a suitable yeast host.
Strain and Pathway Construction
The goal is to engineer a yeast strain that produces the pathway intermediate and expresses the novel enzyme candidate(s).
- Host Selection: Saccharomyces cerevisiae is a preferred host due to its well-characterized genetics, ease of manipulation, and eukaryotic protein processing machinery [80] [70]. For pathways requiring high acetyl-CoA supply, engineering peroxisome assembly in S. cerevisiae can significantly increase precursor availability [103].
- Protocol: Strain Engineering
- Base Strain: Start with a yeast strain (e.g., SynV) previously engineered for the de novo production of the required pathway intermediate(s). For THP production, this would be a strain producing (S)-tetrahydrocolumbamine [80].
- Vector Design: Clone the gene(s) encoding the top-predicted enzyme candidates (e.g., O-methyltransferases for THP production) into a suitable expression vector. Use a strong, inducible promoter (e.g., the GAL promoter system) [80] [70].
- Transformation: Introduce the expression vector(s) into the base strain using standard yeast transformation protocols (e.g., lithium acetate method).
- Screening: Select for transformants on appropriate dropout media and confirm integration/expression via colony PCR and/or analytical methods.
Cultivation and Metabolite Analysis
This protocol details the cultivation of engineered strains and the detection of the target compound.
Materials:
- Engineered S. cerevisiae strain(s) and base strain control.
- Appropriate synthetic complete (SC) dropout medium with 2% glucose.
- Induction medium (e.g., SC with 2% galactose).
- Organic solvents for extraction (e.g., ethyl acetate, methanol).
- Analytical standards for the target compound and pathway intermediates.
Protocol: Production and Detection
- Pre-culture: Inoculate a single colony into 5 mL of SC dropout medium with glucose. Incubate at 30°C with shaking (250 rpm) for 24-48 hours.
- Induction: Dilute the pre-culture to a standard OD600 (e.g., 0.05-0.1) in induction medium to activate the heterologous pathway. Incubate for 3-7 days at 30°C with shaking.
- Metabolite Extraction:
- Centrifuge culture samples (e.g., 1 mL) to separate cells and supernatant.
- Extract the supernatant with an equal volume of ethyl acetate by vortexing for 10-15 minutes.
- Centrifuge to separate phases, collect the organic (upper) layer, and evaporate to dryness under a gentle nitrogen stream.
- Reconstitute the dried extract in a suitable solvent (e.g., methanol) for analysis.
- Analysis and Validation:
- Analyze the extracts using Liquid Chromatography-Mass Spectrometry (LC-MS/MS).
- Identify the target compound (e.g., THP) by comparing its retention time and mass fragmentation pattern to an authentic commercial standard.
- Quantify production titers using a standard curve generated from the analytical standard.
Diagram 2: Experimental workflow for pathway derivative validation.
The Scientist's Toolkit: Research Reagent Solutions
The following table details key reagents and resources essential for implementing this workflow.
Table 3: Essential Research Reagents and Resources
| Item | Function/Description | Example/Source |
|---|---|---|
| BNICE.ch [80] | Computational tool for generating novel biochemical reactions and expanding metabolic networks. | EPFL; Generates a network of derivatives from core pathway. |
| BridgIT [80] | Enzyme prediction tool that identifies candidate enzymes for a novel reaction by structural similarity. | EPFL; Identifies O-methyltransferases for THP production. |
| S. cerevisiae SynV Chassis [103] | A genetically stable yeast host strain with precise recombination features, suitable for natural product production. | Laboratory strain; Serves as the microbial factory. |
| GAL Promoter System [103] | A strong, inducible promoter system for controlling the expression of heterologous genes in yeast. | Standard biological part; Induced with galactose. |
| LC-MS/MS System | Analytical platform for detecting, identifying, and quantifying small molecules in complex extracts. | e.g., UHPLC coupled to triple quadrupole MS; Confirms de novo synthesis. |
| Analytical Standards | Pure chemical compounds used as references for identifying and quantifying metabolites via LC-MS. | Commercial suppliers (e.g., Sigma-Aldrich); Essential for validation. |
Concluding Remarks
The integration of computational pathway expansion with robust experimental validation in yeast provides a powerful and systematic framework for accessing novel natural product derivatives. This workflow, from in-silico prediction to de novo biosynthesis, effectively bridges the gap between bioinformatics and metabolic engineering. By leveraging tools like BNICE.ch for cheminformatic prospecting and BridgIT for enzyme candidate prediction, researchers can efficiently navigate the vast biochemical space and prioritize feasible, high-value targets. Subsequent strain construction and analytical protocols enable the translation of these predictions into tangible microbial production systems. As the number of reconstructed heterologous pathways continues to grow, this structured approach will be instrumental in unlocking the full potential of PNPs and their derivatives for drug discovery and development.
Assessing Economic Viability and Scalability for Industrial Translation
Application Note
Economic Context for Heterologous Protein Production in Yeast
The global market for heterologous proteins, including biopharmaceuticals and industrial enzymes, represents a multi-billion-dollar industry, with the medicinal protein sector alone projected to reach approximately USD 400 billion by 2025 [4] [11]. Saccharomyces cerevisiae is a predominant microbial cell factory for producing these proteins, accounting for about one-sixth of all pharmaceuticals licensed for human use [4]. However, the economic viability of production processes is universally challenged by cellular burden, where resource competition between the host cell and the heterologous pathway reduces final protein titers and process efficiency [104]. Simultaneously, the language services market, which supports the global documentation, regulatory compliance, and commercialization of these biotechnological products, is itself a major industry, valued at an estimated USD 71.7 billion in 2024 and projected to grow to USD 75.7 billion in 2025 [105]. Assessing the economic viability and scalability of the "industrial translation" pipeline—from strain construction to market delivery—requires an integrated analysis of both biological and linguistic-economic factors.
Quantitative Market and Production Data
The tables below summarize key quantitative data essential for evaluating the economic landscape of heterologous protein production and its supporting translation services.
Table 1: Key Metrics for the Language Services Industry (Supporting Sector)
| Metric | 2024 Value | 2025 Projection | Key Trend / Note |
|---|---|---|---|
| Global Market Size | USD 71.7 billion [105] | USD 75.7 billion [105] | Projected to reach USD 92.3 billion by 2029 [105] |
| Machine Translation (MT) Market | USD 678 million [106] | USD 706 million [106] | Projected to reach USD 995 million by 2032 [106] |
| Cost Reduction via MTPE | - | 30-50% [106] | Machine Translation Post-Editing offers significant savings [106] |
| Top Industry Challenge | Price pressure / Increasing revenue [105] | - | Reported as a top-three business challenge for LSPs [105] |
Table 2: Representative Heterologous Protein Titers in S. cerevisiae
| Protein Type | Example Product | Titer / Activity | Production Scale | Reference |
|---|---|---|---|---|
| Medicinal Protein | Transferrin | 2.33 g/L | Fed-batch, 10 L bioreactor [4] | |
| Medicinal Protein | Antithrombin III | 312 mg/L | Fed-batch, 5 L bioreactor [4] | |
| Industrial Enzyme | Lipase | 11,000 U/L | Fed-batch, 5 L bioreactor [4] | |
| Industrial Enzyme | Laccase3 | 1176.04 U/L | Batch, shake flask [4] | |
| Food Protein | Brazzein | 9 mg/L | Batch, shake flask [4] |
Protocols
This section outlines detailed methodologies for assessing economic viability and scalability, integrating both experimental and market-analysis workflows.
Protocol 1: Techno-Economic Analysis (TEA) of a Yeast-Based Production Process
Objective: To quantitatively evaluate the economic feasibility and identify key cost drivers for the production of a heterologous protein from a reconstructed pathway in yeast.
Materials:
- Strain Engineering Data: Plasmid maps, genomic integration data, and fermentation performance metrics (e.g., titer, yield, productivity).
- Process Modeling Software: SuperPro Designer, Aspen Plus, or open-source TEA tools.
- Cost Databases: Vendor quotes for raw materials, equipment, and utilities.
Procedure:
- Define Process Basis: Establish the production capacity (e.g., kg of protein/year) and the overall process flow diagram.
- Model Upstream Processing (USP):
- Input strain-specific parameters: maximum growth rate (μmax), product yield on substrate (Yp/s), and final product titer (from Table 2) [4].
- Model the bioreactor operation (e.g., batch, fed-batch) and calculate consumption rates for carbon source, nitrogen, and other nutrients.
- Model Downstream Processing (DSP):
- Design the purification train based on the protein's localization (intracellular vs. secreted). Secretion, a key advantage of S. cerevisiae, significantly reduces DSP complexity and cost [4] [11].
- Include unit operations such as centrifugation, filtration, chromatography, and formulation. The number and complexity of these steps are major cost drivers.
- Capital and Operating Cost Estimation:
- Estimate the Total Capital Investment (TCI), including equipment and facility costs.
- Estimate the Total Operating Cost, encompassing raw materials, labor, utilities, and waste disposal.
- Economic Profitability Analysis:
Protocol 2: Scalability Assessment via Fermentation Scale-Up
Objective: To translate a laboratory-scale protein production process into a pilot or industrial-scale operation while maintaining or improving yield.
Materials:
- Strain: Engineered S. cerevisiae production strain.
- Bioreactors: Lab-scale (e.g., 1 L), pilot-scale (e.g., 100 L), and production-scale systems.
- Analytical Equipment: HPLC, spectrophotometer, cell counter.
Procedure:
- Bench-Scale Optimization:
- In lab-scale bioreactors, optimize conditions to mitigate cellular burden. This can be achieved by using strong, tunable promoters and optimizing gene copy number to balance expression and host fitness [4] [11].
- Monitor metrics like the specific growth rate and the Respiratory Quotient (RQ) to understand metabolic activity.
- Pilot-Scale Translation:
- Scale up the process using scale-down models and constant criteria such as Volumetric Oxygen Transfer Coefficient (kLa) or Power per Unit Volume (P/V) to maintain consistent physiology and productivity.
- Implement Dynamic Process Control strategies (e.g., dissolved oxygen, pH, feed rate control) to handle the heterogeneity of large-scale bioreactors.
- Process Performance Validation:
- At the pilot scale, measure the final product titer, volumetric productivity, and product quality (e.g., via SDS-PAGE, mass spectrometry).
- Compare these results with bench-scale data to calculate the scale-up efficiency.
Protocol 3: Market Localization and Compliance Strategy
Objective: To ensure the technical documentation and labeling for a yeast-derived therapeutic or enzyme are accurately translated and comply with target market regulations, supporting successful commercialization.
Materials:
- Source Documents: Technical manuals, clinical trial protocols, drug substance specifications, and safety data sheets.
- Regulatory Guidelines: FDA, EMA, and other target market regulatory body requirements.
- Language Technology: Translation Management System (TMS), Computer-Assisted Translation (CAT) tools with Translation Memory (TM), and terminology management databases [108].
Procedure:
- Pre-Translation Analysis:
- In collaboration with Subject Matter Experts (SMEs), create a bilingual glossary of critical technical and regulatory terms to ensure consistency [108] [109].
- Classify content types to determine the appropriate translation method (e.g., human translation for regulatory submissions, MTPE for internal technical documents) [106].
- Controlled Translation Execution:
- Utilize a TMS to enforce adherence to the approved glossary and style guide.
- For certain content, employ a Machine Translation Post-Editing (MTPE) workflow to achieve a 30-50% cost reduction while maintaining accuracy [106]. Human post-editing is critical to correct errors and ensure nuanced technical meaning is preserved.
- Quality Assurance and Compliance:
- Conduct rigorous quality control checks, including proofreading by a second linguist and in-country regulatory experts.
- Ensure the final translated output complies with relevant standards such as ISO 17100 for translation services and industry-specific regulations like the EU Medical Device Regulation (MDR) [108] [109].
Visualizations
Integrated Scalability Assessment Workflow
This diagram outlines the multi-stage protocol for scaling up production and localizing documentation.
Integrated Scalability Assessment Workflow
Economic Viability Analysis Framework
This diagram illustrates the logical flow of the Techno-Economic Analysis (TEA), highlighting the key inputs and decision points.
Economic Viability Analysis Framework
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents and Tools for Economic and Scalability Assessment
| Category | Item / Solution | Function / Explanation | Relevance to Viability & Scalability |
|---|---|---|---|
| Strain Engineering | Codon-Optimized Gene Synthesis | Enhances translation efficiency in yeast; mitigates burden by matching host codon bias [4] [11]. | Increases protein titer, a primary driver of economic viability. |
| Strain Engineering | Tunable Promoter Systems (e.g., pGAL1, synthetic promoters) | Allows precise control of heterologous gene expression to balance product yield and cellular burden [4]. | Enables optimization of metabolic flux, improving process robustness at scale. |
| Process Analytics | Metabolomics Kits (e.g., GC-MS, LC-MS sample prep) | Quantifies intracellular metabolites to identify bottlenecks in central metabolism under burden [104]. | Provides data for in-silico models and guides targeted strain re-engineering. |
| Process Analytics | Bioanalyzer / HPLC Systems | Measures key performance indicators (KPIs) like product titer and substrate consumption in real-time. | Essential for collecting accurate data for Techno-Economic Analysis (TEA). |
| Market & Compliance | Translation Management System (TMS) | Centralized platform for managing multilingual terminology, translation memories, and workflows [108]. | Ensures consistency and reduces cost/time for global regulatory submissions. |
| Market & Compliance | Regulatory Intelligence Databases | Provide up-to-date requirements from agencies (FDA, EMA) for target markets. | Mitigates regulatory risk, a critical factor for the economic viability of drug development. |
Conclusion
Heterologous pathway reconstruction in yeast has evolved from a simple gene transfer exercise to a sophisticated discipline integrating systems and synthetic biology. The convergence of advanced genome editing, dynamic regulation strategies, and powerful computational models now enables the precise design and optimization of yeast cell factories. Future directions will focus on further automating the design-build-test-learn cycle, expanding the use of non-conventional yeasts with unique metabolic capabilities, and de-risking the scale-up process for clinical manufacturing. For drug development professionals, these advancements promise to establish yeast as a resilient and versatile platform for the sustainable and on-demand production of even the most complex plant-derived pharmaceuticals and new-to-nature therapeutics, ultimately accelerating the pipeline from biological discovery to clinical application.
References
- 1. Heterologous expression [https://en.wikipedia.org/wiki/Heterologous_expression]
- 2. Heterologous Metabolic Pathways: Strategies for Optimal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7385092/]
- 3. Recent developments towards the heterologous ... [https://www.sciencedirect.com/science/article/abs/pii/S095816690500162X]
- 4. Engineering strategies for enhanced heterologous protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10801990/]
- 5. Burden Imposed by Heterologous Protein Production in ... [https://www.frontiersin.org/journals/fungal-biology/articles/10.3389/ffunb.2022.827704/full]
- 6. Engineering yeast for the production of plant terpenoids using ... [https://pubs.rsc.org/en/content/articlehtml/2023/np/d3np00005b]
- 7. Transforming non-conventional yeasts into key players in ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2025.1600187/full]
- 8. Yeast Heterologous Expression Systems for the Study of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10342074/]
- 9. Yeast as C1 cell factory: Transforming methanol and ... [https://www.sciencedirect.com/science/article/pii/S0734975025001272]
- 10. Yeast-based heterologous production of the Colletochlorin ... [https://www.sciencedirect.com/science/article/pii/S1096717623001428]
- 11. Engineering strategies for enhanced heterologous protein ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-024-02299-z]
- 12. Step-by-step optimization of a heterologous pathway for de ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11316701/]
- 13. De Novo Biosynthesis of Asperosaponin VI in ... [https://www.sciencedirect.com/science/article/pii/S2405805X25001188]
- 14. Reprogramming yeast metabolism for customized starch ... [https://www.nature.com/articles/s41467-025-58067-z]
- 15. Transforming non-conventional yeasts into key players in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12081427/]
- 16. Current Challenges and Opportunities in Non-native ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2020.594061/full]
- 17. Comparison of Yeasts as Hosts for Recombinant Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6027275/]
- 18. Recent progress on heterologous protein production in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11087369/]
- 19. Rewiring of metabolic pathways in yeasts for sustainable ... [https://www.sciencedirect.com/science/article/abs/pii/S0960852423000949]
- 20. Comprehensive comparison of Yarrowia lipolytica and ... [https://www.nature.com/articles/s41598-020-58683-3]
- 21. Metabolic reprogramming and computation-aided protein ... [https://www.sciencedirect.com/science/article/pii/S2405805X25000675]
- 22. computational methods for biosynthetic pathway design [https://pmc.ncbi.nlm.nih.gov/articles/PMC12159830/]
- 23. Review of Machine Learning Methods for the Prediction ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8247443/]
- 24. Designing pathways for bioproducing complex chemicals ... [https://www.nature.com/articles/s41467-025-59827-7]
- 25. Machine learning for metabolic pathway optimization [https://www.sciencedirect.com/science/article/pii/S2001037023001447]
- 26. Identifying promoters to enhance heterologous gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12296805/]
- 27. Chaperone overexpression boosts heterologous small ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-025-02728-7]
- 28. Summary of U.S. FDA GRAS Updates in the Third Quarter ... [https://www.cirs-group.com/en/food/summary-of-us-fda-gras-updates-in-the-third-quarter-of-2025]
- 29. Customized yeast cell factories for biopharmaceuticals [https://pmc.ncbi.nlm.nih.gov/articles/PMC8246677/]
- 30. High throughput mutagenesis and screening for yeast ... [https://jbioleng.biomedcentral.com/articles/10.1186/s13036-022-00315-7]
- 31. A high‐throughput screening method for the discovery of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9996697/]
- 32. High-throughput platform for yeast morphological profiling ... [https://www.nature.com/articles/s41540-022-00212-1]
- 33. Recent trends in the production of proteins by precision ... [https://www.sciencedirect.com/science/article/pii/S295019462500319X]
- 34. Angel Yeast Showcases Microbial Protein Innovations at FI ... [https://en.angelyeast.com/news/ye-savory/angel-yeast-showcases-microbial-protein-innovations-at-fi-africa-2025,-driving-sustainable-food-solutions-in-mea.html]
- 35. Recent advances in genetic engineering and chemical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11963765/]
- 36. Genome Editing Systems Across Yeast Species - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7744358/]
- 37. Multiplex Genome Editing in Yeast by CRISPR/Cas9 [https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2021.719148/full]
- 38. A systematic comparison of CRISPR-Cas9 allele editing in ... [https://www.nature.com/articles/s41598-025-19115-2]
- 39. Optimizing the CRISPR/Cas9 system for gene editing in ... [https://www.sciencedirect.com/science/article/pii/S2667370325000050]
- 40. Conditional guide RNA deactivation by mRNA and small ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12332140/]
- 41. The editable landscape of the yeast genome reveals ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12577706/]
- 42. Design of highly functional genome editors by modelling ... [https://www.nature.com/articles/s41586-025-09298-z]
- 43. Binary vector copy number engineering improves ... [https://www.nature.com/articles/s41587-024-02462-2]
- 44. 3.0 Strategies for Yeast Genetic Improvement in Brewing ... [https://www.mdpi.com/2306-5710/11/4/100]
- 45. CRISPR-GPT for agentic automation of gene-editing ... [https://www.nature.com/articles/s41551-025-01463-z]
- 46. Advanced Approaches: PCR, Traditional Cloning, and ... [https://blog.edvotek.com/2025/08/06/advanced-approaches-pcr-traditional-cloning-and-golden-gate-assembly-compared/]
- 47. Combinatorial optimization of gene expression through ... [https://www.nature.com/articles/s41467-024-44997-7]
- 48. Plant Promoters and Terminators for High-Precision ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10328392/]
- 49. Exchange of endogenous and heterogeneous yeast ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7736810/]
- 50. Use of High Capacity Terminators in Saccharomyces ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3769427/]
- 51. Designing strong inducible synthetic promoters in yeasts [https://www.nature.com/articles/s41467-024-54865-z]
- 52. Mix and Match: Promoters and Terminators for Tuning ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2022.876316/full]
- 53. Development of Terminator–Promoter Bifunctional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10298055/]
- 54. Improved protein glycosylation enabled heterologous ... [https://www.sciencedirect.com/science/article/pii/S2214030122000244]
- 55. Metabolic engineering of Saccharomyces cerevisiae for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11191272/]
- 56. Reconstitution of a 10-gene pathway for synthesis of the ... [https://www.nature.com/articles/ncomms4283]
- 57. Development and Perspective of Production of Terpenoids ... [https://www.sciepublish.com/article/pii/131]
- 58. Yeast metabolism adaptation for efficient terpenoids ... [https://www.nature.com/articles/s41467-024-54298-8]
- 59. Yeast-Based Biosynthesis of Natural Products From Xylose [https://pmc.ncbi.nlm.nih.gov/articles/PMC7886706/]
- 60. Engineering yeasts for xylose metabolism [https://www.sciencedirect.com/science/article/abs/pii/S0958166906000668]
- 61. Compartmentalization of metabolic pathways in yeast ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3659820/]
- 62. Compartmentalization engineering of yeasts to overcome ... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2023.1132244/full]
- 63. Compartmentalization and transporter engineering strategies ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-022-01819-z]
- 64. Optimizing spatial and temporal control of yeast metabolic ... [https://escholarship.org/uc/item/64w384ps]
- 65. A Yeast Metabolite Extraction Protocol Optimised for Time ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3430680/]
- 66. Metabolic Engineering of Saccharomyces cerevisiae - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC98985/]
- 67. Metabolic engineering for compartmentalized biosynthesis ... [https://www.sciencedirect.com/science/article/pii/S0944501324002167]
- 68. In vivo pathway optimization in yeast via loxpsym-mediated ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12448870/]
- 69. Comparative Analysis of Codon Optimization Tools [https://pmc.ncbi.nlm.nih.gov/articles/PMC12010093/]
- 70. Engineering Saccharomyces cerevisiae for efficient ... [https://www.sciencedirect.com/science/article/pii/S2667370323000541]
- 71. DNA and RNA codon tables [https://en.wikipedia.org/wiki/DNA_and_RNA_codon_tables]
- 72. Chemically modified tRNA enhances the translation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12373798/]
- 73. Deep generative optimization of mRNA codon sequences ... [https://www.nature.com/articles/s41467-025-64894-x]
- 74. Codon Optimization Program from EnCor Biotechnology Inc. [https://files.encorbio.com/protocols/Codon.htm]
- 75. Rewiring yeast metabolism to synthesize products beyond ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7736362/]
- 76. Rewiring carbon metabolism in yeast for high level ... [https://www.nature.com/articles/s41467-019-12961-5]
- 77. Yeast metabolic chassis designs for diverse ... [https://www.nature.com/articles/srep29694]
- 78. Yeast as a cell factory: current state and perspectives - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4486425/]
- 79. YeastFab: the design and construction of standard ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4513847/]
- 80. A computational workflow for the expansion of ... [https://www.nature.com/articles/s41467-021-22022-5]
- 81. Metabolic and Evolutionary Engineering of Food Yeasts [https://www.mdpi.com/2227-9717/13/6/1852]
- 82. Advances in adaptive laboratory evolution applications for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12391446/]
- 83. Metabolic engineering and adaptive laboratory evolution of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12320376/]
- 84. Adaptive Laboratory Evolution of Yeasts for Aroma ... [https://www.mdpi.com/2311-5637/8/8/372]
- 85. Physiology of yeasts in relation to biomass yields [https://pubmed.ncbi.nlm.nih.gov/1807201/]
- 86. System-Level Insights into Yeast by Thermodynamic... Metabolism [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1002415]
- 87. Metabolic engineering of Saccharomyces cerevisiae for co ... [https://biotechnologyforbiofuels.biomedcentral.com/articles/10.1186/s13068-025-02685-8]
- 88. Elucidating the itaconic acid pathway dynamics in ... [https://www.sciencedirect.com/science/article/abs/pii/S1096717625001831]
- 89. Engineering Saccharomyces cerevisiae for growth on ... [https://pubmed.ncbi.nlm.nih.gov/39873813/]
- 90. Integration of a multi-step heterologous pathway in ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-019-1257-z]
- 91. MULTI-SCULPT: Multiplex Integration via Selective, CRISPR ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9288493/]
- 92. A Standardized Protocol for Assuring the Validity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10093877/]
- 93. Integrated omics of Saccharomyces cerevisiae CENPK2 ... [https://www.nature.com/articles/s41540-024-00382-0]
- 94. Design of tables for the presentation and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10346464/]
- 95. TRYing to evaluate production costs in microbial ... [https://www.sciencedirect.com/science/article/pii/S0167779924001197]
- 96. Comparing the fermentation performance of Escherichia coli ... [https://biotechnologyforbiofuels.biomedcentral.com/articles/10.1186/1754-6834-3-11]
- 97. Performance and robustness analysis reveals phenotypic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10618107/]
- 98. Brettanomyces yeasts — From spoilage organisms to ... [https://www.sciencedirect.com/science/article/pii/S0168160515001865]
- 99. Fermentation profiles of the yeast Brettanomyces ... [https://pubmed.ncbi.nlm.nih.gov/32889766/]
- 100. Recent advances in metabolic engineering of ... [https://www.sciencedirect.com/science/article/abs/pii/S1096717618300028]
- 101. Computational tools and resources for designing new ... [https://www.sciencedirect.com/science/article/pii/S0958166922000568]
- 102. Computational Tools for Metabolic Engineering - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3361690/]
- 103. Engineering yeast peroxisome assembly enables the ... [https://www.nature.com/articles/s41467-025-65444-1]
- 104. Burden Imposed by Heterologous Protein Production in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10512257/]
- 105. The 2025 Nimdzi 100 [https://www.nimdzi.com/nimdzi-100-2025/]
- 106. Translation Trends in 2025 [https://www.tomedes.com/translator-hub/translation-industry-trends]
- 107. Economic Viability Analysis → Term [https://climate.sustainability-directory.com/term/economic-viability-analysis/]
- 108. Understanding Technical Translation Methods: Industries, ... [https://seatongue.com/blog/business/understanding-technical-translation-methods-industries-approaches-and-regulations/]
- 109. the Crucial Importance of Accuracy in Translation [https://www.versioninternationale.com/en/blog/technical-documentation-translation-industrial/]