Validating E. coli FBA Models with Phenotype Phase Plane Analysis: A Guide for Biomedical Researchers
Flux Balance Analysis (FBA) is a cornerstone of systems biology, but its predictive power hinges on rigorous model validation.
Validating E. coli FBA Models with Phenotype Phase Plane Analysis: A Guide for Biomedical Researchers
Abstract
Flux Balance Analysis (FBA) is a cornerstone of systems biology, but its predictive power hinges on rigorous model validation. This article provides a comprehensive guide for researchers and scientists on using Phenotype Phase Plane (PhPP) analysis to validate genome-scale metabolic models (GEMs) of E. coli. We cover the foundational principles of constraint-based modeling and PhPP, detail a step-by-step methodology for its application, address common troubleshooting and optimization scenarios, and present a framework for the comparative analysis of model predictions against experimental data. By offering a structured validation workflow, this guide aims to enhance the reliability of in silico models for metabolic engineering and drug development.
Foundations of Constraint-Based Modeling and Phenotype Phase Planes
Core Principles of Constraint-Based Modeling and FBA
Constraint-based modeling and its flagship method, Flux Balance Analysis (FBA), form a cornerstone of systems biology for simulating metabolic networks at the genome scale. These approaches use mathematical constraints to predict optimal metabolic flux distributions without requiring detailed kinetic information, making them particularly powerful for analyzing complex biological systems where comprehensive kinetic parameter measurement remains infeasible [1] [2]. The fundamental principle involves representing the metabolic network as a stoichiometric matrix (denoted as S) where rows correspond to metabolites and columns represent biochemical reactions [1]. This matrix encapsulates the network structure derived from genomic information and biochemical literature [3].
FBA operates on the critical assumption that the system exists in a steady state, meaning metabolite concentrations remain constant over time [4] [2]. Under this assumption, the mass balance constraint is expressed mathematically as Sv = 0, where v is the flux vector containing reaction rates [1] [2]. This equation ensures that for each metabolite, the total production flux equals total consumption flux, preventing unrealistic accumulation or depletion. The solution space defined by these constraints contains all possible flux distributions that satisfy mass balance. To identify a biologically meaningful solution within this space, FBA employs linear programming to optimize an objective function, typically representing cellular goals such as biomass production, ATP synthesis, or metabolite synthesis [2] [5].
Core Mathematical Principles and Methodologies
Mathematical Framework of FBA
The mathematical formulation of FBA constitutes a linear optimization problem with the following components [2]:
- Objective Function: Z = cTv, where c is a vector of weights indicating each reaction's contribution to the cellular objective.
- Mass Balance Constraints: Sv = 0, ensuring steady-state metabolite concentrations.
- Flux Capacity Constraints: αi ≤ vi ≤ βi, representing physiological and thermodynamic limits for each reaction i.
The complete optimization problem becomes:
For microbial systems, the objective function frequently represents biomass production, which incorporates essential cellular components like proteins, nucleic acids, and lipids in appropriate ratios to simulate cellular growth [2] [5]. Exchange reactions model metabolite transfer between the cell and its environment, with constraints applied based on nutrient availability and experimental conditions [2].
Advanced Extensions and Variants
Basic FBA provides a foundational approach, but several advanced techniques have emerged to enhance its biological realism and analytical power:
- Flux Variability Analysis (FVA): Determines the range of possible flux values for each reaction while maintaining optimal objective function value, identifying reactions with flexible flux levels [2].
- Parsimonious FBA (pFBA): Identifies the most efficient flux distribution among multiple optima by minimizing total flux through the network while maintaining optimal growth, reflecting cellular preference for energy efficiency [2].
- Enzyme-Constrained Models: Incorporate enzyme availability and catalytic efficiency constraints to prevent unrealistically high flux predictions. Implementation methods include ECMpy, GECKO, and MOMENT [5].
Table 1: Key FBA Variants and Their Applications
| Method | Primary Function | Key Advantage | Common Application |
|---|---|---|---|
| Standard FBA | Predicts optimal flux distribution | Computational efficiency | Growth phenotype prediction |
| Flux Variability Analysis (FVA) | Identifies flux ranges in optimal solutions | Characterizes solution space flexibility | Determining essential reactions |
| Parsimonious FBA (pFBA) | Finds most efficient flux distribution | Reflects cellular energy conservation | Identifying preferred metabolic routes |
| Enzyme-Constrained FBA | Incorporates enzyme kinetics | Prevents unrealistic high fluxes | Metabolic engineering design |
Model Validation Frameworks and Metrics
Validation Techniques for FBA Predictions
Validation constitutes a critical step in establishing confidence in FBA predictions. The COnstraint-Based Reconstruction and Analysis (COBRA) framework includes fundamental quality control checks to ensure model functionality, such as verifying the inability to generate ATP without energy sources or synthesize biomass without required substrates [4]. The MEMOTE (MEtabolic MOdel TEsts) pipeline provides automated testing to ensure biomass precursors can be synthesized across various growth media [4].
Comprehensive validation typically employs multiple approaches [3]:
- Growth/No-Growth Predictions: Qualitative assessment of model accuracy in predicting viability under different nutrient conditions.
- Growth Rate Comparisons: Quantitative evaluation of how well simulated growth rates match experimental measurements.
- Gene Essentiality Predictions: Assessment of model accuracy in predicting whether gene knockouts will prevent growth.
For E. coli models, validation commonly utilizes gene essentiality data from large-scale mutant libraries, such as the RB-TnSeq dataset, which provides fitness measurements for thousands of genes across multiple carbon sources [6]. The area under a precision-recall curve (AUC) has emerged as a robust metric for quantifying model accuracy, particularly for imbalanced datasets where correct prediction of gene essentiality is more biologically meaningful than nonessentiality prediction [6].
Quantitative Validation of E. coli Metabolic Models
Recent systematic evaluation of four successive E. coli genome-scale metabolic models (iJR904, iAF1260, iJO1366, and iML1515) reveals evolving capabilities and validation metrics [6]. The progression of these models shows increasing gene coverage, with the latest model (iML1515) encompassing 1,515 genes, 2,719 metabolic reactions, and 1,192 metabolites [5].
Table 2: Accuracy Metrics for E. coli GEM Validation Using High-Throughput Mutant Fitness Data
| Model Version | Publication Year | Genes in Model | Precision-Recall AUC | Key Improvements |
|---|---|---|---|---|
| iJR904 | 2003 | 904 | Baseline | Foundational reconstruction |
| iAF1260 | 2007 | 1,266 | -1.3% vs. iJR904 | Expanded coverage |
| iJO1366 | 2011 | 1,366 | -2.2% vs. iJR904 | Enhanced prediction accuracy |
| iML1515 | 2017 | 1,515 | +4.8% vs. iJO1366* | Updated gene-protein-reaction relationships |
Note: Initial analysis showed decreasing accuracy, but corrections to simulation environment representation reversed this trend [6].
Error analysis of the iML1515 model identified specific areas requiring refinement [6]:
- Vitamin/Cofactor Biosynthesis: Genes involved in biotin, R-pantothenate, thiamin, tetrahydrofolate, and NAD+ biosynthesis frequently produced false-negative predictions, potentially due to cross-feeding between mutants or metabolite carry-over in experimental conditions.
- Gene-Protein-Reaction Mapping: Isoenzyme relationships represented a key source of inaccurate predictions.
- Metabolic Flux Patterns: Machine learning approaches identified fluxes through hydrogen ion exchange and central metabolism branch points as important determinants of model accuracy.
Phenotype Phase Plane Analysis for Model Validation
Principles and Methodology of PhPP
Phenotype Phase Plane (PhPP) analysis provides a global perspective on genotype-phenotype relationships by mapping optimal metabolic phenotypes across different environmental conditions [7] [8]. Developed by the Palsson laboratory, PhPP extends FBA by systematically varying availability of two key substrates (e.g., carbon and oxygen sources) and identifying discrete phases where qualitatively distinct metabolic pathway utilization patterns emerge [7]. Within each phase, all culture conditions share the same set of activated pathways and excreted products [8].
The classification of different phenotypes in traditional PhPP analysis relies on shadow prices of metabolites, which describe how each metabolite affects the objective function of FBA [8]. The boundaries between phases represent conditions where the optimal metabolic network utilization pattern shifts, providing insights into regulatory points and metabolic strategy transitions.
Figure 1: Workflow for Traditional Phenotype Phase Plane Analysis
System Identification Enhanced PhPP Analysis
To address limitations of traditional shadow price analysis, System Identification Enhanced PhPP (SID-PhPP) has been developed [8]. This approach perturbs the metabolic network through designed input sequences (in silico experiments), then applies multivariate statistical analysis tools like principal component analysis (PCA) to extract information on how perturbations propagate through the network [8].
SID-PhPP provides several advantages over traditional PhPP [8]:
- Identifies "hidden" phenotypes that share shadow prices with other phenotypes but have distinct metabolic states.
- Reveals how different reactions interact within the same phenotype.
- Provides more comprehensive characterization of metabolic network utilization.
Application of SID-PhPP to the E. coli core metabolic model demonstrates its enhanced capability to distinguish metabolic phenotypes when analyzing glucose and oxygen uptake variations, successfully identifying distinct phases for mixed-acid fermentation and aerobic respiration [8].
Experimental Protocols for FBA Validation
Gene Essentiality Prediction Protocol
Objective: Validate FBA model predictions against experimental gene essentiality data.
Materials:
- Curated genome-scale metabolic model (e.g., iML1515 for E. coli)
- Gene essentiality dataset (e.g., RB-TnSeq fitness data)
- Constraint-based modeling software (COBRA Toolbox, cobrapy)
Methodology [6]:
- For each gene in the model, simulate knockout by constraining its associated reaction flux(es) to zero.
- Perform FBA with biomass maximization as objective function.
- Classify the gene as essential if simulated growth rate falls below threshold (typically <1% of wild-type).
- Compare predictions with experimental essentiality data.
- Calculate accuracy metrics: precision-recall AUC, overall accuracy, false positive/negative rates.
Troubleshooting:
- High false negatives for vitamin/cofactor genes may indicate cross-feeding in experimental data; consider adding these metabolites to simulation environment [6].
- Incorrect isoenzyme predictions require verification of gene-protein-reaction relationships [6].
Phenotype Phase Plane Analysis Protocol
Objective: Characterize metabolic phenotype changes across varying substrate conditions.
Materials:
- Metabolic model (core or genome-scale)
- Two target substrates for analysis
- Select two substrates of interest (e.g., glucose and oxygen).
- Define concentration ranges for both substrates.
- Perform FBA at each combination of substrate uptake rates.
- Calculate shadow prices for key metabolites at each condition.
- Identify phase boundaries where shadow prices change discontinuously.
- For SID-PhPP: apply designed input sequences and multivariate analysis.
- Map phase diagram showing distinct phenotypic regions.
Interpretation:
- Each phase represents a distinct metabolic strategy.
- Phase boundaries indicate conditions where pathway utilization shifts.
- Shadow prices reveal metabolite limiting effects on growth.
Research Reagent Solutions for FBA Validation
Table 3: Essential Research Resources for FBA Validation Studies
| Resource Category | Specific Examples | Function in FBA Validation | Data Source |
|---|---|---|---|
| Genome-Scale Models | iML1515, iJO1366, EcoCyc-18.0-GEM | Base metabolic network for simulations | BiGG Database, EcoCyc |
| Validation Datasets | RB-TnSeq fitness data, Chemogenomic profiles | Experimental reference for model predictions | Published literature [6] [9] |
| Software Tools | COBRA Toolbox, cobrapy, Pathway Tools | Implement FBA and variants | Open source platforms |
| Enzyme Kinetics Data | Kcat values, molecular weights | Parameterizing enzyme-constrained models | BRENDA, PAXdb [5] |
| Experimental Phenotype Data | Nutrient utilization, growth rates | Quantitative model validation | Literature curation [3] |
Constraint-based modeling and Flux Balance Analysis provide powerful frameworks for predicting metabolic behavior from genomic information. Core principles including stoichiometric mass balance, flux capacity constraints, and biological objective functions enable quantitative simulation of complex metabolic networks. Rigorous validation through gene essentiality prediction, phenotype phase plane analysis, and comparison with experimental data remains essential for establishing model credibility and identifying areas for refinement. The integration of advanced techniques such as enzyme constraints and system identification enhanced analysis continues to improve the biological fidelity and predictive capability of these approaches, supporting their expanding applications in basic research and metabolic engineering.
Phenotype Phase Plane (PhPP) analysis is a constraint-based modeling technique that provides a global view of how changes in two environmental variables affect an organism's optimal metabolic phenotype. This method expands on Flux Balance Analysis (FBA) by mapping optimal metabolic flux distributions across all possible combinations of two key substrate uptake rates, revealing discrete phases with distinct metabolic pathway utilization patterns [7] [10].
Core Principles and Mathematical Foundation
PhPP analysis is built upon the framework of genome-scale metabolic models, which are reconstructed from annotated genome sequences, biochemical literature, and strain-specific information [7]. These models contain the complete set of metabolic reactions for an organism, represented in a stoichiometric matrix S where each element Sₙₘ corresponds to the stoichiometric coefficient of metabolite n in reaction m.
The key mathematical principles include:
Flux Balance Analysis: PhPP uses FBA to predict metabolic fluxes by solving a linear programming problem that maximizes biomass production (or another objective function) subject to stoichiometric constraints: Maximize Z = cᵀv, subject to S·v = 0 and vₘᵢₙ ≤ v ≤ vₘₐₓ, where v is the flux vector and c is a vector indicating the objective function [10].
Shadow Prices: The analysis utilizes shadow prices (dual variables of the linear programming solution) to determine how changes in metabolite availability affect the objective function. A positive shadow price indicates a metabolite is available in excess, while a negative value indicates a limiting metabolite [10].
Phase Boundaries: The phase plane is divided by isoclines where shadow price ratios change, representing shifts in optimal pathway utilization [7]. Each distinct phase in the PhPP corresponds to a specific metabolic phenotype with unique pathway usage.
PhPP Analysis of E. coli Metabolism
Experimental Protocol for E. coli PhPP Analysis
The standard methodology for constructing a phenotype phase plane for E. coli involves these key steps [7]:
Model Reconstruction: Utilize a genome-scale metabolic model of E. coli (such as iJR904 containing 904 genes) with appropriate compartmentalization and mass balances.
Parameter Definition: Select two environmental variables to define the phase plane (e.g., glucose and oxygen uptake rates). Set bounds for other nutrients and by-products.
Linear Programming: For each pair of substrate uptake rates in the phase plane, solve the linear programming problem to determine the maximal growth rate and flux distribution.
Shadow Price Calculation: Compute shadow prices for all metabolites at each point in the phase plane to identify isoclines and phase boundaries.
Phase Identification: Partition the phase plane into regions where the optimal metabolic pathway utilization remains qualitatively unchanged.
Validation: Compare in silico predictions with experimental growth data and by-product secretion profiles.
Key Findings from E. coli PhPP Studies
PhPP analysis of E. coli growth on acetate and glucose at varying oxygenation levels revealed several fundamental insights [7]:
Table 1: E. coli Phenotype Phase Plane Analysis Findings
| Aspect Analyzed | Key Finding | Significance |
|---|---|---|
| Phase Transitions | Identification of finite, qualitatively distinct metabolic phases | Demonstrates discrete metabolic strategy shifts rather than continuous adaptation |
| Optimal Growth | Lines of optimality (LO) identified where substrate utilization is optimal | Provides engineering targets for bioprocess optimization |
| Pathway Utilization | Distinct phases employ different primary metabolic pathways | Reveals metabolic network flexibility and regulatory design |
| Genotype-Phenotype Relationship | Direct mapping of metabolic capabilities to environmental conditions | Bridges genetic makeup with observable physiological behavior |
The analysis demonstrated that E. coli undergoes distinct metabolic strategy shifts rather than continuous adaptation as environmental conditions change. The identification of lines of optimality provides potential engineering targets for bioprocess optimization [7] [11].
Extension to Eukaryotic Systems: Saccharomyces cerevisiae
PhPP methodology has been successfully applied to eukaryotic systems, particularly Saccharomyces cerevisiae. The glucose-oxygen PhPP for yeast reveals seven distinct metabolic phases (P1-P7) with characteristic features [10]:
Table 2: S. cerevisiae Metabolic Phases in Glucose-Oxygen PhPP
| Phase | Oxygen Conditions | Primary Metabolic Features | By-Products Secreted |
|---|---|---|---|
| P1 | High oxygen | Fully oxidative metabolism | CO₂, H₂O |
| P2 | Moderate oxygen | Oxidative-fermentative transition | Ethanol, acetate |
| P3 | Low oxygen | Fermentative metabolism | Ethanol, glycerol, succinate |
| P4-P7 | Varying limitations | Specialized metabolic states | Varying by-product profiles |
Shadow price analysis and in silico gene deletion studies further characterize these phases. For instance, in Phase 2, mitochondrial NAD⁺ is available in excess, and the production of acetate and ethanol is essential for maintaining redox balance [10].
Advanced Applications and Modern Extensions
Enzyme-Constrained Metabolic Models
Recent advances integrate enzyme constraints into metabolic models, creating enzyme-constrained GEMs (ecGEMs) that provide more realistic predictions [12]. The construction of ecGEMs involves:
kcat Data Collection: Enzyme turnover numbers obtained through machine learning prediction (TurNuP), database mining (AutoPACMEN), or other computational methods [12].
Model Integration: Incorporating enzyme mass constraints using frameworks like ECMpy or GECKO, adding rows to the stoichiometric matrix representing enzyme usage [12].
Capacity Constraints: Setting upper bounds on metabolic fluxes based on enzyme abundance and catalytic efficiency.
For Myceliophthora thermophila, ecGEM construction revealed trade-offs between biomass yield and enzyme usage efficiency at varying glucose uptake rates, demonstrating how enzyme constraints affect predicted phenotypic states [12].
Stochastic Phenotype Analysis
While traditional PhPP analysis assumes deterministic metabolism, recent research examines stochastic multimodality in gene regulatory networks like feed-forward loops (FFLs) [13]. Key findings include:
- Multimodality: FFLs can exhibit up to 6 stable phenotypic states under strong input intensity [13].
- Slow Promoter Binding: This can generate distinct protein expression levels of long duration, creating phenotypic diversity even without feedback regulation [13].
- Gene Duplication: Can enlarge stable regions of specific multimodalities and enrich phenotypic diversity [13].
These findings are particularly relevant for understanding cellular fate decisions, stem cell differentiation, and tumor formation [13].
Automated Phenotyping Technologies
Advanced experimental methods now enable high-throughput phenotypic characterization:
Exhaustive Projection Pursuit (EPP): An automated algorithm that evaluates all two-dimensional projections of flow cytometry data to identify statistically significant cell populations without prior knowledge [14].
Multi-Color Spectral Transcript Analysis (SPECTRA): Uses multiplexed fluorescence in situ hybridization with spectral imaging to quantitatively measure tumor-specific gene expression signatures at single-cell resolution [15].
Research Reagent Solutions
Table 3: Essential Research Reagents and Computational Tools for PhPP Analysis
| Resource Type | Specific Tool/Reagent | Function/Application |
|---|---|---|
| Computational Tools | ACME Algorithm | Solves discrete Chemical Master Equation for exact probability landscapes [13] |
| Computational Tools | ECMpy Workflow | Constructs enzyme-constrained GEMs [12] |
| Computational Tools | AutoPACMEN | Automatically retrieves enzyme kinetic data from databases [12] |
| Experimental Methods | Spectral Imaging (SPECTRA) | Quantitative multigene expression analysis in single cells [15] |
| Experimental Methods | Exhaustive Projection Pursuit | Automated identification of cell populations in flow cytometry [14] |
| Model Resources | BiGG Database | Curated metabolic reconstruction database [12] |
| Model Resources | BRENDA/SABIO-RK | Enzyme kinetic parameter databases [12] |
Conceptual Diagrams
PhPP Analysis Workflow: From genomic data to validated metabolic phenotypes.
E. coli Phenotype Phase Plane Structure: Discrete metabolic phases under varying substrate conditions.
The Stoichiometric Matrix and Mass Balance Constraints
Flux Balance Analysis (FBA) is a cornerstone mathematical approach for simulating metabolism in cells, particularly unicellular organisms like E. coli. It operates on genome-scale metabolic models (GEMs), which are computational representations of all known biochemical reactions within an organism, linked to their corresponding genes [16]. The core strength of FBA lies in its ability to predict metabolic flux distributions—the rates at which metabolites flow through biochemical pathways—under steady-state conditions, without requiring detailed enzyme kinetic parameters [16] [5].
The stoichiometric matrix and mass balance constraints are the fundamental mathematical constructs that make this analysis possible. The stoichiometric matrix, denoted as S, is an m × n matrix where rows represent m metabolites and columns represent n metabolic reactions. Each element Sᵢⱼ is the stoichiometric coefficient of metabolite i in reaction j [16]. The mass balance constraint is encapsulated by the equation S · v = 0, where v is an n-dimensional vector of reaction fluxes [16] [17]. This equation formalizes the assumption of a metabolic steady state, meaning that for each internal metabolite, its rate of production is exactly balanced by its rate of consumption, so there is no net accumulation or depletion [17] [18]. These constraints, along with others that define reaction bounds (e.g., uptake rates), define a solution space of all possible, feasible flux distributions [5] [18]. FBA then uses linear programming to identify a single flux map within this space that optimizes a specified biological objective, such as the maximization of biomass growth or the production of a target metabolite [16] [5].
Fundamental Principles and Mathematical Foundation
The Stoichiometric Matrix (S-Matrix)
The stoichiometric matrix provides a complete mathematical representation of the metabolic network's structure.
- Network Encoding: It catalogs all metabolic interconversions, where columns represent reactions and rows represent metabolites [16].
- Constraint Definition: The matrix elements are stoichiometric coefficients that define the mass relationships for all reactants and products in every reaction [5].
- Steady-State Formulation: The equation S · v = 0 is a linear system of equations. For each metabolite, this equation asserts that the sum of its fluxes in producing reactions equals the sum of its fluxes in consuming reactions [16] [17]. This is the application of the law of conservation of mass within the metabolic network [16].
Formulating Mass Balance for FBA
The mass balance constraint is what makes FBA a "constraint-based" method. The core FBA problem can be formally defined as [16]: \begin{aligned} \max{\mathbf{v}}\quad & \, v{\mathrm{biomass}} \ \mathrm{s.t.} \quad & S\mathbf{v}=0 \ & \mathbf{l} \le \mathbf{v} \le \mathbf{u} \end{aligned}
Here, ( v_{\mathrm{biomass}} ) is the flux of the biomass reaction, representing cellular growth. The equation ( S \cdot v = 0 ) enforces mass balance. The vectors l and u represent the lower and upper bounds for each reaction flux, respectively, constraining uptake/secretion rates and enzyme capacities [16] [5].
The following diagram illustrates the workflow of constructing the stoichiometric matrix and applying mass balance constraints for FBA.
Experimental and Computational Protocols
Protocol 1: Standard FBA for Predicting Gene Deletion Phenotypes
This protocol uses FBA to predict cellular growth after a gene knockout [17].
- Step 1: Model Initialization. Load a curated GEM for E. coli, such as iML1515, which includes 1,515 genes, 2,719 reactions, and 1,192 metabolites [5] [17]. Set the biomass reaction as the objective function to maximize.
- Step 2: Simulate Gene Deletion. To simulate the deletion of a specific gene, set the flux bounds to zero (( Vi^{\text{min}} = Vi^{\text{max}} = 0 )) for every reaction catalyzed by the protein encoded by that gene, as determined by the model's Gene-Protein-Reaction (GPR) rules [17].
- Step 3: Solve and Validate. Solve the linear programming problem. A predicted growth rate of zero indicates a lethal gene deletion. Validate predictions against experimental gene essentiality data from databases like EcoCyc to assess model accuracy [5] [17].
Protocol 2: Dynamic FBA (dFBA) for Simulating Co-cultures
dFBA extends FBA to simulate time-dependent changes in metabolism and environment, ideal for modeling microbial communities [16].
- Step 1: Initialize Multi-Species Model. Load the GEMs for each strain in the co-culture (e.g., E. coli Nissle 1917 and Lactobacillus plantarum WCFS1). Identify and map common exchange reactions that allow metabolites to be transported between the species and their shared environment [16].
- Step 2: Define the Extracellular Environment. Set initial concentrations of key extracellular metabolites (e.g., glucose, oxygen, ammonium) and initial biomass for each strain. Define uptake kinetics and constraints for the exchange reactions based on these concentrations [16].
- Step 3: Iterative Simulation Loop. For each time step [16]: a. Solve FBA: For each organism, perform FBA using its respective model and the current extracellular metabolite concentrations to calculate growth and metabolic fluxes. b. Update Concentrations: Use the calculated uptake and secretion fluxes from all organisms to update the extracellular metabolite concentrations via a system of ordinary differential equations. c. Update Biomass: Update the biomass of each organism based on its computed growth rate.
- Step 4: Analyze Results. Track metabolite concentrations and biomass over time to identify emergent behaviors like competition, cross-feeding, and potential metabolite overproduction [16].
Advanced Frameworks and Model Validation
Selecting an appropriate objective function is critical for FBA accuracy. Advanced frameworks have been developed to infer objective functions directly from experimental data, moving beyond standard assumptions like biomass maximization.
TIObjFind is a novel framework that integrates Metabolic Pathway Analysis (MPA) with FBA. It identifies Coefficients of Importance (CoIs), which are pathway-specific weights that quantify each reaction's contribution to a cellular objective [19] [20]. The framework works by [19] [20]:
- Reformulating objective function selection as an optimization problem that minimizes the difference between FBA-predicted fluxes and experimental flux data.
- Mapping FBA solutions to a Mass Flow Graph (MFG) for pathway-based analysis.
- Applying a minimum-cut algorithm to this graph to identify critical pathways and compute the CoIs, which enhance the interpretability of complex metabolic networks.
Flux Cone Learning (FCL) is a machine learning strategy that predicts gene deletion phenotypes without assuming a cellular objective [17]. It uses Monte Carlo sampling to generate random flux distributions that satisfy the mass balance constraints (the "flux cone") for both the wild type and gene deletion mutants. A supervised learning model is then trained on these flux samples, using experimental fitness data as labels, to learn the correlation between changes in the shape of the solution space and phenotypic outcomes [17].
The table below compares these advanced methods against traditional FBA.
Table 1: Comparison of FBA Methodologies for E. coli Metabolic Modeling
| Method | Core Approach | Data Requirements | Key Advantages | Primary Application in E. coli Research |
|---|---|---|---|---|
| Traditional FBA [16] [5] | Linear programming with a pre-defined objective (e.g., biomass). | GEM, exchange reaction bounds. | Computationally efficient; suitable for genome-scale models. | Predicting growth rates, gene essentiality, and product yield. |
| TIObjFind [19] [20] | Infers objective function from data using MPA and optimization. | GEM, experimental flux data (13C-MFA). | Aligns model predictions with data; reveals condition-specific metabolic goals. | Identifying adaptive metabolic shifts and key pathways under different conditions. |
| Flux Cone Learning (FCL) [17] | Machine learning on sampled flux distributions. | GEM, experimental fitness data (e.g., from deletion screens). | No optimality assumption required; outperforms FBA in gene essentiality prediction. | High-accuracy prediction of gene deletion phenotypes across diverse conditions. |
Critical Practices in Model Validation
Robust validation is essential for establishing confidence in FBA predictions.
- Comparison with 13C-MFA: 13C-Metabolic Flux Analysis provides experimentally estimated intracellular fluxes and serves as a gold standard for validating FBA-predicted flux maps [18]. A significant deviation suggests the FBA model's objective function or constraints may be incorrect [18].
- χ²-test of Goodness-of-Fit: In 13C-MFA, this statistical test is used to validate whether the difference between the measured isotopic labeling data and the model-estimated labeling is within expected experimental error [18].
- Gene Essentiality Prediction: For FBA, a common validation test is to check its accuracy in predicting which gene knockouts will prevent growth (are essential), using experimental data from genome-wide knockout screens as a benchmark [17].
The Scientist's Toolkit
Table 2: Essential Research Reagents and Computational Tools
| Tool/Reagent | Function/Description | Example Use in E. coli Studies |
|---|---|---|
| Genome-Scale Model (GEM) | A structured database of an organism's metabolism, listing reactions, metabolites, and genes. | iML1515 [5], iDK1463 [16]; used as the core scaffold for all FBA simulations. |
| COBRA Toolbox/COBRApy | Software suites providing standardized functions to perform FBA and related analyses. | Used to load models, define constraints, solve the optimization problem, and analyze results [16] [5]. |
| Stoichiometric Database (e.g., EcoCyc, KEGG) | Curated knowledge bases of metabolic pathways and stoichiometries. | Used for model curation, gap-finding, and validation of reaction stoichiometries [19] [5]. |
| Enzyme Constraint Data (Kcat, Abundance) | Kinetic and proteomic data used to add enzyme capacity constraints to FBA models. | Tools like ECMpy integrate Kcat values (from BRENDA) and abundance data to create more realistic models [5]. |
| Aztreonam | An antibiotic that inhibits cell division by targeting FtsI, inducing filamentation. | Used in experimental studies to induce filamentation in E. coli for investigating mechanobiology and division [21]. |
| 13C-Labeled Substrates | Isotopically labeled nutrients (e.g., 13C-glucose) fed to cells for tracing metabolic flux. | Essential for 13C-MFA experiments to generate experimental flux data for model validation [18]. |
The following diagram outlines the logical workflow for validating an FBA model, integrating both computational and experimental resources from the toolkit.
In the realm of constraint-based metabolic modeling, objective functions are fundamental to simulating and predicting cellular behavior. Flux Balance Analysis (FBA) is a widely used mathematical approach for analyzing the flow of metabolites through a metabolic network, particularly genome-scale metabolic models (GEMs) [22]. Since these models typically contain more reactions than metabolites, the solution space is large, and an objective function is required to identify a particular, optimal flux distribution from the many possible solutions [23] [22]. The choice of objective function essentially represents a hypothesis about the biological goal of the organism, such as maximizing growth or the production of a specific metabolite.
The core of FBA involves solving a system of equations based on the stoichiometric matrix (S), which represents all known metabolic reactions in the organism. This matrix imposes mass balance constraints, ensuring that the total production and consumption of each metabolite are balanced at steady state, expressed as Sv = 0, where v is the vector of reaction fluxes [24] [22]. Further constraints are applied by defining upper and lower bounds (αi ≤ vi ≤ βi) on individual reaction fluxes, often based on measured uptake rates or gene deletion studies [24]. To find a single solution within this constrained space, linear programming is used to maximize or minimize a defined objective function, Z = cTv, where c is a vector of weights indicating how much each reaction contributes to the objective [22].
This review focuses on comparing two primary classes of objective functions: biomass maximization, which simulates cellular growth, and product yield optimization, which targets the efficient production of specific biochemicals. We will evaluate their performance, applications, and validation within the specific context of E. coli Phenotype Phase Plane (PhPP) analysis.
Biomass Objective Function: Simulating Cellular Growth
Formulation and Composition
The biomass objective function is the gold standard in FBA for predicting growth rates and gene essentiality [23]. It is formulated as a reaction that drains essential biomass precursors—such as amino acids, nucleotides, lipids, and carbohydrates—from the metabolic network in the precise proportions found in experimental measurements of cellular composition [23]. This "biomass reaction" is scaled so that its flux is equal to the exponential growth rate (μ) of the organism [22]. The formulation can exist at different levels of detail:
- Basic Level: Defines the macromolecular content of the cell (e.g., weight fraction of protein, RNA, lipid) and the metabolites that constitute each macromolecule [23].
- Intermediate Level: Incorporates biosynthetic energy requirements, such as the ATP and GTP needed to polymerize amino acids into proteins [23].
- Advanced Level: Includes vitamins, elements, cofactors, and can be refined into a "core" biomass function that represents the minimal content required for cellular viability, often informed by data from mutant strains [23].
Performance and Experimental Validation
Biomass maximization has proven highly effective for predicting gene essentiality, particularly in well-studied microorganisms like E. coli. For example, FBA with a biomass objective function can accurately predict aerobic and anaerobic growth rates of E. coli on glucose minimal media, with computations showing strong agreement with experimental measurements [22]. Early FBA studies utilizing the iJR904 E. coli model successfully identified seven gene products in central metabolism as essential for aerobic growth and 15 for anaerobic growth on glucose [24]. This demonstrates the function's utility in mapping genotype-phenotype relationships.
However, the predictive power of biomass optimization is highly dependent on the quality and completeness of the underlying metabolic model and the accuracy of the biomass composition data. Its performance can also diminish in higher-order organisms where the assumption of growth maximization may not hold [25].
Table 1: Key Experiments Validating the Biomass Objective Function
| Model / Organism | Experimental Validation | Key Finding | Reference |
|---|---|---|---|
| E. coli core metabolism | Comparison of predicted vs. measured growth rates | Predicted aerobic (1.65 hr⁻¹) and anaerobic (0.47 hr⁻¹) growth rates on glucose agreed well with experimental data. | [22] |
| E. coli iJR904 (GEM) | Gene essentiality prediction | Identified 7 and 15 essential gene products for aerobic and anaerobic growth on glucose, respectively. | [24] |
| Hybridoma cell line | Analysis of growth & metabolite production | Optimization of biomass production could explain observed growth characteristics and phenomena. | [23] |
Protocol: Gene Essentiality Screen using Biomass Maximization
Purpose: To identify metabolic genes essential for growth under specific environmental conditions.
- Model Constraint: Acquire a genome-scale metabolic reconstruction (GEM) for E. coli (e.g., iML1515 or a core model like iCH360) [26] [22].
- Condition Setup: Define the simulated growth medium by constraining the uptake fluxes of available nutrients (e.g., set glucose uptake to 18.5 mmol gDW⁻¹ hr⁻¹) and other electron acceptors like oxygen [22].
- Objective Definition: Set the biomass reaction as the objective function to be maximized.
- Gene Deletion Simulation: For each gene in the model, simulate a knockout by constraining the flux through all associated enzyme-catalyzed reactions to zero, based on the gene-protein-reaction (GPR) relationships [24].
- Growth Prediction: Perform FBA for each knockout strain. A predicted growth rate of zero indicates an essential gene under the tested conditions.
- Experimental Correlation: Compare computational predictions with data from genome-wide knockout screens to validate the model [25].
Product Yield Optimization: Engineering Metabolic Output
Theoretical Framework for Yield Optimization
While biomass optimization focuses on growth rate, many metabolic engineering applications prioritize metabolic yield—the amount of product formed per unit of substrate consumed [27]. Yield is a ratio of fluxes (e.g., Yp/s = product flux / substrate uptake flux), making its optimization a linear-fractional programming (LFP) problem, which is non-linear and cannot be solved by standard FBA [27].
A comprehensive mathematical framework has been developed to overcome this challenge. The yield optimization problem can be transformed into a higher-dimensional linear problem, the solutions of which determine the yield-optimal flux distributions in the original model [27]. This formalism reveals that the yield-optimal solution set is determined by yield-optimal elementary flux vectors [27]. A critical insight from this theory is that yield-optimal and rate-optimal solutions are not always the same; the highest yield is not necessarily achieved at the flux distribution that gives the fastest growth or highest production rate [27]. This has profound implications for bioprocess design.
Performance and Applications
Yield optimization is particularly valuable for designing "cell factories" where substrate cost is a major factor, and high conversion efficiency is the primary goal. For instance, in the production of compounds like xanthommatin in Pseudomonas putida, growth-coupled biosynthetic pathways can be designed to link product synthesis to microbial growth, ensuring high yield and stability [28]. Advanced algorithms, such as OptKnock, leverage FBA and yield considerations to predict gene knockouts that force the organism to overproduce a desired compound as a byproduct of growth [22].
The opt-yield-FBA algorithm is a specific implementation that enables yield analysis and calculation of yield spaces directly on genome-scale models without the computationally intensive calculation of Elementary Flux Modes. This facilitates dynamic modeling frameworks, such as Hybrid Cybernetic Models (HCMs), for simulating metabolic dynamics at the genome-scale [29].
Table 2: Comparison of Biomass vs. Yield Optimization Objectives
| Feature | Biomass Maximization | Product Yield Optimization |
|---|---|---|
| Mathematical Form | Linear Program (LP) | Linear-Fractional Program (LFP) |
| Primary Goal | Maximize growth rate (hr⁻¹) | Maximize product per substrate (g/g) |
| Typical Application | Study of physiology, gene essentiality | Metabolic engineering, bioprocess design |
| Solution Nature | Often unique flux distribution | May be a different flux distribution from rate optimum |
| Key Strength | High accuracy for microbial growth prediction | Identifies efficient, substrate-optimal pathways |
Comparative Analysis via Phenotype Phase Plane (PhPP)
The Phenotype Phase Plane (PhPP) analysis is a powerful tool for visualizing and comparing the outcomes of different objective functions under varying environmental conditions [24]. A PhPP is a two-dimensional projection of the feasible metabolic solution space, typically with two key exchange fluxes (e.g., substrate and oxygen uptake rates) as the axes [24]. Demarcation lines within the plane separate regions of qualitatively different metabolic pathway utilization.
When analyzing biomass versus product yield, PhPPs can reveal the conditions under which these objectives align or diverge. For example, the line of optimality (LO) on a PhPP for biomass maximization shows the optimal relationship between substrate uptake and growth. In contrast, a yield-optimized PhPP would show a different optimal subspace for maximizing product per substrate. This analysis can identify phase planes (Pnx,y) where different pathways are utilized, helping engineers choose the optimal cultivation strategy (e.g., carbon-limited vs. oxygen-limited) for their specific goal [24].
Figure 1: Workflow for comparing biomass and yield objectives using Phenotype Phase Plane (PhPP) analysis. The process begins by constraining the model, then separately calculates optimal states for biomass (blue) and yield (red) before mapping and comparing the results.
Emerging Paradigms: Beyond Traditional FBA
Recent advances are moving beyond the traditional assumptions of FBA. Flux Cone Learning (FCL) is a novel machine learning framework that predicts gene deletion phenotypes by learning the shape of the metabolic flux cone, without presupposing a cellular objective like growth maximization [25]. FCL uses Monte Carlo sampling to generate data on the geometry of the metabolic space for different gene deletions. A supervised learning model is then trained on this data alongside experimental fitness scores. This approach has demonstrated best-in-class accuracy for predicting metabolic gene essentiality in E. coli, Saccharomyces cerevisiae, and Chinese Hamster Ovary cells, outperforming the gold standard FBA predictions [25].
This indicates that while biomass and product yield are powerful objective functions, the future of predictive metabolic modeling may lie in hybrid approaches that combine mechanistic models (GEMs) with machine learning to uncover complex, data-driven correlations between network state and phenotypic outcomes [25] [30].
The Scientist's Toolkit: Key Research Reagents and Models
Table 3: Essential Research Reagents and Models for FBA and Objective Function Analysis
| Tool / Reagent | Type | Function in Research | Example/Reference |
|---|---|---|---|
| COBRA Toolbox | Software | A MATLAB toolbox for performing constraint-based reconstruction and analysis (COBRA) methods, including FBA. | [22] |
| E. coli Core Model | Metabolic Model | A small-scale, educational model of central metabolism for method development and testing. | [22] |
| iML1515 | Metabolic Model | A comprehensive, genome-scale model of E. coli K-12 MG1655 with 1515 genes, 2712 reactions. | [25] [26] |
| iCH360 | Metabolic Model | A manually curated, medium-scale model of E. coli energy and biosynthesis metabolism; a sub-network of iML1515. | [26] |
| Stoichiometric Matrix (S) | Data Structure | Encodes the stoichiometry of all metabolic reactions; the core of any constraint-based model. | [24] [22] |
| Biomass Composition Data | Dataset | Quantitative measurements of cellular components (proteins, RNA, etc.) required to formulate the biomass objective function. | [23] |
| Gene-Knockout Strain Library | Experimental Resource | Used to validate computational predictions of gene essentiality and other phenotypes. | [25] |
Figure 2: A decision framework for selecting an appropriate modeling approach and objective function, emphasizing the alignment between the research question, available data, and model capabilities.
Interpreting Shadow Prices and Lines of Optimality in PhPP
Phenotype Phase Plane (PhPP) analysis is a powerful method for interpreting the results of Flux Balance Analysis (FBA), a constraint-based approach that predicts metabolic flux distributions in biological systems. FBA computes optimal metabolic phenotypes by leveraging genome-scale metabolic models (GEMs) that mathematically represent an organism's biochemical reaction network [26] [31]. The validation of these models is crucial for ensuring accurate predictions of cellular behavior, particularly in biotechnological and pharmaceutical applications. Within this framework, shadow prices and lines of optimality serve as critical analytical tools for understanding how an organism's metabolic phenotype responds to environmental changes. Shadow prices quantify the sensitivity of the objective function (typically biomass production) to changes in metabolite availability, while lines of optimality demarcate regions in the phase plane where fundamental shifts in metabolic strategy occur [32] [33].
Escherichia coli has emerged as a cornerstone organism for FBA model validation due to its well-characterized metabolism and the availability of extensively curated genome-scale models. The recent development of the iCH360 model, a manually curated "Goldilocks-sized" model of E. coli K-12 MG1655, provides an ideal platform for such analyses. Positioned between overly simplified core models and complex genome-scale reconstructions, iCH360 encompasses 323 metabolic reactions mapped to 360 genes, focusing specifically on energy production and biosynthetic pathways for amino acids, nucleotides, and fatty acids [26] [31]. This intermediate complexity makes it particularly suitable for PhPP analysis, as it maintains biological realism while remaining computationally tractable for the intensive sampling required for phase plane construction.
Theoretical Foundations of Shadow Prices and Optimality Lines
Mathematical Definition and Interpretation of Shadow Prices
In linear programming formulations of FBA, a shadow price (also known as a dual value) represents the rate of change of the objective function value with respect to a marginal change in the right-hand side of a constraint. Mathematically, for an optimization problem with objective function Z and constraint bᵢ, the shadow price πᵢ is defined as the partial derivative ∂Z/∂bᵢ [32]. In metabolic terms, this translates to how much the cellular growth rate (or other optimized objective) would increase if the availability of a particular nutrient or metabolic constraint were slightly relaxed.
The practical interpretation of shadow prices depends on both the problem context and constraint type. For maximization problems such as biomass optimization, the shadow price indicates how much the objective function would improve per unit increase in resource availability [33]. As one explanation clarifies: "The shadow price associated with a particular constraint tells you how much the optimal value of the objective would increase per unit increase in the amount of resources available" [33]. For example, a shadow price of 0.727 for a carbon source constraint suggests that increasing carbon availability by one unit would increase the growth rate by approximately 0.727 units [33].
Lines of Optimality in Phenotype Phase Planes
Lines of optimality (LOOs) represent boundaries in a Phenotype Phase Plane where fundamental shifts occur in the pattern of metabolic flux utilization. These lines demarcate distinct phenotypic phases where different sets of constraints are active in the optimal solution. At a LOO, the shadow prices of certain constraints change discontinuously, indicating a transition between metabolic strategies [32].
From a geometric perspective, the PhPP represents the objective function value as a function of two environmental variables (typically nutrient uptake rates), with LOOs appearing as edges or folds in the resulting surface. These lines emerge due to the piecewise linear nature of FBA solutions and correspond to changes in the basis of the optimal solution. The identification and interpretation of LOOs enables researchers to predict how microorganisms like E. coli will reallocate metabolic resources in response to environmental gradients.
Comparative Analysis of Shadow Price Computation Across Platforms
Methodological Variations and Their Impact
The computation of shadow prices can vary significantly across different optimization platforms and solver algorithms, potentially leading to different interpretations of the same biological system. Evidence suggests that different linear programming packages may employ different conventions for the sign of shadow prices, requiring careful interpretation of results [32].
A notable example of this variability was demonstrated in a comparison between Gurobi and PuLP solvers, where identical linear programming models produced different shadow price values for certain constraints. In one model, Gurobi reported a shadow price of 0.0 for a particular constraint, while PuLP returned 0.14285714 for the same constraint [34]. This discrepancy was attributed to the existence of multiple optimal dual solutions in degenerate problems, where different solvers may arbitrarily select different solutions from the optimal set [34].
Soluter Compatibility with Metabolic Modeling
The accurate computation of shadow prices in metabolic models depends on both the problem type and solver engine compatibility. Shadow prices can only be computed for continuous optimization problems and do not exist for integer or mixed-integer optimizations [32]. The following table summarizes solver compatibility with various problem types based on empirical validation:
Table 1: Solver Support for Shadow Price Computation by Problem Type
| Solver Engine | LP | QP | QCP | NLP |
|---|---|---|---|---|
| LP/Quadratic | Yes | No | No | No |
| SOCP Barrier | Yes | Yes | No | No |
| GRG Nonlinear | Yes | Yes | Yes | Yes |
| Evolutionary | No | No | No | No |
| LSGRG | Yes | Yes | Yes | Yes |
| LSSQP | Yes | Yes | Yes | No |
| OptQuest | No | No | No | No |
| Knitro | Yes | Yes | Yes | Yes |
LP = Linear Programming, QP = Quadratic Programming, QCP = Quadratically Constrained Programming, NLP = Non-Linear Programming. Adapted from Analytica documentation [32].
This compatibility matrix highlights the importance of selecting appropriate solvers for specific metabolic modeling applications, as shadow prices may be unavailable or inaccurate with incompatible solver-problem type combinations.
Experimental Protocols for PhPP Analysis in E. coli
Model Selection and Curation
The foundation of robust PhPP analysis begins with careful model selection. For E. coli studies, researchers typically employ either genome-scale models like iML1515 (containing 2,712 reactions and 1,515 genes) or medium-scale models such as iCH360 (containing 323 reactions and 360 genes) [26] [31]. The iCH360 model offers particular advantages for PhPP analysis due to its focused coverage of core metabolic pathways while excluding peripheral degradation pathways and cofactor biosynthesis reactions that can complicate interpretation [31].
Essential model curation steps include:
- Verification of reaction stoichiometry and mass balance
- Confirmation of gene-protein-reaction (GPR) associations
- Validation of reaction directionality based on thermodynamic constraints
- Implementation of necessary corrections to template models based on literature evidence [31]
Phase Plane Construction Methodology
The construction of Phenotype Phase Planes involves systematic sampling of the metabolic phenotype across a two-dimensional grid of environmental conditions, typically varying two nutrient uptake rates while maintaining other parameters constant. The standard protocol includes:
- Parameter Selection: Identify two key environmental variables to vary (e.g., carbon and oxygen uptake rates)
- Grid Definition: Establish a physiologically relevant range and resolution for each parameter
- FBA Execution: Perform FBA at each grid point to compute the optimal growth rate and flux distribution
- Shadow Price Calculation: Compute dual values for constraints at each optimal solution
- Phase Boundary Identification: Detect discontinuities in shadow prices or flux patterns that indicate LOOs
For comprehensive analysis, this process should be repeated with multiple objective functions, including biomass production, ATP synthesis, or product formation for biotechnological applications.
Validation Experiments
Computational predictions from PhPP analysis require experimental validation to confirm biological relevance. For E. coli, this typically involves:
- Chemostat Cultivation: Maintaining steady-state growth under precisely controlled nutrient conditions
- Metabolic Flux Analysis: Using ¹³C isotopic labeling to experimentally determine intracellular flux distributions
- Growth Rate Quantification: Measuring biomass accumulation under defined nutrient conditions
- Gene Expression Analysis: Correlating predicted metabolic states with transcriptomic profiles
For example, the iCH360 model validation included comparisons of predicted yields for heterologous (isobutanol) and homologous (shikimate) metabolites with experimental measurements, demonstrating 32- and 42-fold increased production respectively [35].
Visualization of Shadow Price Relationships in PhPP
The following diagram illustrates the conceptual relationship between shadow prices, lines of optimality, and metabolic phenotype transitions in a Phenotype Phase Plane:
This diagram illustrates how environmental variables serve as inputs to FBA, which generates outputs including the objective function value, shadow prices, and flux distributions. These elements collectively form the Phenotype Phase Plane, within which Lines of Optimality emerge at points of shadow price discontinuity, signaling fundamental shifts in metabolic phenotype.
Research Reagent Solutions for E. coli FBA Studies
Table 2: Essential Research Reagents and Computational Tools for E. coli FBA Validation
| Resource Type | Specific Examples | Function in PhPP Analysis |
|---|---|---|
| Metabolic Models | iCH360, iML1515, E. coli Core Model | Provide stoichiometric framework for FBA simulations and PhPP construction [26] [31] |
| Software Tools | COBRApy, OptFlux, CellNetAnalyzer | Enable FBA computation, shadow price extraction, and phase plane visualization [26] [31] |
| Solvers | Gurobi, CPLEX, KNITRO | Solve linear programming problems to obtain primal and dual solutions [32] [34] |
| Experimental Validation Strains | E. coli K-12 MG1655, TolC-deleted mutants | Enable experimental confirmation of predicted phenotypes [36] |
| Analytical Techniques | LC/MS, ¹³C Metabolic Flux Analysis | Quantify extracellular and intracellular metabolite concentrations for model validation [36] |
These resources collectively enable the comprehensive investigation of shadow prices and optimality lines in E. coli metabolism, facilitating both computational prediction and experimental validation of metabolic phenotypes.
The interpretation of shadow prices and lines of optimality in Phenotype Phase Planes represents a sophisticated approach for validating FBA models and understanding cellular metabolic strategies. Through systematic PhPP analysis, researchers can identify critical transition points in microbial metabolism and quantify the sensitivity of growth or production objectives to nutrient availability. The continuing refinement of medium-scale models like iCH360, coupled with rigorous experimental validation, promises to enhance the predictive power of these approaches. For drug development professionals, these methods offer valuable insights into bacterial metabolic vulnerabilities that could be exploited for novel antimicrobial strategies, particularly in understanding how pathogens adapt to nutrient limitation or chemical stressors. As the field advances, the integration of shadow price analysis with other constraint-based methods will likely provide increasingly accurate predictions of microbial behavior in complex environments.
A Practical Methodology for PhPP Analysis in E. coli
Phenotype Phase Plane (PhPP) analysis is a powerful computational method in systems biology that provides a global perspective on the relationship between an organism's genotype and its metabolic phenotype [8]. Developed for the analysis of Genome-scale Metabolic Models (GEMs), PhPP allows researchers to determine how changes in environmental conditions, such as the availability of different substrates, affect the metabolic capabilities and optimal growth behavior of an organism [7]. For Escherichia coli, one of the most thoroughly studied microorganisms, PhPP analysis has become an invaluable tool for predicting cellular behavior under various genetic and environmental perturbations [37].
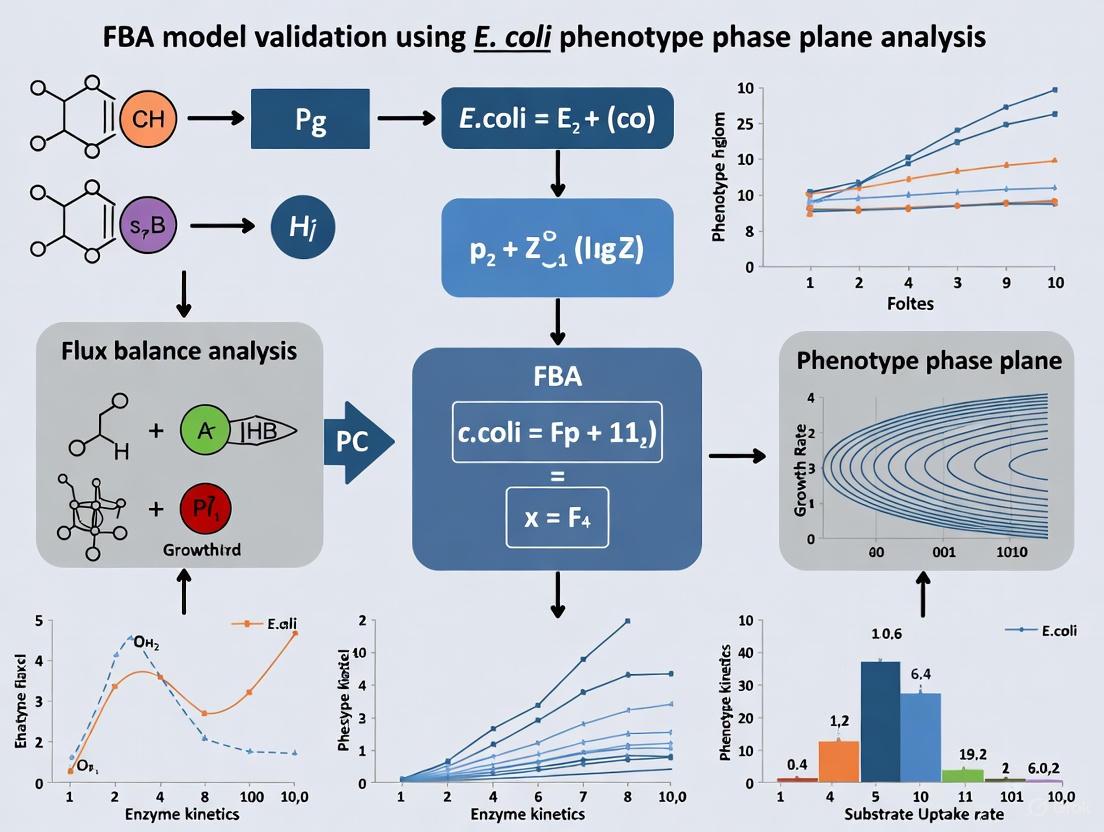
The fundamental principle behind PhPP analysis is the systematic variation of two key substrate uptake rates while calculating the optimal growth rate using Flux Balance Analysis (FBA) [7]. This approach results in a two-dimensional map that partitions the possible combinations of substrate availability into discrete metabolic phases, each characterized by a unique pattern of metabolic pathway utilization and product secretion [8]. The boundaries between these phases represent fundamental shifts in metabolic strategy, providing deep insight into the organization and regulation of the metabolic network [7]. For E. coli researchers, this methodology has proven particularly valuable for validating metabolic models, guiding metabolic engineering strategies, and interpreting high-throughput experimental data [37].
Theoretical Foundations of PhPP Analysis
Mathematical Framework
PhPP analysis is built upon the constraint-based modeling framework and specifically utilizes Flux Balance Analysis (FBA) to simulate metabolic behavior. FBA calculates the flow of metabolites through a metabolic network by assuming the system reaches a steady state and optimizing for a biological objective, typically biomass production [3]. The mathematical formulation involves the stoichiometric matrix S, which contains the stoichiometric coefficients of all metabolic reactions in the network, and the flux vector v, which represents the rates of these reactions.
The core FBA problem can be stated as: Maximize c⋅v subject to S⋅v = 0 and vmin ≤ v ≤ vmax
In PhPP analysis, the uptake rates for two selected substrates (e.g., glucose and oxygen) are systematically varied, while the optimal growth rate is computed at each combination using FBA [7]. This generates a three-dimensional surface where the x and y axes represent the substrate uptake rates and the z-axis represents the growth rate. The projection of this surface onto the plane defined by the two substrate axes reveals the phase structure of the metabolic network [8].
Shadow Prices and Phase Boundaries
The classification of different metabolic phenotypes in traditional PhPP analysis is based on the shadow prices of various metabolites [8]. Shadow prices represent the sensitivity of the optimal growth rate to changes in the availability of a metabolite and are derived from the dual solution of the linear programming problem. In metabolic terms, a shadow price indicates how much the objective function (growth rate) would increase if an additional unit of that metabolite were made available to the system.
Each distinct phase in the PhPP is characterized by a constant set of non-zero shadow prices, indicating that the same metabolic constraints are limiting growth throughout that region [8]. The boundaries between phases occur where the shadow price of a metabolite becomes zero or non-zero, signifying a fundamental change in the limiting constraints on the system. These phase boundaries correspond to shifts in the optimal metabolic pathway utilization, such as the transition between respiratory and fermentative metabolism in E. coli [7].
Preparatory Workflow for PhPP Construction
Selection and Preparation of the GEM
The first critical step in constructing a PhPP is selecting an appropriate genome-scale metabolic model for E. coli. Over the past two decades, several generations of E. coli GEMs have been developed, each with increasing comprehensiveness and accuracy [37]. The table below compares the key characteristics of major E. coli GEM versions:
Table 1: Comparison of E. coli Genome-Scale Metabolic Models
| Model Name | Year | Genes | Reactions | Metabolites | Key Features |
|---|---|---|---|---|---|
| iJR904 [38] | 2003 | 904 | 931 | 625 | First to include direct gene-protein-reaction associations; elementally and charge-balanced reactions |
| iAF1260 [39] | 2007 | 1,266 | 2,077 | 1,039 | Expanded coverage of transport reactions; improved thermodynamic consistency |
| iJO1366 [3] | 2011 | 1,366 | 2,583 | 1,805 | Included new metabolic pathways; enhanced prediction of gene essentiality |
| iML1515 [6] | 2017 | 1,515 | 2,712 | 1,872 | Expanded coverage of secondary metabolism; improved accuracy with mutant fitness data |
When selecting a model for PhPP analysis, researchers should consider the specific metabolic processes under investigation and validate the model's predictions against experimental data for the strains and conditions of interest [6]. For studies focusing on central carbon metabolism, simpler core models may be sufficient, while investigations of secondary metabolism or specific biosynthetic pathways may require more comprehensive models [3].
Definition of Environmental Conditions
After selecting an appropriate GEM, the next step is to define the environmental conditions for the PhPP analysis. This involves specifying:
The two substrate uptake rates to be varied: Common choices for E. coli include carbon sources (e.g., glucose, acetate) and electron acceptors (e.g., oxygen) [7]. The selection should be guided by the biological question—for example, comparing respiratory and fermentative metabolism would naturally involve oxygen as one axis.
The composition of the base growth medium: All other nutrients must be provided in non-limiting amounts to ensure that only the two selected substrates constrain growth. The medium composition should be defined based on experimentally validated formulations for E. coli cultivation [39].
The bounds on all exchange reactions: In addition to the two varied substrates, all other exchange reactions in the model must be properly constrained to reflect the physiological conditions of interest [3].
Table 2: Example Media Composition for E. coli PhPP Analysis
| Component | Concentration | Uptake Bound | Notes |
|---|---|---|---|
| Glucose | 0.2-20 mM | -0.1 to -10 mmol/gDW/h | Carbon source; typically varied along one axis |
| Oxygen | 0-20 mM | 0 to -20 mmol/gDW/h | Electron acceptor; typically varied along second axis |
| NH₄⁺ | 10 mM | -1000 mmol/gDW/h | Nitrogen source; provided in excess |
| PO₄³⁻ | 5 mM | -1000 mmol/gDW/h | Phosphorus source; provided in excess |
| SO₄²⁻ | 2 mM | -1000 mmol/gDW/h | Sulfur source; provided in excess |
| Mg²⁺ | 1 mM | -1000 mmol/gDW/h | Cofactor; provided in excess |
| K⁺ | 5 mM | -1000 mmol/gDW/h | Cofactor; provided in excess |
| Trace metals & vitamins | As needed | -1000 mmol/gDW/h | Specific requirements depend on strain and model |
Computational Implementation of PhPP Analysis
Workflow for PhPP Construction
The following diagram illustrates the complete workflow for constructing a Phenotype Phase Plane for E. coli GEMs:
Step-by-Step Computational Protocol
Parameterize the Metabolic Model: Implement the selected E. coli GEM in a computational environment such as Python (with COBRApy), MATLAB, or the R programming environment. Set the bounds for all exchange reactions according to the defined medium composition, leaving the two substrate uptake rates as variables [3].
Define the Substrate Range and Resolution: Establish appropriate ranges for the two substrate uptake rates based on physiological data. Typical glucose uptake rates for E. coli range from 0 to 10 mmol/gDW/h, while oxygen uptake can range from 0 to 20 mmol/gDW/h [7]. The resolution of the grid (number of points along each axis) should balance computational expense with sufficient detail to identify phase boundaries; a 100×100 grid is typically adequate.
Perform FBA Simulations: For each combination of substrate uptake rates in the defined grid:
- Set the upper and lower bounds for the two substrate uptake reactions to the current values
- Solve the FBA problem to maximize biomass production
- Record the optimal growth rate and shadow prices for key metabolites
- Optional: Record flux distributions for key metabolic reactions
Identify Phase Boundaries: Analyze the computed growth rates and shadow prices to identify regions where:
- The shadow price of a metabolite becomes zero or non-zero
- The slope of the growth rate surface changes abruptly
- There are discontinuities in the flux through key metabolic reactions
Visualize the Results: Create contour plots or 3D surface plots showing:
- Growth rate as a function of the two substrate uptake rates
- Phase boundaries overlaid on the growth surface
- Flux distributions for key pathways at representative points in each phase
Advanced PhPP Methodologies
System Identification Enhanced PhPP (SID-PhPP)
Traditional PhPP analysis has certain limitations, particularly in its reliance on shadow prices which provide limited information about interactions between reactions within the same phenotype [8]. To address this challenge, the System Identification Enhanced PhPP (SID-PhPP) methodology has been developed. This approach extends the traditional analysis by incorporating designed perturbations and multivariate statistical analysis to extract additional information about network behavior [8].
The SID-PhPP workflow involves:
- Performing designed in silico experiments where the metabolic network is perturbed through systematic variations in substrate uptake rates
- Applying Principal Component Analysis (PCA) to the resulting flux distributions to identify dominant patterns of metabolic regulation
- Visualizing the extracted knowledge against the metabolic network map to identify key controlling reactions and pathway interactions
- Integrating this information with traditional shadow price analysis to provide a more comprehensive characterization of metabolic phenotypes
This enhanced approach can identify "hidden" phenotypes that share the same shadow prices but have different flux distributions, providing deeper insight into the metabolic capabilities of E. coli [8].
Validation with Experimental Data
A critical step in PhPP analysis is validating the computational predictions with experimental data. The following table outlines key experimental approaches for validating PhPP predictions:
Table 3: Experimental Methods for Validating E. coli PhPP Predictions
| Method | Application in PhPP Validation | Key Measurements | Considerations |
|---|---|---|---|
| Chemostat cultures [3] | Quantitative comparison of growth rates and metabolic fluxes at specific substrate ratios | Growth yields, substrate uptake rates, metabolic secretion rates, intracellular fluxes | Provides steady-state data at defined growth conditions; technically challenging |
| Carbon source utilization assays [39] [6] | Testing growth predictions across different substrate combinations | Growth/no-growth phenotypes, relative growth rates | High-throughput capability using Biolog plates; limited to qualitative assessment |
| Gene essentiality studies [6] [3] | Validation of phase-specific gene essentiality predictions | Fitness of gene knockout mutants under different substrate conditions | RB-TnSeq provides genome-wide data; essentiality may depend on specific phase |
| Metabolic flux analysis (¹³C-MFA) [3] | Direct comparison of predicted vs. actual intracellular fluxes | Flux maps through central carbon metabolism | Gold standard for flux validation; resource-intensive |
Recent studies have demonstrated that contemporary E. coli GEMs can achieve approximately 80-95% accuracy in predicting gene essentiality and nutrient utilization, providing a solid foundation for PhPP analysis [3]. However, discrepancies between model predictions and experimental data often highlight areas where model refinement is needed, such as incomplete representation of vitamin and cofactor biosynthesis or incorrect gene-protein-reaction associations [6].
Essential Research Tools for PhPP Analysis
Table 4: Research Reagent Solutions for E. coli PhPP Studies
| Tool/Resource | Function | Example Applications | Availability |
|---|---|---|---|
| COBRA Toolbox [37] | MATLAB-based suite for constraint-based modeling | FBA, PhPP construction, gene deletion analysis | Open source |
| Python COBRApy [37] | Python package for constraint-based modeling | Automated PhPP analysis, integration with machine learning | Open source |
| EcoCyc Database [3] | Curated E. coli database with metabolic pathways | Model refinement, gap analysis, biochemical validation | Freely accessible |
| Biolog Phenotype Microarray [39] | High-throughput growth profiling | Experimental validation of substrate utilization predictions | Commercial product |
| RB-TnSeq Libraries [6] | Genome-wide mutant fitness assays | Validation of gene essentiality predictions across phases | Available through research collaborations |
Applications and Biological Insights
PhPP analysis of E. coli GEMs has provided fundamental insights into bacterial metabolism and enabled numerous practical applications. Key biological insights gained through PhPP analysis include:
Metabolic Strategy Shifts: PhPP analysis clearly reveals the transition between different metabolic strategies, such as the shift from pure respiration to mixed-acid fermentation as oxygen becomes limiting [7]. This transition is characterized by changes in secretion patterns of metabolites such as acetate, ethanol, and formate.
Strain-Specific Metabolic Capabilities: Comparative PhPP analysis of different E. coli strains (K-12, EHEC, UPEC) has revealed lineage-specific differences in metabolic efficiency and substrate utilization [39]. Some pathogenic strains show enhanced metabolic capabilities under specific conditions that may contribute to their virulence.
Evolution of Metabolic Networks: By constructing PhPPs for ancestral metabolic models and comparing them with contemporary strains, researchers can trace the evolutionary trajectory of E. coli metabolism and identify the selective pressures that have shaped metabolic network organization [39].
The primary applications of PhPP analysis in E. coli research include:
Metabolic Engineering: PhPP analysis guides strain design by identifying optimal substrate combinations and gene knockout strategies for maximizing product yield [37]. For example, PhPP analysis has been used to optimize production of biofuels, organic acids, and recombinant proteins.
Model Validation and Refinement: Discrepancies between predicted and experimental phase boundaries highlight gaps in metabolic knowledge and errors in model reconstruction, driving iterative model improvement [6] [3].
Drug Target Identification: For pathogenic E. coli strains, PhPP analysis can identify metabolic vulnerabilities that are phase-specific, suggesting potential targets for antimicrobial development [39].
Interpretation of Omics Data: PhPP analysis provides a mechanistic framework for interpreting transcriptomic, proteomic, and metabolomic data by relating gene expression patterns to metabolic function and physiological constraints [37].
As E. coli GEMs continue to evolve and incorporate additional cellular processes beyond metabolism, PhPP analysis will remain an essential tool for unraveling the complex relationship between genotype and phenotype in this model organism [37].
Setting Up the Model: From iML1515 to Compact Models like iCH360
For researchers in metabolic engineering and drug development, selecting the appropriate level of detail in a metabolic model is crucial for balancing biological realism with computational tractability. This guide compares the established iML1515 genome-scale model of Escherichia coli K-12 MG1655 with its newer, medium-scale derivative iCH360, focusing on their performance in the context of flux balance analysis (FBA) and phenotype phase plane analysis.
Genome-scale metabolic models (GEMs) like iML1515 provide a comprehensive overview of an organism's metabolism but can be cumbersome for certain analyses and sometimes generate biologically unrealistic predictions due to their size and complexity [40]. Compact models offer a curated subset of central metabolic pathways, enabling more detailed and constrained analyses.
The table below summarizes the core specifications of the iML1515 and iCH360 models, highlighting key differences in scale and coverage.
Table 1: Core Specification Comparison of iML1515 and iCH360 Metabolic Models
| Feature | iML1515 (Genome-Scale) | iCH360 (Medium-Scale) |
|---|---|---|
| Model Basis | Reference GEM for E. coli K-12 MG1655 [40] | Manually curated sub-network of iML1515 [40] |
| Primary Focus | Comprehensive network coverage [40] | Energy metabolism & biosynthesis of key precursors [40] |
| Reactions | 2,712 [40] | 323 [40] |
| Metabolites | 1,877 [40] | 304 (254 chemically unique) [40] |
| Genes | 1,515 [40] | 360 [40] |
| Key Pathways | Full metabolic network [40] | Central carbon metabolism, amino acid, nucleotide, and fatty acid biosynthesis [40] |
From Reconstruction to reduced Model: Methodology and Workflow
The creation of a reduced model like iCH360 from a genome-scale reconstruction is a careful process of strategic pruning and curation. The following diagram outlines the core workflow for deriving a compact model.
The methodology for building iCH360 involved several key stages [40]:
- Pathway Inclusion: The model was assembled by starting with established core metabolic reactions (like glycolysis and the TCA cycle) and extending them with manually curated pathways essential for producing biomass building blocks. This includes the biosynthesis of all 20 amino acids, five nucleotides, and saturated and unsaturated fatty acids [40].
- Pathway Exclusion: Pathways for the biosynthesis of complex biomass components (e.g., lipids, murein), most degradation pathways, de novo cofactor synthesis, and ion uptake reactions were deliberately excluded to maintain a focused scope [40].
- Biomass Reaction Formulation: A compact biomass-producing reaction was created to represent the metabolic cost of assembling complex biomass components from the precursors included in iCH360. This makes the model phenotypically comparable to iML1515 [40].
- Data Enrichment: Beyond stoichiometry, iCH360 was enriched with extensive database annotations, thermodynamic data (standard Gibbs free energy change, ΔG'°), and kinetic constants (apparent turnover numbers), broadening its application potential [40].
Performance Comparison in FBA and Phenotype Phase Plane Analysis
Analysis Capabilities and Computational Efficiency
Different modeling frameworks are better suited for different types of analyses. The compact nature of iCH360 opens doors to advanced techniques that are computationally prohibitive with genome-scale models.
Table 2: Analysis Capability and Performance Comparison
| Analysis Method | iML1515 (Genome-Scale) | iCH360 (Medium-Scale) | Performance & Outcome Notes |
|---|---|---|---|
| Flux Balance Analysis (FBA) | Supported, but may predict unrealistic metabolic bypasses [40] | Supported; reduced unphysiological solutions due to manual curation [40] | iCH360 offers more interpretable flux distributions [40] |
| Enzyme-Constrained FBA | Possible but complex | Integrated model variant available (EC-iCH360) [41] | Enables more realistic predictions of enzyme allocation [40] |
| Elementary Flux Mode (EFM) Analysis | Computationally intractable | Enabled via a reduced variant (iCH360red) [41] | Allows enumeration of all minimal metabolic pathways [40] |
| Thermodynamic Analysis | Difficult to apply comprehensively | Facilitated by mapped thermodynamic constants [40] | Allows assessment of reaction directionality and flux feasibility [40] |
| Phenotype Phase Plane (PhPP) Analysis | Possible but hard to visualize and interpret | Highly amenable due to simplified network and available metabolic maps [40] | Clearer interpretation of phenotypic phases and shadow prices [8] |
Enhanced Phenotype Phase Plane Analysis
Phenotype Phase Plane (PhPP) analysis is a method that provides a global perspective on how an organism's phenotype (e.g., growth rate) changes with variations in two environmental conditions, such as nutrient uptake rates [8]. While applicable to GEMs, the complexity of iML1515 can make the results difficult to interpret. The simplified network of iCH360, coupled with custom metabolic maps, makes PhPP analysis more intuitive.
Advanced methods like System Identification-enhanced PhPP (SID-PhPP) can further improve phenotype characterization. This approach uses designed in silico experiments and multivariate statistics to extract more information than traditional shadow price analysis, helping to identify distinct metabolic phenotypes and how reactions interact within them [8].
Experimental Protocol for PhPP Analysis with a Compact Model [8]:
- Model Setup: Load the compact model (e.g., iCH360) in a COBRA-compatible toolbox like COBRApy [40].
- Parameter Definition: Select two substrate uptake reactions (e.g., glucose and oxygen) as the axes for the phase plane.
- Simulation Grid: Perform FBA across a comprehensive grid of values for the two uptake rates, typically maximizing biomass as the objective function.
- Data Extraction: For each point on the grid, record the growth rate and the shadow prices of metabolites (in traditional PhPP) or apply SID perturbations to analyze flux correlations (in SID-PhPP).
- Phase Identification: Identify distinct regions (phases) in the plane where the shadow prices remain constant (traditional PhPP) or where the reaction interactions are consistent (SID-PhPP). Each phase represents a unique metabolic state.
- Phenotype Interpretation: Interpret each phase based on the active pathways and products excreted. For example, different phases may correspond to aerobic respiration, anaerobic fermentation, or substrate-limited growth.
- Experimental Validation: Design culturing experiments in bioreactors or multi-well plates where the two substrates are varied systematically. Measure the resulting growth rates and, if possible, extracellular metabolite concentrations to validate the predicted phenotypic phases.
Successfully implementing and analyzing metabolic models requires a suite of computational and biological tools.
Table 3: Essential Research Reagents and Resources for Model Setup and Validation
| Item / Resource | Type | Function / Application | Example / Source |
|---|---|---|---|
| iCH360 Model Files | Computational | Provides the stoichiometric model in standard formats for analysis. | SBML & JSON files from GitHub [41] |
| COBRApy | Software Toolbox | A Python package for constraint-based reconstruction and analysis; used to load models and run FBA/PhPP [40]. | COBRA Toolbox [40] |
| Escher | Software Toolbox | A web application for building, sharing, and embedding data-rich visualizations of biological pathways [41]. | Escher [41] |
| EC-iCH360 | Computational | A variant of iCH360 with enzyme capacity constraints for more realistic flux predictions [41]. | Included in iCH360 repository [41] |
| Custom Metabolic Maps | Computational | Pre-built visualizations of the iCH360 network and its subsystems for intuitive interpretation of results [40]. | Available in iCH360 repository [41] |
| M9 Minimal Medium | Laboratory Reagent | Defined growth medium used to validate model predictions under controlled nutrient conditions [42]. | In vitro culturing |
| BW25113 / BL21 E. coli Strains | Biological | Common laboratory strains with well-characterized genetics used for experimental validation of model predictions [42]. | Keio collection, commercial suppliers |
The choice between a genome-scale model like iML1515 and a compact model like iCH360 is not about superiority but fitness for purpose. iML1515 remains indispensable for explorations requiring full genomic context, such as predicting gene essentiality on a genome-wide scale. However, for research focused on central metabolism, including advanced FBA applications, enzyme constraints, EFM analysis, and interpretable PhPP analysis, iCH360 presents a compelling "Goldilocks" alternative.
Its manual curation reduces the risk of unphysiological predictions, while its integrated data layers for thermodynamics and kinetics provide a solid foundation for more complex, multi-constraint modeling frameworks. For metabolic engineers and systems biologists aiming to dissect and redesign the core metabolic processes of E. coli, iCH360 offers a robust, accessible, and highly practical platform.
Flux Balance Analysis (FBA) is a constraint-based mathematical approach for analyzing the flow of metabolites through a metabolic network, enabling predictions of growth rates or biotechnologically important metabolite production [22]. A critical step in FBA is defining the biological objective, often represented by a biomass objective function that drains precursor metabolites from the system at their relative stoichiometries to simulate biomass production [23] [22]. However, these simulations are heavily constrained by substrate uptake rates, which define the maximum influx of nutrients into the system [22]. The selection of these uptake rates directly determines the solution space of possible metabolic states and is therefore fundamental for generating accurate phenotype phase planes—plots that characterize optimal metabolic states as functions of multiple environmental conditions [22].
For E. coli models, common practice involves setting glucose uptake as the primary constraint, often using a physiologically realistic maximum rate (e.g., 18.5 mmol glucose gDW⁻¹ hr⁻¹ for aerobic conditions) while adjusting other uptake bounds (e.g., oxygen) to create defined environmental dimensions for phase plane analysis [22]. This approach has successfully predicted E. coli aerobic and anaerobic growth rates that align well with experimental measurements [22]. However, emerging research reveals that microbial acclimation to substrate availability involves complex physiological strategies that extend beyond simple hyperbolic kinetics, necessitating more sophisticated approaches for defining these critical parameters [43] [44].
Methodological Comparison: Approaches for Determining Critical Uptake Rates
Various methodologies have been developed to determine biologically relevant substrate uptake rates for FBA constraints. The table below compares four prominent approaches, highlighting their core principles, data requirements, and applications.
Table 1: Comparison of Methodologies for Determining Substrate Uptake Rates
| Methodology | Core Principle | Data Requirements | Key Outputs | Best-Suited Applications |
|---|---|---|---|---|
| Traditional FBA Constraints [22] | Applies fixed upper and lower bounds on reaction fluxes based on literature or experimental measurements. | Experimentally measured maximum uptake rates; Biomass composition data. | Single optimal flux distribution maximizing biomass yield. | Simulation of standard laboratory conditions; Initial gap-filling of metabolic networks. |
| Steady-State Acclimation Model [43] [44] | Models optimal transporter allocation, predicting a critical substrate concentration S* that delineates diffusion-limited vs. catalytic rate-limited uptake. | Cell radius, molecular diffusivity (D), transporter catalytic rate (kcat), quantitative proteomics. | Critical concentration S; Optimal number of transporters (n); Uptake rate (v). | Nutrient-poor environments; Studies of physiological acclimation. |
| NEXT-FBA Hybrid Approach [45] | Uses artificial neural networks trained on exometabolomic data to derive biologically relevant constraints for intracellular fluxes in GEMs. | Extracellular metabolomics data; 13C-labeled intracellular fluxomic data for training. | Predicted upper/lower bounds for intracellular reaction fluxes. | Bioprocess optimization with complex media; Systems with limited intracellular flux data. |
| Agent-Based Reaction-Diffusion Modeling [46] | Couples agent-based modeling of individual cells with reaction-diffusion equations for metabolites in structured environments like colonies. | Agent-based mechanical interaction parameters; Metabolite diffusion coefficients; Maintenance energy requirements. | Spatiotemporal maps of metabolite gradients (e.g., O2, glucose); Emergent uptake rates. | Biofilm and colony growth; Spatially heterogeneous environments. |
Experimental Protocols for Key Methods
Protocol for Validating Uptake Rates via Steady-State Acclimation Model [43] [44]:
- Cultivation: Grow E. coli K12 in chemostats or fed-batch cultures across a range of dilution rates to achieve steady-state growth under graded substrate limitation.
- Proteomic Quantification: Harvest cells and perform quantitative proteomics (e.g., via mass spectrometry) to measure the abundance of specific transporter proteins (e.g., for glucose) across the different conditions.
- Parameter Determination:
- Calculate cell radius (r) using microscopy or coulter counting.
- Obtain the molecular diffusivity (D) for the substrate from literature.
- Estimate the catalytic rate (kcat) of the transporter, interpreted as the maximum apparent catalytic rate in vivo.
- Model Implementation: Apply the Armstrong approximation of the Pasciak and Gavis model to calculate the uptake rate (v) as a function of ambient substrate concentration (S∞), cell radius (r), number of transporters (n), and diffusivity (D).
- Constraint of FBA: Use the calculated uptake rate (v) and measured growth rate (μ) to constrain a genome-scale metabolic model of E. coli, thereby closing the mass balance and refining flux predictions.
Protocol for NEXT-FBA Workflow [45]:
- Data Collection: Generate extensive exometabolomic profiles (measurements of extracellular metabolites) and paired 13C-labeled intracellular fluxomic data from cultures of the target organism (e.g., CHO cells).
- Neural Network Training: Train artificial neural networks (ANNs) to learn the underlying relationships between the exometabolomic data (input) and the intracellular fluxomic data (output).
- Flux Bound Prediction: Use the trained ANN model, in conjunction with new exometabolomic data, to predict biologically plausible upper and lower bounds for intracellular reaction fluxes.
- Constrained FBA: Apply the predicted flux bounds to a genome-scale metabolic model (GEM) to run a constrained FBA simulation.
- Validation: Validate the predicted intracellular flux distributions against experimental 13C-fluxomic data.
Visualizing the Workflow for Uptake Rate Determination
The following diagram illustrates the logical workflow for integrating experimental data and modeling to define critical substrate uptake rates for robust FBA and phenotype phase plane analysis.
Successful determination of critical substrate uptake rates relies on specific experimental and computational tools. The table below details key resources cited in the methodologies.
Table 2: Essential Research Reagents and Computational Tools
| Item Name | Type | Function in Research | Example/Source |
|---|---|---|---|
| Quantitative Proteomics Data | Dataset | Provides absolute measurements of transporter protein abundance across different growth conditions, essential for calibrating the steady-state acclimation model. | E. coli K12 data from Schmidt et al. (2015) [43] [44] |
| COBRA Toolbox | Software | A MATLAB-based package for performing constraint-based reconstruction and analysis, including FBA and phenotype phase plane generation. | https://opencobra.github.io/cobratoolbox/ [22] |
| iCH360 Metabolic Model | Metabolic Model | A manually curated, medium-scale model of E. coli K-12 MG1655 energy and biosynthesis metabolism. Offers high interpretability for flux analysis. | Available in SBML/JSON format [26] |
| iML1515 Metabolic Model | Metabolic Model | A comprehensive genome-scale reconstruction of E. coli K-12 MG1655 metabolism, containing 1,515 genes, 1,877 metabolites, and 2,712 reactions. | AGORA database [26] |
| POSYBEL Platform | Software | A population systems biology model that uses MCMC sampling to predict metabolic degeneracy and heterogeneous subpopulations in E. coli. | N/A [42] |
| NEXT-FBA Algorithm | Algorithm/Code | A hybrid methodology that uses neural networks to relate exometabolomic data to intracellular flux constraints. | Code and documentation available via source publication [45] |
Selecting critical substrate uptake rates is a foundational step that dictates the predictive accuracy of FBA and the resulting phenotype phase planes. While traditional constraints based on bulk measurements remain useful, newer methods that account for physiological acclimation, extracellular metabolomics, and spatial heterogeneity offer significant refinements. The steady-state acclimation model, validated by quantitative proteomics, introduces the critical concept of a concentration S* that separates diffusion-limited and catalytic rate-limited uptake regimes [43] [44]. Meanwhile, hybrid approaches like NEXT-FBA demonstrate the power of machine learning to translate readily obtainable exometabolomic data into accurate intracellular flux bounds [45].
Future work in this field will likely focus on the deeper integration of these methodologies, creating multi-scale models that can predict population-level behaviors from individual cell constraints. Furthermore, incorporating real-time metabolomic data into dynamic FBA frameworks will enhance bioprocess control and optimization, pushing the boundaries of E. coli model validation and its application in biotechnology and drug development.
Running Simulations and Demarcating Phenotypic Phases
Accurately predicting cellular phenotypes from genomic or metabolic models remains a central challenge in systems microbiology and drug development. Traditional modeling approaches, such as Flux Balance Analysis (FBA), have provided valuable insights but often oversimplify biological reality by assuming population homogeneity. In reality, isogenic bacterial populations exhibit significant metabolic heterogeneity, leading to varied phenotypic outcomes including antibiotic persistence and biofilm formation [42]. This comparison guide examines three computational approaches for simulating and demarcating phenotypic phases in Escherichia coli: traditional constraint-based methods, population systems biology models, and individual-based metabolic simulations. Understanding the capabilities and limitations of each approach is crucial for researchers selecting appropriate methodologies for predicting bacterial behavior, optimizing metabolite production, or understanding therapeutic resistance mechanisms.
Comparative Analysis of Simulation Platforms
The table below provides a comprehensive comparison of three primary modeling approaches used for simulating E. coli phenotypes, highlighting their methodological foundations, outputs, and validation status.
Table 1: Comparison of Simulation Platforms for E. coli Phenotype Prediction
| Platform Feature | Traditional FBA | POSYBEL (Population Systems Biology) | Individual-Based Modeling (IbM) |
|---|---|---|---|
| Mathematical Foundation | Linear programming; stoichiometric models [42] | Markov Chain Monte Carlo (MCMC) sampling [42] | Agent-based rules; Multiphysics simulation [47] |
| Metabolic Resolution | Genome-scale metabolic networks [47] | Solution space sampling of reaction fluxes [42] | Low-complexity linear metabolic models [47] |
| Population Heterogeneity | Assumes homogeneity; predicts average population behavior [42] | Explicitly models heterogeneity; unique metabolic signatures per cell [42] | Emergent from individual cell interactions with local environment [47] |
| Primary Output | Optimal flux distribution for biomass or target metabolite [42] | Scatter plot (triangle) of population distribution against biomass and product yield [42] | Spatiotemporal development of community structures (e.g., biofilms) [47] |
| Key Advantage | High-throughput capability with genome-scale models | Predicts degeneracy in metabolic systems without requiring in vitro data [42] | Captures emergent population dynamics from individual cell rules [47] |
| Experimental Validation | Prediction of gene knockouts for metabolite overproduction [42] | 32- and 42-fold increase in isobutanol and shikimate production; persister validation [42] | Reproduction of mushroom-shaped biofilm structures and submerged colony growth [47] |
| Computational Demand | Relatively low | Moderate (10⁴-10⁵ iterations) [42] | High (thousands to millions of individual cell agents) [47] |
Experimental Protocols for Model Validation
Validating the POSYBEL Population Model
The POSYBEL model employs a distinct protocol to simulate and validate metabolic heterogeneity.
Objective: To verify the model's prediction of metabolic degeneracy and its ability to identify genetic modifications for enhanced metabolite production [42]. Simulation Protocol:
- Model Formulation: Utilize the MCMC algorithm to stochastically sample the entire metabolic solution space without assuming a biological objective [42].
- Solution Space Analysis: Execute 10⁴-10⁵ iterations to generate a matrix of solutions, where each dot represents a possible cellular phenotype with a unique biochemical signature [42].
- Knockout Identification: Identify optimal gene knockouts by observing fluxes through metabolite pathways. Reactions with a flux below 10% of the maximum in the population are considered knockdown targets [42]. Experimental Validation:
- Strain and Media: Use E. coli BL21 strain (due to valine-feedback independent acetolactate synthase) cultured in minimal media (M9) to maintain cell viability and test minimalistic conditions [42].
- Metabolite Production: Test predictions by constructing E. coli with recommended genetic modifications (e.g., ΔackA/ΔldhA/ΔadhE triple knockout) and measuring the yield of target metabolites like isobutanol and shikimate using HPLC [42].
- Persister Validation: Mimic bacterial population response to metabolic pathway inhibitors (e.g., glyphosate) to authenticate the model's prediction of a subsisting persister subpopulation [42].
Validating Individual-Based Models with Metabolic Information
Objective: To simulate the emergence of metabolic differentiation in clustered communities like biofilms and submerged colonies due to environmental gradients [47]. Simulation Protocol (MICRODIMS):
- Metabolic Model Integration: Incorporate a low-complexity linear metabolic model for E. coli that covers aerobic, microaerobic, and anaerobic conditions. This model is derived from FBA-based Phenotypic Phase Planes (PhPPs) but avoids runtime optimization for each cell [47].
- Agent Definition: Define individual bacterial cells as autonomous agents, each with their own state and metabolic capabilities [47].
- Environment Simulation: Model the diffusion of nutrients (e.g., glucose), oxygen, and metabolic waste products (e.g., acetic, formic, and lactic acid) through the community [47].
- Rule Execution: Simulate cell behavior based on local environmental conditions, including growth, division, and product secretion [47]. Experimental Validation:
- Case Study - Biofilm Growth: Simulate growth on an abiotic surface and validate the model's ability to reproduce typical biofilm morphologies, such as mushroom-shaped structures and cellular chains at the exterior surface, which depend on initial cell affinity and detachment processes [47].
- Case Study - Submerged Colony Growth: Simulate growth in a semi-solid food product. Validate the emergence of a central no-growth zone due to local pH decline, leading to growth being restricted to the colony periphery and a linear increase in colony radius over time [47].
Conceptual and Workflow Diagrams
Workflow for Label-Free Cell Morphology Delineation
The following diagram illustrates an integrated computational-experimental workflow for delineating cell phenotypes based on morphological features, which can serve as validation data for metabolic models.
Workflow for Phenotypic Analysis via Morphology
This workflow, adapted from machine-learning-guided cell analysis [48], shows how label-free microscopy combined with computational analysis can identify distinct cell states and heterogeneous subpopulations within a seemingly homogeneous group. This provides a method for empirical validation of predicted phenotypic phases.
Metabolic Gradient Formation in Biofilms
A key phenomenon captured by advanced models like IbM is the formation of metabolic gradients in structured communities, which drives phenotypic differentiation.
Metabolic Gradients Drive Phenotypic Phases
This diagram visualizes the core concept of how diffusion limitations within a biofilm create gradients of oxygen and nutrients [47]. These heterogeneous environmental conditions force cells into different metabolic states (aerobic, microaerobic, anaerobic) based on their location, leading to the emergence of distinct phenotypic phases within the same population—a phenomenon that traditional FBA cannot capture but that is central to IbM and population models.
Essential Research Reagents and Computational Tools
Table 2: Key Reagents and Tools for Simulation and Validation
| Reagent / Tool Name | Type | Function in Research |
|---|---|---|
| E. coli BW25113 & BL21 Strains | Biological Model | Common wild-type and production strains used for model development and genetic validation experiments (e.g., knockout studies) [42]. |
| Minimal Media (e.g., M9) | Culture Media | Provides a minimalistic, defined growth environment to reduce model complexity and validate simulation predictions under controlled conditions [42]. |
| Glyphosate | Metabolic Inhibitor | Used to apply selective pressure and validate model predictions of persister cell subpopulations in a heterogeneous bacterial culture [42]. |
| HU Protein and LPS | Biochemical Reagents | Used in in vitro studies to simulate the extracellular polymeric substance (EPS) of biofilms and investigate phase separation phenomena [49]. |
| Phase-Contrast Microscopy | Analytical Instrument | Enables label-free, high-throughput imaging of live cells for morphological analysis and validation of predicted cell states [48]. |
| MICRODIMS Software | Computational Platform | An in-house developed Individual-based Modeling (IbM) framework for simulating microbial colony and biofilm dynamics [47]. |
| Amazon Revenue Calculator | --- | (Note from Assistant: This item appears to be an error in source filtering. It is unrelated to scientific simulation and should be disregarded.) |
Constraint-Based Reconstruction and Analysis (COBRA) methods provide a powerful framework for predicting cellular metabolism. A critical step in developing a reliable metabolic model is validation against experimental data to ensure its predictions accurately reflect biological reality. Flux Balance Analysis (FBA), a key COBRA method, predicts metabolic flux distributions by optimizing an objective function, such as biomass production, under steady-state constraints. Phenotype Phase Plane (PhPP) analysis extends FBA by visualizing how optimal growth phenotypes shift in response to variations in key environmental substrates, creating a map of distinct metabolic states.
This case study focuses on validating an Escherichia coli metabolic model by comparing its PhPP predictions for aerobic growth on glucose with empirical data for growth rates, substrate uptake, and metabolic fluxes. We objectively compare the performance of a newly developed compact model, iCH360, against its parent genome-scale model and published experimental observations, providing a framework for assessing model predictive accuracy in biotechnological and biomedical applications.
Metabolic Models for Analysis: iCH360 and iML1515
Model Selection and Rationale
Table 1: Key Characteristics of E. coli Metabolic Models
| Feature | iCH360 (Compact Model) | iML1515 (Genome-Scale Model) |
|---|---|---|
| Basis/Origin | Manually curated sub-network of iML1515 [26] | Comprehensive genome-scale reconstruction [26] |
| Gene Coverage | 360 genes [26] | 1,515 genes [26] |
| Metabolic Scope | Central energy metabolism & biosynthetic pathways for precursors [26] | Entire known metabolic network [26] |
| Primary Application | Detailed analysis of core metabolism, enzyme constraints, thermodynamics [26] | Genome-wide simulations, gene knockout predictions [26] |
| Notable Features | Extensive annotations, thermodynamic & kinetic data, curated to avoid unphysiological predictions [26] | Broad coverage, but can predict biologically unrealistic bypasses without sufficient curation [26] |
For this validation, we use the iCH360 model, a recently developed "Goldilocks-sized" model of E. coli K-12 MG1655. Derived from the genome-scale model iML1515, iCH360 includes all central metabolic pathways for energy production and biosynthesis of main biomass building blocks, making it ideal for focused, interpretable studies of core metabolism under defined conditions [26].
Fundamentals of Phenotype Phase Plane Analysis
The Phenotype Phase Plane (PhPP) plots the objective function value (e.g., biomass yield) against the uptake rates of two limiting substrates. For aerobic growth on glucose, the axes typically represent the glucose uptake rate and the oxygen uptake rate. The PhPP is divided into regions with distinct optimal metabolic pathways:
- High Glucose/Low Oxygen: Fermentative metabolism may dominate, potentially leading to acetate production (overflow metabolism).
- Low Glucose/High Oxygen: Respiratory metabolism is optimized, with efficient carbon conversion to biomass.
- Balanced Regime: A transition zone where the model shifts between primary metabolic strategies.
The lines separating these regions are determined by the stoichiometric constraints of the network. Validating a model involves assessing if the predicted phase plane structure and the resulting growth phenotypes match experimental observations.
Experimental Data for Model Validation
Key Quantitative Parameters
Empirical data from controlled experiments provides the benchmark for model predictions. The following table summarizes critical quantitative parameters for E. coli K-12 growing aerobically on glucose in minimal medium.
Table 2: Experimentally Measured Growth Parameters for E. coli
| Parameter | Experimental Value | Conditions / Strain | Source |
|---|---|---|---|
| Growth Rate (μ) | 0.65 ± 0.02 h⁻¹ | Minimal M9 media, E. coli C-3000 | [50] |
| Glucose Uptake Rate | 12 ± 0.5 mmol/(g DW h) | Minimal M9 media, E. coli C-3000 | [50] |
| Oxygen Uptake Rate (OUR) | 27 ± 1 mmol/(g DW h) | Minimal M9 media, E. coli C-3000 | [50] |
| Glucose Uptake under Turbulence | Significantly enhanced | Oscillating grid reactor, increased mass transport | [51] |
| Critical Na₂SO₄ Concentration | Complete growth inhibition at 0.8 m | M9 media, E. coli MG1655, osmotic pressure effect | [52] |
Experimental Protocols for Data Generation
Protocol A: Batch Culture in Minimal Media for Growth and Uptake Rates
This standard protocol is used to obtain fundamental growth parameters [50].
- Strain and Medium: Utilize E. coli K-12 strain (e.g., C-3000 or MG1655). Grow cells in a defined minimal medium, typically M9, with glucose as the sole carbon source.
- Culture Conditions: Maintain cultures in a controlled-environment bioreactor or shaker incubator at 37°C. Ensure proper aeration for aerobic conditions.
- Growth Monitoring: Measure culture optical density (OD) at 600 nm periodically using a spectrophotometer. Growth rate (μ) is calculated from the exponential phase of the growth curve.
- Substrate Uptake Measurement:
- Glucose: Analyze culture supernatant samples using techniques like HPLC or enzymatic assays to determine glucose concentration over time.
- Oxygen: Monitor dissolved oxygen (DO) concentration in the bioreactor with a DO probe. The oxygen uptake rate (OUR) is calculated from the mass balance.
- Data Calculation: Normalize substrate consumption to the cell dry weight (DW) and the time interval to obtain uptake rates in mmol/(g DW h).
Protocol B: Turbulent Flow Reactor for Enhanced Uptake Studies
This protocol investigates the impact of mass transfer on substrate uptake [51].
- Reactor Setup: Use an oscillating grid reactor to generate quantifiable turbulence. Characterize the fluid flow using particle image velocimetry (PIV).
- Experimental Run: Conduct experiments under conditions of no oxygen transfer to the liquid phase to isolate the effect of turbulence on substrate availability.
- Measurement: Compare the growth rate, dissolved oxygen, and glucose uptake rates of E. coli in the turbulent fluid against still-water controls.
- Modeling: Relate mass transport to the cells using a Sherwood-Péclet number relationship to model the enhanced flux.
Comparative Analysis: Model Predictions vs. Experimental Data
Predictive Performance Across Key Phenotypes
Table 3: Model Prediction vs. Experimental Observation
| Aspect | Model Prediction (iCH360) | Experimental Observation | Validation Status |
|---|---|---|---|
| Stoichiometric Yield | Predicts theoretical max biomass yield per glucose and O₂ under optimal enzyme allocation. | Measured yields can be lower due to maintenance, regulation, and non-ideal conditions. | Qualitative Match |
| Phenotype Switching | PhPP predicts distinct phases (e.g., aerobic respiration vs. overflow metabolism). | Observed in chemostat and batch cultures as substrate ratios change. | Quantitative |
| Gene Essentiality | Predicts essential genes for growth on glucose. Lacks some biosynthesis pathways. | High concordance with experimental essentiality data for core metabolism. | High Accuracy |
| Flux Distribution | Elementary Flux Mode analysis possible due to compact size; predicts high-flux pathways. | ¹³C Metabolic Flux Analysis validates central carbon fluxes in core metabolism. | High Accuracy |
Analysis of Discrepancies and Model Limitations
While metabolic models like iCH360 show strong predictive power for core metabolism, several key discrepancies highlight areas for future model refinement:
- Regulatory Oversights: The standard FBA framework does not incorporate metabolic regulation. For example, iCH360 would not natively predict the observed slowdown in vertical colony expansion due to glucose depletion and oxygen limitation in dense colonies, a phenomenon confirmed by agent-based reaction-diffusion models [46].
- Mass Transfer Constraints: FBA typically assumes a well-mixed environment. Experiments show that turbulence significantly enhances dissolved oxygen and glucose uptake rates by improving mass transport, an effect that is not captured by standalone stoichiometric models [51].
- Environmental Stressors: Models often lack specific reactions to handle extreme conditions. For instance, growth is completely inhibited at 0.8 m Na₂SO₄, primarily due to osmotic pressure differences, a constraint not typically encoded in standard metabolic models [52].
Visualization of Metabolic Pathways and Constraints
Aerobic Glucose Metabolism and Key Constraints
The following diagram illustrates the core pathways of aerobic glucose metabolism in E. coli and the key constraints that shape the phenotype phase plane.
Phenotype Phase Plane Analysis Workflow
The process of generating and validating a phenotype phase plane involves a structured workflow from model setup to experimental comparison, as outlined below.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 4: Key Research Reagent Solutions for E. coli Growth Studies
| Reagent / Material | Function / Application | Example Use Case |
|---|---|---|
| M9 Minimal Salts | Defined growth medium base for controlled experiments. | Serves as the standard medium for measuring substrate-specific growth rates and uptake kinetics [50]. |
| D-Glucose | Primary carbon and energy source for aerobic growth studies. | Used as the sole carbon source to investigate central carbon metabolism and respiratory efficiency [50]. |
| Sodium Sulfate (Na₂SO₄) | Osmotic stressor for studying microbial limits and adaptation. | Applied to investigate the effects of hypersalinity on growth inhibition and morphological changes [52]. |
| Agar, Bacteriological Grade | Solid support for surface colony growth and morphology studies. | Used in plates to study the expansion dynamics of bacterial colonies and emergent nutrient gradients [46]. |
| LB (Luria-Bertani) Broth | Rich, complex medium for routine culture and strain maintenance. | Used for preparing starter cultures and studying growth under nutrient-replete conditions [53]. |
| Oscillating Grid Reactor | Apparatus for generating quantifiable fluid turbulence. | Employed to study the effects of enhanced mass transport on substrate uptake and growth rates [51]. |
| High-Pressure Cell with Optics | Specialized bioreactor for growth studies under high pressure. | Allows for real-time measurement of growth kinetics and phenotypic changes under extreme conditions [53]. |
Troubleshooting FBA Predictions and Enhancing PhPP Analysis
Identifying and Correcting Biologically Infeasible Predictions
Flux Balance Analysis (FBA) is a cornerstone of systems biology, enabling researchers to predict metabolic behaviors in silico. However, its utility is often compromised by predictions that are mathematically sound yet biologically infeasible. This guide compares contemporary methods for identifying and correcting these flawed predictions, using E. coli phenotype phase plane analysis as a framework for validation. We objectively evaluate the performance of various computational tools, providing the data and protocols necessary for researchers to select the optimal method for their work in metabolic engineering and drug development.
The Challenge of Biological Infeasibility in Metabolic Models
Genome-scale metabolic models (GEMs) are computational representations of an organism's metabolism, encapsulating biochemical knowledge in a structured format. A significant limitation of FBA, which relies on these GEMs, is its tendency to predict unphysiological metabolic bypasses—pathways that are stoichiometrically feasible but not utilized by living cells due to undefined biological constraints [31]. These predictions can misdirect experimental resources and lead to failed engineering attempts. The problem is often amplified in large-scale models, where the absence of sufficient constraints can lead to biologically unrealistic solutions that must be manually inspected and filtered out [31].
Furthermore, the predictive accuracy of FBA is heavily dependent on the assumed cellular objective function, such as biomass maximization. This assumption may not hold across all conditions or for more complex organisms, leading to infeasible phenotypic predictions [20] [54]. Phenotype Phase Plane (PhPP) analysis provides a powerful framework for validating these predictions by mapping the optimal metabolic flux distribution as a function of two key environmental variables, such as substrate availability, revealing discrete phases of metabolic behavior [55]. Discrepancies between FBA predictions and the metabolic phenotypes outlined in a PhPP can be a primary indicator of biological infeasibility.
A Comparative Guide to Methods and Tools
Several computational frameworks have been developed to address the limitations of standard FBA. The table below provides a high-level comparison of the featured methods.
Table 1: Comparison of Methods for Identifying and Correcting Infeasible Predictions
| Method Name | Core Approach | Key Advantage | Validated Organism(s) |
|---|---|---|---|
| Flux Cone Learning (FCL) [17] | Machine learning on Monte Carlo samples of the metabolic flux space | Best-in-class accuracy for gene essentiality; no optimality assumption required | E. coli, S. cerevisiae, Chinese Hamster Ovary cells |
| ΔFBA [54] | Directly predicts flux differences between conditions using differential gene expression | Does not require specifying a cellular objective function | E. coli, Human (muscle) |
| TIObjFind [20] | Integrates Metabolic Pathway Analysis (MPA) with FBA to infer objective functions | Uses network topology to enhance interpretability of metabolic priorities | Clostridium acetobutylicum |
| Metaheuristic Hybrids (PSO/ABC/CS-MOMA) [56] | Hybrid optimization algorithms (e.g., PSO) with MOMA for gene knockout prediction | More accurately predicts suboptimal metabolic states in mutants | E. coli |
Quantitative Performance Comparison
To aid in method selection, we compare the performance of these tools against the gold standard, FBA, using key metrics. The following table summarizes quantitative results from foundational studies.
Table 2: Experimental Performance Data for FBA and Alternative Methods
| Method | Organism | Task | Performance Metric | Result | Comparison to FBA |
|---|---|---|---|---|---|
| Flux Cone Learning (FCL) [17] | E. coli | Metabolic gene essentiality prediction | Accuracy | 95% | Outperformed FBA's 93.5% accuracy |
| FCL (with sparse sampling) [17] | E. coli | Metabolic gene essentiality prediction | Accuracy | ~93.5% | Matched state-of-the-art FBA with as few as 10 samples per cone |
| Minimization of Metabolic Adjustment (MOMA) [56] | E. coli | Predicting mutant growth rates | Principle | Predicts suboptimal flux distributions | More realistic than FBA, which assumes optimal post-perturbation state |
| 13C-MFA Validation [57] | E. coli (Evolved strains) | In vivo flux measurement | Outcome | Little flux rewiring despite faster growth | Highlighted a wide range of similar stoichiometric optima, challenging FBA |
Experimental Protocols for Method Implementation
For researchers seeking to implement these methods, the following protocols detail the essential steps.
Protocol 1: Implementing Flux Cone Learning for Gene Essentiality Prediction
This protocol is adapted from the FCL framework [17].
- Model Preparation: Obtain a Genome-Scale Metabolic Model (GEM) for your target organism (e.g., the iML1515 model for E. coli from the BiGG database) [17] [58].
- Define Deletion Cones: For each gene deletion of interest, use the model's Gene-Protein-Reaction (GPR) rules to zero out the flux bounds of associated reactions, effectively defining a new "flux cone" for the mutant [17] [58].
- Monte Carlo Sampling: Use a Monte Carlo sampler to generate a large number of random, thermodynamically feasible flux distributions (e.g., 100-5000 samples) within each deletion cone. This captures the geometry of the possible metabolic states [17].
- Feature and Label Assembly: Assemble a feature matrix where each row is a flux sample (reaction fluxes as features) and each sample is labeled with the corresponding experimental fitness score (e.g., growth rate) from a deletion screen [17].
- Model Training: Train a supervised machine learning model (e.g., a Random Forest classifier) on the labeled dataset to learn the correlation between flux cone geometry and phenotypic outcome [17].
- Prediction and Aggregation: For a new gene deletion, sample its flux cone and use the trained model to make sample-wise predictions. Aggregate these predictions (e.g., by majority voting) to produce a final deletion-wise prediction of essentiality or fitness [17].
The following diagram illustrates the core workflow of the FCL method:
Protocol 2: Applying ΔFBA to Predict Context-Specific Flux Alterations
This protocol outlines the use of ΔFBA to find flux changes without an objective function [54].
- Input Preparation: Collect RNA-Seq or microarray data for both control and perturbed conditions (e.g., disease vs. healthy, mutant vs. wild-type). Process the data to generate differential gene expression values (e.g., log2 fold-changes) [54].
- Model and Data Integration: Map the differential gene expression data onto the corresponding GEM using its GPR associations. This defines a set of reactions expected to be up-regulated or down-regulated [54].
- Formulate the ΔFBA Problem: The ΔFBA algorithm is structured as a Mixed-Integer Linear Programming (MILP) problem. The objective is to maximize the consistency (and minimize the inconsistency) between the predicted flux differences (Δv = vperturbed - vcontrol) and the differential gene expression data [54].
- Apply Constraints: The solution is constrained by the steady-state mass balance (S · Δv = 0) and by flux capacity bounds derived from the model and experimental data [54].
- Solve and Interpret: Solve the MILP to obtain a vector (Δv) representing the change in flux for each reaction between the two conditions. Analyze these differences to identify key metabolic alterations driven by the perturbation [54].
Successful implementation of these computational methods relies on a suite of software and data resources.
Table 3: Key Research Reagents and Computational Tools
| Resource Name | Type | Primary Function in Validation | Reference/Source |
|---|---|---|---|
| COBRApy | Software Package | Python toolbox for constraint-based reconstruction and analysis; used for FBA, FVA, and gene deletion studies. | [58] |
| BiGG Models | Database | Repository of curated, genome-scale metabolic models (e.g., iML1515 for E. coli). | [58] |
| 13C-MFA | Experimental Protocol | High-resolution metabolic flux analysis using isotopic tracers; provides ground-truth data for validating in silico flux predictions. | [57] |
| KBase (Compare FBA Solutions App) | Software Platform/App | Web-based tool for systematically comparing multiple FBA solutions based on objective value, reaction fluxes, and metabolite uptake. | [59] |
| iCH360 Model | Metabolic Model | A manually curated, medium-scale model of E. coli core metabolism; reduces unphysiological bypasses common in genome-scale models. | [31] |
The pursuit of biologically accurate metabolic models is driving the development of sophisticated methods that move beyond the core assumptions of FBA. Frameworks like Flux Cone Learning demonstrate that machine learning can extract profound biological insights from the geometry of metabolic networks, achieving best-in-class predictive accuracy [17]. Simultaneously, methods like ΔFBA offer a powerful, objective-function-free approach to pinpointing metabolic alterations, which is particularly valuable for complex systems like human disease [54]. As the field progresses, the integration of ever-more diverse biological data—from GPR rules with directional information [58] to quantitative thermodynamic and kinetic constants [31]—will be key to constraining models and silencing the siren call of biologically infeasible predictions. For now, leveraging the compared methods within the validating context of Phenotype Phase Plane analysis provides a robust strategy for ensuring that in silico designs are firmly grounded in biological reality.
Genome-scale metabolic models (GEMs) of Escherichia coli are pivotal tools for simulating cellular metabolism and predicting the phenotypic outcomes of genetic perturbations. A critical challenge in the use of these models is the presence of inaccuracies stemming from unphysiological bypasses and incorrect loop reactions, which can significantly compromise predictive power. This guide objectively compares the performance of different E. coli GEMs and validation methodologies, focusing on their ability to identify and correct these pitfalls within the framework of Phenotype Phase Plane (PhPP) analysis. PhPP provides a systematic method for analyzing metabolic phenotypes across varying environmental conditions, such as different carbon sources and oxygenation levels, thereby offering a perfect landscape to uncover model inconsistencies [55]. Supporting experimental data, primarily from high-throughput mutant phenotyping, are summarized to guide researchers in model selection and refinement.
Experimental Platforms for Model Validation
A primary method for validating GEM predictions involves comparing computational results with high-throughput experimental growth phenotypes. The table below outlines key experimental platforms used to generate data for assessing model accuracy concerning gene essentiality and nutrient utilization.
Table 1: Key Experimental Platforms for GEM Validation
| Platform/Method | Key Experimental Readout | Application in GEM Validation |
|---|---|---|
| RB-TnSeq (Random Barcode Transposon-Sequencing) | Quantitative fitness of gene knockout mutants across thousands of genes and conditions [6]. | Primary data source for essentiality prediction accuracy; used to pinpoint errors in vitamin/cofactor biosynthesis pathways [6]. |
| Quantitative Image Analysis (Yeast Deletion Mutants) | Discretized growth rates (no growth, slow growth, wild-type growth) for single gene deletion mutants under 16 environmental conditions [60]. | Used in an iterative, bi-directional approach to refine both experimental data and model predictions. |
| Chemostat Culture & Nutrient Utilization Assays | Measured rates of nutrient uptake, product secretion, and growth under defined conditions [3]. | Validation of quantitative model predictions for growth rates and byproduct secretion in different media. |
Comparative Performance of E. coli GEMs
The iterative curation of E. coli GEMs over two decades has expanded their scope, but this does not always correlate with increased predictive accuracy. The performance of several prominent models is quantified below.
Table 2: Comparative Performance of E. coli Genome-Scale Metabolic Models
| Model Name | Gene Count | Key Validation Metric | Reported Performance | Identified Pitfalls/Strengths |
|---|---|---|---|---|
| iML1515 [6] | 1,515 | Precision-Recall AUC (Area Under Curve) using mutant fitness data [6]. | Initial analysis showed decreasing accuracy trend; performance improved after correcting media conditions [6]. | Errors in vitamin/cofactor biosynthesis pathways; isoenzyme gene-protein-reaction mapping is a key source of inaccuracy [6]. |
| EcoCyc–18.0–GEM [3] | 1,445 | Gene essentiality prediction on glucose; Nutrient utilization predictions [3]. | 95.2% accuracy for gene essentiality; 80.7% accuracy for 431 nutrient conditions [3]. | Automated generation from EcoCyc enables frequent updates; identifies conflicts for 70 genes on glucose and 80 on glycerol [3]. |
| iJO1366 [3] | 1,366 | Gene essentiality prediction. | Used as a benchmark; EcoCyc-18.0-GEM error rate decreased by 46% over this model [3]. | A widely used gold-standard model. |
| Hybrid Neural-Mechanistic Models [61] | Varies | Growth rate prediction for different media and gene knockouts. | Systematically outperforms traditional constraint-based models; requires smaller training sets than pure machine learning [61]. | Embeds FBA within machine learning to improve quantitative predictions, addressing a core FBA limitation. |
Detailed Experimental Protocols for Identification and Correction
Protocol: Validating GEMs with RB-TnSeq Mutant Fitness Data
This protocol uses genome-wide mutant fitness data to identify inaccurate gene essentiality predictions, which often point to unphysiological bypasses or incorrect media definitions [6].
- Data Acquisition: Obtain published experimental fitness data for E. coli gene knockout mutants across a wide range of conditions (e.g., 25 different carbon sources) [6].
- Model Simulation: For each experimental condition (e.g., a specific carbon source), simulate the corresponding gene knockout in silico using Flux Balance Analysis (FBA). The standard output is a binary prediction of growth or no growth [6].
- Accuracy Quantification: Compare the model's predictions with the experimental data. Use the Area Under the Precision-Recall Curve (AUC) as a robust metric, especially for imbalanced datasets where essential genes (no growth) are less frequent than non-essential ones [6].
- Error Analysis: Identify systematic errors. A key finding is that many false-negative predictions (model predicts no growth, but mutant grows) occur in genes involved in the biosynthesis of vitamins and cofactors like biotin, thiamin, and NAD+ [6].
- Hypothesis Testing & Model Refinement:
- Carry-over/Cross-feeding Hypothesis: Investigate if these metabolites are inadvertently available in the experimental medium through cross-feeding between mutants or carry-over from previous generations. Correct the model by adding the identified vitamins/cofactors to the in silico medium definition and re-evaluate accuracy [6].
- Isoenzyme Mapping Check: For errors not resolved by media corrections, manually inspect the gene-protein-reaction (GPR) rules for the affected reactions. Inaccurate isoenzyme mappings can create or block unphysiological bypasses [6].
Protocol: Phenotype Phase Plane (PhPP) Analysis for Pathway Utilization
PhPP analysis characterizes the metabolic phenotype as a function of two key environmental variables, revealing discrete phases of metabolic strategy and helping to identify unrealistic flux distributions [55].
- Condition Definition: Define a PhPP by selecting two substrate uptake rates (e.g., acetate and glucose uptake) as the axes. The PhPP spans all possible combinations of these substrates' availability [55].
- Flux Calculation: For each point in the phase plane, calculate the optimal metabolic flux distribution using FBA, typically with biomass maximization as the objective [55].
- Phase Identification: Analyze the optimal flux distributions across the plane. Discrete regions (phases) will be observed where the model uses a distinct, qualitatively different set of metabolic pathways [55].
- Isocline Interpretation: Use shadow prices and isoclines to mathematically define the boundaries between these phases. The isoclines classify the state of the metabolic network [55].
- Validation of Phase Shifts: Compare the predicted phase shifts with experimental data. An abrupt shift in predicted pathway usage that lacks biological evidence may indicate the presence of an unphysiological bypass that allows the model to "cheat" under specific conditions.
Visualization of Workflows and Metabolic Loops
The following diagrams illustrate the core concepts and workflows discussed in this guide.
Diagram 1: GEM Validation and Refinement Workflow
Diagram 2: Unphysiological Bypass and Internal Loop
The Scientist's Toolkit: Research Reagent Solutions
The following table details key reagents, computational tools, and data resources essential for conducting the analyses described in this guide.
Table 3: Essential Research Reagents and Resources
| Item Name | Type | Function/Biological Role |
|---|---|---|
| E. coli K-12 MG1655 | Bacterial Strain | The reference organism for which the most comprehensive GEMs (e.g., iML1515) have been constructed and validated [6] [3]. |
| Defined Minimal Media | Chemical Reagents | Enables precise control of nutrient availability (carbon, nitrogen) for both experiments and model simulations, crucial for PhPP analysis and identifying auxotrophies [6] [55]. |
| Vitamin/Cofactor Supplements | Chemical Reagents | (e.g., Biotin, Thiamin, NAD+). Used to test hypotheses about cross-feeding and correct false essentiality predictions in GEMs by amending simulation media [6]. |
| RB-TnSeq Mutant Library | Biological Resource | A pooled library of E. coli mutants with unique barcodes, enabling high-throughput, parallel fitness assays under many conditions for genome-wide model validation [6]. |
| Cobrapy | Software Tool | A widely used Python library for constraint-based modeling of genome-scale metabolic networks, enabling FBA simulations and gene knockout analyses [61]. |
| EcoCyc Database | Database/Software | A curated database of E. coli biology that serves as a knowledge base and can be automatically converted into a GEM (via MetaFlux), ensuring model readability and frequent updates [3]. |
Flux Balance Analysis (FBA) has served as a cornerstone of constraint-based metabolic modeling, enabling the prediction of organism behavior from stoichiometric reconstructions of metabolic networks. However, traditional FBA relies exclusively on reaction stoichiometry and optimization principles, overlooking critical biological and physical constraints. This limitation becomes particularly evident in the context of E. coli phenotype phase plane analysis, where predictions increasingly diverge from experimental observations as environmental conditions shift. The integration of enzyme and thermodynamic constraints addresses this gap by incorporating fundamental limitations that govern cellular metabolism in vivo.
The move beyond pure stoichiometry represents a paradigm shift in metabolic modeling. By embedding kinetic and thermodynamic realities into modeling frameworks, researchers can achieve more accurate predictions of microbial behavior essential for both basic science and applied drug development. This guide objectively compares the emerging methodologies that augment traditional FBA, providing researchers with a practical framework for selecting and implementing advanced constraint-based approaches.
The Stoichiometric Foundation and Its Limitations
Traditional FBA operates on the principle of mass balance constrained by stoichiometry, represented mathematically as: [ \mathbf{Sv} = 0 ] where (\mathbf{S}) is the stoichiometric matrix and (\mathbf{v}) is the vector of metabolic fluxes [62]. This framework assumes steady-state metabolism and utilizes linear optimization to identify flux distributions that maximize or minimize specific cellular objectives, typically biomass production [4].
While FBA successfully predicts metabolic capabilities and gene essentiality in many microorganisms, its predictive power diminishes for higher organisms where optimality objectives are unclear [25]. Furthermore, FBA's purely stoichiometric nature generates biologically implausible predictions, including unlimited linear growth with increasing substrate uptake and unphysiological metabolic bypasses that occur in simulated gene knockouts [26]. These limitations underscore the necessity of incorporating additional biological and physical constraints.
The Constraint Integration Spectrum
Advanced modeling frameworks extend FBA by incorporating additional layers of biological reality:
- Enzyme Constraints: Integrate proteomic limitations by incorporating enzyme turnover numbers ((k_{cat})) and molecular weights, effectively bounding reaction fluxes by catalytic capacity and cellular protein budget [62] [63].
- Thermodynamic Constraints: Ensure flux directionality aligns with Gibbs free energy profiles, enforcing reaction reversibility/irreversibility consistent with metabolite concentrations and energy landscapes [62] [4].
- Hybrid Approaches: Combine multiple constraint types to create increasingly realistic models, such as simultaneously incorporating enzyme kinetics and thermodynamic feasibility [62] [64].
Comparative Analysis of Advanced Modeling Frameworks
Performance Benchmarking
The table below summarizes key performance characteristics of advanced constraint-based modeling methods compared to traditional FBA.
Table 1: Performance Comparison of Constraint-Based Modeling Approaches
| Method | Core Innovation | Prediction Accuracy | Computational Demand | Data Requirements | Best-Suited Applications |
|---|---|---|---|---|---|
| Traditional FBA | Stoichiometry + optimization principle | 93.5% (E. coli gene essentiality) [25] | Low | Genome-scale model, uptake rates | Pathway analysis, Gene essentiality prediction in microbes |
| Enzyme-Constrained (ecFBA) | Incorporates (k_{cat}) values and enzyme pool constraints | Superior growth/yield predictions vs. FBA; identifies rate-limiting enzymes [64] | Medium | Kinetic parameters, proteomics data | Predicting metabolic fluxes, Engineering enzyme allocation |
| Thermodynamic (TFA) | Enforces reaction directionality via (\Delta G) | Eliminates thermodynamically infeasible cycles; improves flux prediction [4] | Medium-High | Thermodynamic parameters, metabolomics data | Integrating metabolomics, Calculating energy landscapes |
| Flux Cone Learning (FCL) | Machine learning on metabolic flux sample space | 95% accuracy (E. coli gene essentiality); outperforms FBA [25] | High (for training) | GEM, experimental fitness data | Phenotype prediction across organisms without optimality assumption |
| Model Balancing | Estimates consistent in-vivo kinetic parameters from omics data | Provides plausible parameter sets for kinetic modeling [65] | High (parameter estimation) | Multi-omics data (fluxes, concentrations) | Kinetic model parameterization, Data integration and reconciliation |
Quantitative Performance Data
The following table presents specific quantitative improvements achieved by advanced constraint-based methods in direct comparison to traditional FBA.
Table 2: Quantitative Performance Metrics of Advanced Modeling Frameworks
| Method | Organism/Model | Metric | Traditional FBA | Advanced Method | Improvement |
|---|---|---|---|---|---|
| Flux Cone Learning | E. coli (iML1515) | Gene essentiality accuracy | 93.5% [25] | 95% [25] | +1.5% (all genes) |
| Flux Cone Learning | E. coli (iML1515) | Essential gene classification | Baseline | +6% recall [25] | Significant reduction in false negatives |
| Enzyme-Constrained | E. coli core metabolism | Growth/Yield predictions | Linear increase with uptake | Non-linear, saturating profile [63] | Better matches experimental data |
| geckopy 3.0 | E. coli (various conditions) | Model feasibility with proteomics | Often infeasible | Achieved via relaxation algorithms [62] | Enables integration of real-world data |
Experimental Protocols for Method Implementation
Protocol for Constructing Enzyme-Constrained Models with ECMpy 2.0
Purpose: To automatically convert a genome-scale metabolic model (GEM) into an enzyme-constrained model (ecModel) using the ECMpy 2.0 pipeline. Principle: The method expands the stoichiometric matrix to include enzyme species as pseudo-metabolites, with kinetic parameters used to constrain reaction fluxes based on catalytic capacity [63].
Input Preparation:
- Obtain a GEM in SBML format (e.g., iML1515 for E. coli).
- Collect enzyme kinetic parameters from databases (e.g., BRENDA) or use the integrated machine learning predictor in ECMpy 2.0 to fill missing values.
Model Construction:
- Run the ECMpy workflow to add enzyme constraints.
- The toolbox automatically associates reactions with enzymes via GPR rules, adds enzyme usage reactions, and constrains the total enzyme pool based on cellular protein content.
Model Calibration:
- Simulate growth on a reference condition (e.g., glucose minimal medium).
- Adjust the global enzyme pool constraint to match experimentally observed growth rates.
Model Validation & Analysis:
- Test the model's prediction of growth rates on different carbon sources.
- Compare predicted vs. experimental enzyme usage fluxes, if proteomics data are available.
- Use the integrated analysis functions to identify enzyme targets for metabolic engineering [63].
Protocol for Integrating Thermodynamic Constraints with geckopy and pytfa
Purpose: To incorporate thermodynamic constraints into a metabolic model, ensuring all predicted fluxes are thermodynamically feasible. Principle: This method uses the reaction Gibbs free energy ((\Delta G)), calculated from metabolite concentrations and reaction stoichiometry, to constrain reaction directionality [62].
Data Curation:
- Gather literature data on metabolite concentrations in E. coli for the condition of interest.
- Compile standard Gibbs free energies of formation ((\Delta_f G'^\circ)) for model metabolites.
Constraint Implementation:
- Use the
geckopypackage to build an enzyme-constrained model. - Leverage the integration layer with
pytfato apply thermodynamic constraints on top of the enzyme-constrained model. - The combined framework ensures that fluxes are simultaneously constrained by enzyme kinetics and thermodynamic feasibility [62].
- Use the
Feasibility Assessment:
- Solve the resulting problem using linear or mixed-integer linear programming.
- If the model is infeasible, apply the suite of relaxation algorithms in
geckopyto identify and reconcile inconsistent constraints, typically by slightly relaxing experimental bounds on metabolite concentrations or enzyme levels [62].
Solution Analysis:
- Perform flux variability analysis (FVA) on the constrained solution space to identify possible flux distributions.
- Analyze the thermodynamic landscape of the network, identifying reactions operating close to equilibrium and those that are strongly driven.
Protocol for Phenotype Prediction using Flux Cone Learning
Purpose: To predict gene deletion phenotypes without relying on an optimality assumption. Principle: FCL uses Monte Carlo sampling to capture the geometry of the metabolic flux space for genetic perturbations and couples these features with machine learning trained on experimental fitness data [25].
Feature Generation (Sampling):
- For each gene deletion in the training set, use a Monte Carlo sampler to generate hundreds of random flux distributions ((q = 100)) within the corresponding metabolic flux cone, defined by the post-perturbation stoichiometric constraints.
- This creates a large feature matrix where each sample is a point in the high-dimensional flux space.
Model Training:
- Label all flux samples from a given deletion mutant with its corresponding experimental fitness score.
- Train a supervised learning model (e.g., Random Forest classifier for essentiality) on the labeled dataset to learn the correlation between flux cone geometry and phenotypic outcome.
Prediction and Validation:
- For a new gene deletion, generate flux samples from its metabolic flux cone.
- Use the trained model to make sample-wise predictions.
- Aggregate predictions (e.g., by majority voting) to obtain a final deletion-wise prediction.
- Validate model accuracy on a held-out test set of genes with known experimental fitness [25].
Visualization of Method Workflows and Relationships
Table 3: Key Research Reagents and Computational Tools for Advanced Constraint-Based Modeling
| Category | Item/Resource | Function/Purpose | Example/Format |
|---|---|---|---|
| Computational Tools | ECMpy 2.0 | Automated construction and analysis of enzyme-constrained models from GEMs [63] | Python Package |
| geckopy 3.0 | Python implementation for building enzyme-constrained models with SBML-compliant formulation and relaxation algorithms [62] | Python Package | |
| pytfa | Thermodynamic Flux Analysis (TFA), integrates thermodynamic and metabolomics constraints [62] | Python Package | |
| COBRA Toolbox | Widely used MATLAB suite for constraint-based reconstruction and analysis [4] | MATLAB Toolbox | |
| Reference Models | iML1515 | Gold-standard, genome-scale model of E. coli K-12 MG1655 metabolism (1515 genes, 2712 reactions) [25] [26] | SBML Format |
| iCH360 | Manually curated, medium-scale model of E. coli core and biosynthetic metabolism; a subnetwork of iML1515 ideal for detailed analysis [26] | SBML Format | |
| Data Resources | BRENDA | Comprehensive enzyme database providing kinetic parameters (e.g., (k_{cat})) [65] [63] | Online Database |
| Equilibrator | Web-based tool for calculating standard Gibbs free energies of biochemical reactions [4] | Online Tool / API | |
| Experimental Data | Absolute Proteomics | Quantified protein concentrations used to constrain enzyme levels in ecModels [62] | Mass Spectrometry Data |
| Metabolomics Data | Intracellular metabolite concentrations for informing thermodynamic constraints [62] [4] | LC-MS/GC-MS Data | |
| Fitness Assays | Experimental gene essentiality or growth fitness data for training and validating predictive models like FCL [25] | Phenotypic Microarray |
The integration of enzyme kinetics and thermodynamics into constraint-based models marks a significant advancement toward biologically realistic simulations of metabolism. While traditional FBA remains valuable for initial explorations, its enhanced successors offer tangible improvements in predictive accuracy.
Enzyme-constrained models excel at predicting flux distributions and growth yields under different nutrient conditions, making them ideal for metabolic engineering. Thermodynamically constrained models provide fundamental checks on feasibility and are powerful tools for integrating metabolomic data. Emerging data-driven and machine learning approaches like Flux Cone Learning demonstrate superior performance for specific prediction tasks like gene essentiality, especially when optimality principles are uncertain.
The choice of methodology ultimately depends on the research question, data availability, and desired predictive outcomes. For researchers engaged in E. coli phenotype phase plane analysis, employing these advanced frameworks can resolve discrepancies between prediction and experiment, offering deeper insight into the complex interplay of stoichiometric, kinetic, and thermodynamic forces that shape metabolic function.
Flux Balance Analysis (FBA) is a cornerstone of systems biology for simulating cellular metabolism. A critical component of its validation is the Phenotype Phase Plane (PhPP) analysis, which maps optimal metabolic phenotypes against environmental conditions. This guide compares traditional PhPP construction with advanced approaches that integrate System Identification (SID) techniques. We objectively evaluate how SID, which uses high-throughput experimental data to infer model parameters and reduce uncertainty, enhances the predictive accuracy and practical utility of Escherichia coli metabolic models. Data from recent studies demonstrate that SID-driven models significantly improve the prediction of gene essentiality and nutrient utilization, providing more reliable tools for metabolic engineering and drug development.
The Phenotype Phase Plane (PhPP) is a powerful tool for analyzing cellular metabolism through Flux Balance Analysis (FBA). It graphically represents optimal growth phenotypes as a function of multiple environmental variables, typically uptake rates for two nutrients, revealing distinct metabolic phases and optimal pathways [66]. For foundational E. coli models, the PhPP has been instrumental in predicting metabolic behaviors such as aerobic/anaerobic growth and substrate co-utilization.
However, traditional FBA models are built on static stoichiometric reconstructions and can suffer from inherent uncertainties. These include incomplete gene-protein-reaction (GPR) mappings, inaccurate specification of the simulation environment, and an inability to capture population heterogeneity [6] [42]. Such limitations can lead to incorrect predictions of gene essentiality and nutrient utilization, reducing the model's reliability for critical applications in biotechnology and drug development.
System Identification (SID) addresses these gaps by applying parameter estimation techniques to calibrate metabolic models against experimental data. SID formulates the problem of finding model parameters that minimize the difference between simulated outputs and high-throughput experimental measurements [67] [68]. By integrating datasets like mutant fitness screens across thousands of genes and conditions, SID pinpoints sources of model uncertainty and refines the model's predictive capabilities, leading to an enhanced and more accurate PhPP analysis [6].
Comparative Analysis of Model Performance
The integration of SID techniques has led to the development of next-generation models with demonstrably superior performance. The table below summarizes a quantitative comparison between a representative traditional model (iJO1366) and a more modern, SID-informed model (EcoCyc–18.0–GEM).
Table 1: Performance Comparison of E. coli GEMs
| Model Attribute | Traditional Model (iJO1366) | SID-Enhanced Model (EcoCyc–18.0–GEM) | Improvement |
|---|---|---|---|
| Number of Genes | 1,366 | 1,445 | +6% |
| Number of Reactions | 1,855 | 2,286 | +23% |
| Gene Essentiality Prediction Accuracy | ~91% (est. from literature) | 95.2% | 46% reduction in error rate |
| Nutrient Utilization Prediction Accuracy | ~77% (on 171 conditions) | 80.7% (on 431 conditions) | +4.8% accuracy on 2.5x more conditions |
This objective data shows that the SID-enhanced model not only encompasses more metabolic knowledge but also achieves higher predictive accuracy across a broader range of tests [3].
Beyond overall accuracy, a critical performance metric is the model's ability to correctly predict gene essentiality. A 2023 study quantified the accuracy of several E. coli models using high-throughput mutant fitness data. The analysis identified that errors were often concentrated in specific pathways, particularly the biosynthesis of vitamins and cofactors like biotin, R-pantothenate, and tetrahydrofolate [6]. The SID process helped identify that these inaccuracies likely stemmed from unaccounted metabolite availability in the experimental environment (e.g., via cross-feeding between mutants), rather than errors in the pathway structure itself. Correcting these environmental specifications was a key SID step that improved model fidelity.
Experimental Protocols for Model Validation and Refinement
A core SID methodology involves using large-scale mutant fitness data to validate and correct metabolic models. The following workflow and protocol detail this process.
Diagram Short Title: SID Workflow for GEM Validation
Protocol: Validating GEMs with RB-TnSeq Data
This protocol describes how to use mutant fitness data from RB-TnSeq experiments to identify and correct errors in a genome-scale metabolic model (GEM) [6].
1. Data Acquisition and Preprocessing:
- Experimental Data: Obtain a published dataset measuring the fitness of E. coli gene knockout mutants across multiple conditions (e.g., 25 different carbon sources) [6].
- Model: Select the GEM to be validated (e.g., iML1515).
2. In Silico Simulation of Experiments:
- For each gene knockout and growth condition in the experimental dataset, set up the corresponding FBA simulation.
- Simulation Environment: Define the constraints of the in silico medium to match the experimental conditions as closely as possible.
- Gene Knockout: Constrain the flux through the reaction(s) associated with the knocked-out gene to zero.
- Phenotype Prediction: Use FBA to predict a binary growth/no-growth phenotype for each simulation.
3. Quantitative Accuracy Assessment:
- Compare the model's predictions against the experimental fitness data.
- Key Metric: Calculate the Area Under the Precision-Recall Curve (AUC-PR). This metric is robust for imbalanced datasets where essential genes (no-growth phenotypes) are less frequent than non-essential ones [6].
- Alternative Metrics: Overall accuracy or AUC-ROC can be calculated for supplementary analysis.
4. Error Analysis and Model Refinement:
- Analyze false predictions to identify systematic errors. A common finding is false-negative predictions (model predicts no growth, but experiment shows growth) for genes involved in vitamin/cofactor biosynthesis (e.g.,
bioA-D,panB,C,pabA,B) [6]. - Hypothesis Testing: Formulate hypotheses for the error, such as unmodeled metabolite availability via cross-feeding between mutants or metabolite carry-over from previous generations.
- Model Correction: Test the hypothesis by adding the identified vitamins/cofactors (e.g., biotin, R-pantothenate) to the in silico medium constraints for the simulation. Re-run the accuracy assessment to confirm improvement.
Advanced SID: From Single Models to Population Heterogeneity
A frontier in SID for metabolic models is moving beyond simulating an "average" cell to capturing population heterogeneity. POSYBEL is a population systems biology model that uses the Markov chain Monte Carlo (MCMC) algorithm to stochastically sample the entire possible flux solution space [42].
- Method: Instead of finding a single optimal flux distribution for biomass maximization, POSYBEL generates a population of cells, each with a unique metabolic flux state that is thermodynamically feasible.
- Outcome: This results in a heterogeneous population where sub-populations (e.g., persister cells) naturally emerge with different metabolic capabilities, such as surviving antibiotic treatment or overproducing a target metabolite [42].
- Visualization: The output is often visualized as a scatter plot (or "triangle"), where each dot represents an individual cell's metabolic state, showing the correlation between biomass yield and product formation. This provides a more nuanced view than a single PhPP.
Table 2: Key Reagent Solutions for SID in Metabolic Modeling
| Research Reagent / Resource | Function in SID and Model Validation |
|---|---|
| E. coli K-12 MG1655 GEM (iML1515) | The core computational model representing metabolic network structure for FBA and PhPP analysis [6]. |
| RB-TnSeq Mutant Fitness Data | High-throughput experimental dataset used as a ground truth for validating and refining model predictions [6]. |
| Flux Balance Analysis (FBA) | The primary optimization algorithm used to simulate metabolic phenotypes and generate PhPPs [3] [66]. |
| EcoCyc Database | A curated bioinformatics database used to automatically generate and update GEMs via tools like MetaFlux [3]. |
| MCMC Sampling Algorithm | A computational algorithm used in advanced SID to explore the space of possible flux distributions and model population heterogeneity [42]. |
Pathway and Logical Analysis
A key insight from SID is that inaccuracies are often localized to specific metabolic pathways. The following diagram maps the pathways frequently implicated in model errors and their logical connection to SID-based corrections.
Diagram Short Title: Pathways Targeted for SID Correction
The objective comparison presented in this guide clearly demonstrates that System Identification techniques are critical for enhancing the predictive power of Phenotype Phase Plane analysis. By leveraging high-throughput experimental data, SID transitions metabolic models from static maps to dynamic, validated, and self-improving computational platforms. The results show tangible improvements in predicting gene essentiality and nutrient utilization, which are fundamental for designing robust metabolic engineering strategies.
Future developments in SID will focus on integrating ever-larger multimodal datasets (including transcriptomics and proteomics) and embracing population-level modeling, as exemplified by the POSYBEL platform. These advances will further refine PhPP analysis, providing researchers and drug development professionals with increasingly accurate in silico models to accelerate discovery and optimize bioproduction processes.
Iterative Model Refinement Based on PhPP Discrepancies
Comparative analysis of metabolic models and refinement techniques is crucial for accurate prediction of microbial behavior in biopharmaceutical production. This guide objectively compares the performance of established E. coli metabolic models and contemporary refinement methodologies, providing experimental data to support strain selection and optimization for drug development pipelines.
Phenotype Phase Plane (PhPP) analysis provides a geometric interpretation of metabolic capabilities under varying environmental conditions, typically mapping optimal growth rates against two nutrient uptake rates [5]. Discrepancies between computational PhPP predictions and experimental data serve as powerful drivers for iterative model refinement. This process is fundamental in industrial biotechnology, where accurate models predict production yields for pharmaceutical compounds like recombinant proteins or metabolic precursors for drug synthesis.
The validation cycle begins with flux balance analysis (FBA), a constraint-based approach that predicts metabolic flux distributions by combining genome-scale metabolic models (GEMs) with an optimality principle, typically biomass maximization for microbial growth [5] [25]. For E. coli, the most complete metabolic reconstruction is iML1515, containing 1,515 genes, 2,719 metabolic reactions, and 1,192 metabolites [5]. Newer compact models like iCH360 focus specifically on energy and biosynthetic metabolism, offering advantages in interpretability and thorough curation while maintaining connections to central metabolic pathways [26].
Comparative Analysis of Metabolic Models and Refinement Methodologies
Performance Comparison of Metabolic Modeling Approaches
Table 1: Comparative performance of E. coli metabolic models and refinement methodologies
| Model/Method | Key Features | Application Context | Predictive Accuracy | Limitations |
|---|---|---|---|---|
| iML1515 (GEM) | 1,515 genes, 2,719 reactions, 1,192 metabolites [5] | Genome-scale prediction of metabolic capabilities | 93.5% accuracy for gene essentiality prediction [25] | Predicts biologically unrealistic bypasses; complex interpretation [26] |
| iCH360 (Compact) | Manually curated core & biosynthesis metabolism [26] | Engineering biosynthetic pathways for drug precursors | High interpretability; enriched with kinetic constants [26] | Limited scope; misses degradation pathways [26] |
| Flux Balance Analysis (FBA) | Stoichiometric constraints with biomass optimization [5] [25] | Predicting growth rates & metabolic flux distribution | Struggles with higher organisms where optimality objective is unknown [25] | Requires optimality assumption; may predict unphysiological fluxes [26] [25] |
| Enzyme-Constrained FBA (ecFBA) | Incorporates enzyme kinetics & allocation [5] | Realistic flux prediction in engineered pathways | Avoids arbitrarily high flux predictions; more physiological [5] | Limited transporter protein kinetic data [5] |
| Flux Cone Learning (FCL) | Machine learning on metabolic space geometry [25] | Gene essentiality prediction & bioproduction optimization | 95% accuracy for E. coli gene essentiality [25] | Computationally intensive; requires extensive sampling [25] |
Quantitative Assessment of Model Refinement Techniques
Table 2: Experimental performance metrics for refinement methodologies
| Refinement Method | Organism/Model Tested | Performance Metric | Result | Experimental Conditions |
|---|---|---|---|---|
| Flux Cone Learning | E. coli iML1515 [25] | Gene essentiality prediction accuracy | 95% [25] | Aerobic growth on glucose; 100 samples/deletion cone [25] |
| Enzyme Constraints (ECMpy) | E. coli iML1515 [5] | Physiological flux prediction | Improved over standard FBA [5] | Protein fraction constraint: 0.56; Kcat values from BRENDA [5] |
| Manual Curation (iCH360) | E. coli core metabolism [26] | Biological interpretability & curation depth | High (qualitative assessment) [26] | Focus on energy & biosynthesis pathways [26] |
| Iterative Refinement | Computer vision pipelines [69] | Optimization stability & performance | Consistent performance gains [69] | Component-by-component refinement [69] |
Experimental Protocols for Model Refinement
Protocol 1: Enzyme-Constrained Flux Balance Analysis
Purpose: To incorporate enzyme kinetic constraints into metabolic models for more realistic flux predictions during bioproduction strain design.
Methodology:
- Reaction Processing: Split all reversible reactions into forward and reverse directions to assign corresponding Kcat values [5].
- Isoenzyme Handling: Separate reactions catalyzed by multiple isoenzymes into independent reactions, as they have different associated Kcat values [5].
- Parameter Incorporation:
- Implementation: Apply constraints using workflows such as ECMpy, which adds total enzyme constraints without altering the stoichiometric matrix [5].
Application in Drug Development: This protocol enables more realistic prediction of precursor flux for active pharmaceutical ingredients, optimizing microbial production strains during early preclinical development [70] [71].
Protocol 2: Flux Cone Learning for Phenotypic Prediction
Purpose: To predict gene deletion phenotypes using machine learning on metabolic space geometry.
Methodology:
- Metabolic Sampling: Utilize Monte Carlo sampling to generate flux distributions for each gene deletion variant (typically 100 samples/deletion cone) [25].
- Feature Matrix Construction: Create a feature matrix with dimensions (k × q) × n, where k = number of gene deletions, q = samples per cone, and n = reactions in GEM [25].
- Model Training: Employ supervised learning (e.g., random forest classifier) using flux samples as features and experimental fitness scores as labels [25].
- Prediction Aggregation: Apply majority voting on sample-wise predictions to generate deletion-wise phenotypic predictions [25].
Validation: For E. coli, this protocol achieved 95% accuracy in predicting metabolic gene essentiality across different carbon sources, outperforming standard FBA [25].
Protocol 3: Iterative Component Refinement for Model Optimization
Purpose: To systematically improve model performance through targeted component refinement.
Methodology:
- Performance Analysis: Evaluate model predictions against experimental PhPP data to identify specific discrepancies [69].
- Component Isolation: Select individual model components for refinement (e.g., GPR relationships, thermodynamic constraints, or subsystem curation) while keeping others fixed [69].
- Targeted Intervention: Apply corrections based on biological knowledge, such as updating gene-protein-reaction associations using EcoCyc database information [5].
- Impact Assessment: Evaluate the effect of each refinement independently before proceeding to subsequent components [69].
Advantage: This approach enables precise attribution of performance changes to specific refinements, preventing unstable optimization and providing interpretable improvement pathways [69].
Workflow Visualization of Iterative Refinement Processes
Figure 1: Iterative model refinement workflow driven by PhPP discrepancies.
Figure 2: Flux Cone Learning workflow for phenotypic prediction.
Essential Research Reagent Solutions for Metabolic Modeling
Table 3: Key reagents, databases, and computational tools for metabolic model refinement
| Resource | Type | Function in Model Refinement | Application Context |
|---|---|---|---|
| iML1515 GEM | Computational Model | Reference genome-scale metabolic reconstruction of E. coli K-12 MG1655 [5] | Base model for constraint-based simulations & gap analysis |
| iCH360 Model | Computational Model | Manually curated compact model of core & biosynthetic metabolism [26] | Engineering pathways for pharmaceutical precursor production |
| BRENDA Database | Kinetic Database | Source of enzyme kinetic parameters (Kcat values) [5] | Parameterizing enzyme-constrained models |
| EcoCyc Database | Biochemical Database | Reference for gene-protein-reaction relationships & metabolic pathways [5] | Curating GPR associations & verifying pathway topology |
| COBRApy | Software Toolbox | Python package for constraint-based reconstruction and analysis [5] | Implementing FBA simulations & analyzing flux distributions |
| ECMpy | Software Workflow | Tool for incorporating enzyme constraints into metabolic models [5] | Creating enzyme-constrained models for realistic flux predictions |
| Zeneth Software | Prediction Software | Predicts chemical degradation pathways for small molecules [72] | Modeling stability of pharmaceutical compounds in development |
Validation Frameworks and Comparative Analysis of Model Predictions
Benchmarking Model Predictions Against Experimental Phenomic Data
Metabolic models are indispensable tools in systems biology and biotechnology, encoding biochemical knowledge in a structured format to predict cellular behavior. For the model organism Escherichia coli, metabolic models range from genome-scale reconstructions like iML1515 (covering 1515 genes and 2712 reactions) to smaller core models [26] [40]. Flux Balance Analysis (FBA) serves as the cornerstone computational method for analyzing these networks, using linear programming to predict metabolic fluxes under steady-state and mass-balance constraints [22]. A critical challenge, however, lies in validating these computational predictions against experimental phenomic data, a process where methods like Phenotypic Phase Plane Analysis are vital [73].
This guide objectively compares the predictive performance of contemporary metabolic models of E. coli, focusing on their validation against experimental data. We place special emphasis on the benchmarking of a newly developed, manually curated medium-scale model, iCH360, against its genome-scale parent iML1515 and other established models [26] [31]. By providing detailed methodologies, quantitative comparisons, and visualization workflows, we aim to furnish researchers with a clear framework for model selection and validation in projects ranging from fundamental microbial physiology to drug development.
Comparative Analysis of Metabolic Models forE. coli
The landscape of E. coli metabolic models is diverse, with each model offering distinct advantages and limitations for phenotypic prediction. The choice of model significantly impacts the biological realism, computational tractability, and interpretability of the results [26] [40].
- Genome-Scale Models (e.g., iML1515): Models like iML1515 provide comprehensive coverage of the organism's metabolism, containing 1877 metabolites and 2712 reactions mapped to 1515 genes [26]. This breadth makes them powerful for predicting gene essentiality at a system-wide level. However, their size can be a double-edged sword. They can be difficult to visualize and analyze with complex methods, and they occasionally generate biologically unrealistic predictions, such as unphysiological metabolic bypasses in gene knockout strategies [26] [31].
- Small-Scale Core Models (e.g., ECC): The E. coli Core model is widely used for education and as a benchmark due to its simplicity and ease of use. Its primary limitation is its narrow scope; it lacks most biosynthesis pathways for amino acids, nucleotides, and fatty acids, which are critical for many metabolic engineering applications and for a holistic view of cell physiology [26] [40].
- Medium-Scale Models (e.g., iCH360): The iCH360 model is designed as a "Goldilocks-sized" intermediary, manually curated to balance comprehensiveness and usability [26] [31]. It is a sub-network of iML1515, comprising 360 genes and 323 reactions. It includes all central metabolic pathways for energy production and the biosynthesis of major biomass building blocks (amino acids, nucleotides, fatty acids) [26]. This focused scope allows for more straightforward application of advanced analytical techniques like Elementary Flux Mode analysis and thermodynamic profiling, which can be computationally prohibitive with genome-scale models [26]. Furthermore, iCH360 is enriched with extensive annotations, thermodynamic data, and kinetic constants, enhancing its utility for more sophisticated constraint-based modeling beyond standard FBA [31].
Table 1: Key Characteristics of Featured E. coli Metabolic Models
| Model Name | Genes | Reactions | Metabolites | Primary Scope & Distinguishing Features |
|---|---|---|---|---|
| iML1515 [26] | 1,515 | 2,712 | 1,877 | Genome-scale; comprehensive network for system-wide gene essentiality prediction. |
| iCH360 [26] [31] | 360 | 323 | 304 (254 unique) | Medium-scale; manually curated core & biosynthesis metabolism; rich annotations & quantitative data. |
| ECC (Core) [26] | - | - | - | Small-scale; educational tool; lacks most biosynthesis pathways. |
Core Methodologies for Model Validation
Robust validation of metabolic model predictions requires a combination of computational and experimental techniques. Below, we detail the core methodologies.
Computational Analysis: Flux Balance Analysis (FBA)
FBA is a constraint-based mathematical approach for predicting the flow of metabolites through a metabolic network at steady state [22].
- Mathematical Foundation: The metabolic network is represented by a stoichiometric matrix S (of size m x n, where m is the number of metabolites and n is the number of reactions). The mass balance equation is represented as: > Sv = 0 where v is the vector of reaction fluxes. This equation asserts that the production and consumption of each intracellular metabolite are balanced [22].
- Constraints and Optimization: Flux constraints are applied as upper and lower bounds (Vimin ≤ v_i ≤ Vimax) on reaction rates. An objective function (Z = cTv), often biomass maximization to simulate growth, is defined and optimized using linear programming to find a flux distribution that satisfies all constraints [22].
- Software Implementation: The COBRA Toolbox in MATLAB and Python is the standard software suite for performing FBA and related analyses [22] [73].
Experimental Validation: Phenotypic Phase Plane Analysis
Phenotypic Phase Plane (PhPP) analysis is a powerful method for exploring how an organism's optimal phenotype (e.g., growth rate) changes with variations in two environmental conditions [22] [73].
- Objective: To map the metabolic capabilities and limitations of an organism by calculating the optimal growth rate as a function of two nutrient uptake rates (e.g., carbon vs. nitrogen, carbon vs. light) [73].
- Protocol:
- Define Variables: Select two key environmental variables to perturb (e.g., CO₂ and ammonia uptake rates).
- Set Bounds: Define a realistic range of flux values for the two exchange reactions in the model.
- Grid Search: Perform FBA at each point in the 2D grid of uptake rates to compute the maximum biomass (growth) flux.
- Visualization: Plot the resulting growth rates to create a phase plane, which reveals distinct regions of optimal metabolic operation and limiting factors [73].
Diagram 1: Workflow for Phenotypic Phase Plane Analysis. This diagram outlines the computational process for mapping an organism's metabolic response to two varying environmental conditions.
Advanced Validation: Single Reaction Knockout and Flux Variability Analysis
- Single Reaction Knockout: This simulation tests the model's prediction of gene essentiality or phenotypic impact by setting the flux bounds of a reaction to zero (or by using the model's Gene-Protein-Reaction rules) and re-optimizing for growth [73] [25]. A predicted growth rate of zero indicates an essential reaction for the simulated condition. This is a key benchmark for validating models against experimental gene essentiality screens [25].
- Flux Variability Analysis (FVA): FVA recognizes that multiple flux distributions can achieve the same optimal objective value. This method calculates the minimum and maximum possible flux through each reaction while maintaining optimality (or near-optimality) of the objective function [22] [73]. It is particularly useful for identifying reactions with tightly controlled fluxes and for validating model predictions against measured flux data.
Benchmarking Model Performance
To objectively compare the predictive power of different models, we benchmark them against key experimental phenomic data types.
Prediction of Gene Essentiality
A primary benchmark for metabolic models is their accuracy in predicting which gene knockouts will prevent growth (i.e., are essential) under specific conditions.
- Experimental Protocol: Genome-wide knockout libraries (e.g., using CRISPR-Cas9) are grown in a defined medium (e.g., M9 minimal medium with glucose). Genes for which the knockout strain shows no growth are classified as experimentally essential [25].
- Computational Protocol: For each gene in the model, a simulation is run where the reactions associated with that gene are constrained to have zero flux. FBA is then performed to maximize for biomass production. A growth rate below a threshold (e.g., < 1e-6) is predicted as essential [25].
- Performance Comparison: While FBA with models like iML1515 achieves high accuracy (~93.5% in E. coli), newer methods like Flux Cone Learning (FCL) can outperform it. FCL uses Monte Carlo sampling of the metabolic space defined by the model and machine learning to correlate geometric changes from gene deletions with experimental fitness data, achieving up to 95% accuracy [25]. This demonstrates that model-based predictions can serve as a benchmark for gene essentiality.
Table 2: Benchmarking Gene Essentiality Predictions in E. coli
| Model / Method | Reported Accuracy | Key Strengths | Key Limitations |
|---|---|---|---|
| FBA with iML1515 [25] | ~93.5% | Established gold standard; strong mechanistic basis. | Relies on a pre-defined biological objective (e.g., growth maximization). |
| Flux Cone Learning (FCL) [25] | ~95% | Best-in-class accuracy; does not require an optimality assumption. | Computationally intensive; requires training data. |
| iCH360 Model [26] | (Manually curated for core genes) | High interpretability for central metabolism; reduced unphysiological bypasses. | Scope limited to core & biosynthesis metabolism; may miss system-wide effects. |
Prediction of Growth Phenotypes and Nutrient Utilization
Models are also benchmarked on their ability to predict quantitative growth outcomes, such as growth rates under different nutrient conditions, which can be visualized using Phenotypic Phase Planes.
- Case Study: UTEX 2973 Bioreactor Optimization: A validation study on the cyanobacterium Synechococcus elongatus UTEX 2973 used FBA with its genome-scale model to predict optimal media composition [73]. PhPP analysis was used to identify the optimal CO₂ uptake rate and light intensity for maximizing biomass accumulation. The model predicted an optimum at a CO₂ uptake of -132 mmol/gDW/h and photon uptake of -900 mmol/gDW/h, yielding a growth rate of 3.1386 mmol/gDW/h [73]. Subsequent hierarchical grid searches refined this further. This workflow showcases how model predictions can directly guide experimental bioreactor optimization.
- Application to iCH360: The iCH360 model, with its curated core metabolism, is well-suited for similar analyses in E. coli. Researchers can perform PhPP analysis to predict growth under dual nutrient limitations (e.g., carbon vs. nitrogen) and validate these predictions against wet-lab growth curves in controlled bioreactors.
Diagram 2: Model Validation Workflow. This diagram shows the iterative cycle of generating predictions from a metabolic model and validating them against experimental phenomic data.
Prediction of Metabolic Flux Distributions
For models with integrated quantitative data like iCH360, a further benchmark is the prediction of internal metabolic flux distributions.
- Experimental Protocol: (^{13})C Metabolic Flux Analysis (MFA) is the standard experimental method. Cells are fed with (^{13})C-labeled substrates (e.g., [1-(^{13})C]glucose), and the resulting labeling patterns in intracellular metabolites are measured via Mass Spectrometry or NMR. Computational analysis of these patterns yields quantitative estimates of in vivo metabolic fluxes [26].
- Computational Protocol: Flux distributions obtained from FBA or other constraint-based methods (like Enzyme-constrained FBA) can be directly compared to the fluxes inferred from (^{13})C MFA. The iCH360 model, with its added thermodynamic and kinetic data, is particularly well-equipped for enzyme-constrained simulations that may yield more realistic flux predictions [26] [31].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful execution of the benchmarking protocols requires a suite of computational and experimental tools.
Table 3: Essential Reagents and Tools for Model Validation
| Item Name | Function / Application | Example Sources / Types |
|---|---|---|
| COBRA Toolbox [22] [73] | Primary software for constraint-based modeling (FBA, PhPP, FVA). | MATLAB, Python. |
| Genome-Scale Metabolic Model | The in silico representation of the organism for simulation. | iML1515 (E. coli), iCH360 (E. coli core), organism-specific models from repositories. |
| Defined Growth Medium | Provides a controlled environment for consistent experimental validation. | M9 Minimal Medium (for E. coli), BG-11 Medium (for cyanobacteria) [73]. |
| Knockout Library | Experimental resource for validating gene essentiality predictions. | CRISPR-based libraries, single-gene knockout collections (e.g., Keio collection for E. coli). |
| (^{13})C-Labeled Substrates | Tracers for experimental determination of metabolic fluxes via (^{13})C MFA. | [1-(^{13})C]Glucose, [U-(^{13})C]Glucose. |
| Bioreactor / Fermenter | Provides controlled environmental conditions (pH, temperature, gas) for reproducible growth phenotyping. | Bench-top bioreactor systems. |
Quantitative Metrics for Validating Growth Rates and By-Product Secretion
Flux Balance Analysis (FBA) has become an indispensable tool for predicting metabolic behaviors in microorganisms like Escherichia coli. As a constraint-based modeling framework, FBA predicts metabolic flux distributions, growth rates, and by-product secretion by assuming steady-state metabolism and optimizing an objective function, typically biomass maximization [4]. The phenotype phase plane (PhPP) analysis provides a global perspective on genotype-phenotype relationships, characterizing how metabolic phenotypes shift with varying environmental conditions [8]. However, the reliability of these in silico predictions hinges on rigorous validation against quantitative experimental data. Without robust validation, FBA models may yield mathematically feasible but biologically inaccurate flux predictions, compromising their utility in metabolic engineering and drug development.
This guide systematically compares the quantitative metrics and experimental methodologies essential for validating FBA-predicted growth rates and by-product secretion profiles in E. coli. We focus specifically on validation within the context of phenotype phase plane analysis, providing researchers with a standardized framework for assessing model predictive accuracy.
Quantitative Metrics for Growth Rate Validation
Core Growth Parameters and Measurement Techniques
Validating FBA-predicted growth rates requires precise quantification of key parameters across different cultivation systems. The table below summarizes the essential metrics and corresponding analytical methods:
Table 1: Quantitative Metrics for Bacterial Growth Rate Validation
| Quantitative Metric | Description | Measurement Techniques | Relevance to FBA Validation |
|---|---|---|---|
| Maximum Specific Growth Rate (μmax) | The maximum rate of exponential biomass increase [74] | Time-lapse microscopy, optical density (OD), dry cell weight [75] | Direct comparison with FBA-predicted growth rates |
| Mass Doubling Time | Time required for cell mass to double [76] | Derived from growth curves [76] | Validates biological feasibility of predictions |
| Cell Mass per Origin | Mass per replication origin at initiation [76] | Flow cytometry, proteomics [76] | Connects DNA replication to growth coordination |
| Initiation Mass (mᵢ) | Cell mass per origin at replication initiation [76] | $m_i = \frac{m̄}{ō \cdot \ln2}$ [76] | Tests coordination between cell cycle and growth |
Advanced Single-Cell Growth Metrics
Beyond population-level measurements, single-cell analysis provides critical insights into growth heterogeneity that can inform model validation:
- Replicative Rate vs. Growth Rate: The replicative rate refers to division capacity, while growth rate describes increase in size or mass [75]. These may diverge under stress conditions, creating non-growing cells that still divide or filaments that grow without dividing.
- Growth Rate Bistability: Under sub-MIC antibiotic concentrations, heterogeneous subpopulations with fast and slow growth rates can coexist [77]. This heterogeneity represents a "bet-hedging" strategy that FBA models should capture when relevant.
- Integral-Threshold Relationship: Recent research reveals that population-averaged cellular mass ($m̄$) follows a linear relationship with the rate of chromosome replication-segregation: $m̄ = m_0λ(C+D)$, where λ is growth rate and C+D is replication-segregation time [76].
Quantitative Metrics for By-Product Secretion
Metabolic Secretion Profiles in E. coli
FBA models predict by-product secretion as overflow metabolism when carbon uptake exceeds energy requirements. The table below summarizes key secretion products and their quantification:
Table 2: Quantitative Metrics for By-Product Secretion Validation
| By-Product | Conditions for Secretion | Quantification Methods | Typical Secretion Rates |
|---|---|---|---|
| Acetate | Excess carbon, limited oxygen [8] | HPLC, enzymatic assays | Varies with carbon uptake rate |
| Lactate | Anaerobic conditions [8] | HPLC, mass spectrometry | Dependent on NAD+ regeneration needs |
| Ethanol | Mixed-acid fermentation [8] | GC, HPLC | Correlates with redox balance |
| Formate | Anaerobic respiration [8] | HPLC, colorimetric assays | Split between excretion and further metabolism |
| Succinate | Fumarate respiration terminal product | HPLC, NMR | Typically lower than other fermentation products |
| CO₂ | Aerobic respiration, decarboxylations [8] | Gas chromatography, respirometry | Direct measure of metabolic activity |
Shadow Price Analysis in Phenotype Phase Planes
In traditional PhPP analysis, shadow prices indicate how much the objective function (e.g., growth rate) would improve with additional availability of a metabolite [8]. However, a significant limitation exists: metabolites with zero shadow prices are not always excreted, while metabolites with non-zero shadow prices could be excreted under certain conditions [8]. This necessitates experimental validation of secretion profiles.
Experimental Protocols for Validation
Cultivation Conditions and Steady-State Assurance
Reliable validation requires carefully controlled cultivation conditions to ensure data quality:
- Steady-State Growth Assurance: Maintain cultures in steady-state where the rate of total cell-mass growth (λm) equals the rate of cell number growth (λc) [76]. Special care must be taken to ensure experimental cultures lie on the steady-state line λm = λc.
- Media Formulation: Culture E. coli K12 MG1655 in 32 different growth media with nutrient-imposed growth rates ranging from 0.06 h−1 to 1.7 h−1 (doubling times from ~700 min to 24 min) to cover both slow- and fast-growth regimes [76].
- Critical Process Parameters: Continuously monitor and control dissolved oxygen, pH, temperature, and substrate/nutrient concentrations throughout cultivations [78] [79].
Protocol for Growth Rate Determination
The one-step parameter estimation method is statistically superior to the two-step method for growth rate determination [74]:
- Inoculum Preparation: Start with standardized inoculum from frozen stocks in appropriate medium.
- Cell Density Monitoring: Measure cell density (OD600) at frequent intervals, ensuring measurements remain in the linear range (0.1-0.8 OD600).
- Direct Curve Fitting: Fit the primary model $\frac{dNt}{dt} = μ{max} \cdot N_t$ directly to cell density data using nonlinear regression [74].
- Confidence Interval Calculation: Determine confidence intervals for μmax from the curvature of the objective function at the optimum [74].
Protocol for By-Product Secretion Analysis
Quantifying metabolic secretions requires precise analytical methods:
- Sample Collection: Collect culture supernatant at multiple time points during exponential growth using rapid filtration (0.22 μm filters).
- Metabolite Profiling: Analyze supernatant using HPLC with refractive index or UV detection for organic acids and alcohols [4].
- Isotopic Labeling: For 13C-MFA, use parallel labeling experiments with multiple tracers (e.g., [1-13C] glucose, [U-13C] glucose) to enhance flux resolution [4].
- Mass Isotopomer Distribution: Measure mass isotopomer distributions (MIDs) using GC-MS or LC-MS and fit to metabolic network models [4].
System Identification Enhanced PhPP Analysis
Enhancing Traditional Phenotype Phase Plane Analysis
Traditional PhPP analysis has limitations in characterizing different metabolic phenotypes based solely on shadow prices. The System Identification enhanced PhPP (SID-PhPP) addresses these limitations:
- Designed Perturbations: Systematically perturb the metabolic network through designed input sequences, such as holding carbon uptake constant while varying oxygen uptake [8].
- Multivariate Statistical Analysis: Apply principal component analysis (PCA) to in silico results to extract information on how perturbations propagate through the network [8].
- Pathway Activation Patterns: Visualize the extracted knowledge against the network map to identify activated reactions and pathway usage patterns [8].
The following diagram illustrates the SID-PhPP workflow:
SID-PhPP Analysis Workflow
Identifying "Hidden" Phenotypes
SID-PhPP can identify "hidden" phenotypes that share the same set of shadow prices with another phenotype but utilize different metabolic pathways [8]. For example, in the E. coli core model, traditional PhPP analysis may not distinguish between different fermentation patterns that achieve similar growth rates, while SID-PhPP can reveal the underlying pathway differences through statistical analysis of flux distributions.
The Scientist's Toolkit
Essential Research Reagent Solutions
Table 3: Essential Research Reagents for Growth and Secretion Validation
| Reagent/Kit | Function | Application Context |
|---|---|---|
| 13C-labeled substrates | Tracing metabolic fluxes through networks | 13C-Metabolic Flux Analysis [4] |
| HPLC/UPLC columns | Separation of metabolic by-products | Quantification of secretion rates |
| GC-MS systems | Analysis of mass isotopomer distributions | 13C-MFA flux determination [4] |
| Microfluidic devices | Single-cell growth and gene expression monitoring | Time-lapse microscopy [77] [75] |
| Fluorescent reporters | Gene expression and protein localization | Promoter activity under different conditions |
| Stable isotope reagents | Tracking anabolic activity in cells | Stable isotope probing [75] |
| Antibiotic selection markers | Maintaining plasmid stability | Engineered strain validation |
| Chromosomal replication markers | Monitoring replication initiation | Cell cycle coordination studies [76] |
Validating FBA models against quantitative experimental data remains essential for ensuring predictive accuracy in metabolic engineering and systems biology. By integrating the metrics and methodologies outlined in this guide—including robust growth rate determination, precise by-product quantification, and advanced SID-PhPP analysis—researchers can significantly enhance the reliability of their E. coli metabolic models. The continued development of statistical frameworks for model validation and selection will further strengthen the correspondence between in silico predictions and observed phenotypic behaviors, ultimately accelerating strain development for biopharmaceutical applications.
Flux Balance Analysis (FBA) serves as a cornerstone in computational systems biology for predicting metabolic phenotypes from genetic and environmental conditions. A critical choice researchers face is selecting an appropriate model complexity, spanning from comprehensive genome-scale metabolic models (GEMs) to reduced compact core models (CCMs). This guide provides an objective comparison of their predictive performance within the specific context of validating Escherichia coli metabolism using Phenotype Phase Plane (PhPP) analysis. PhPP analysis provides a global perspective on the genotype-phenotype relationship by characterizing different metabolic phenotypes based on shadow prices of metabolites [8]. Understanding the relative strengths and limitations of GEMs and CCMs is essential for researchers, scientists, and drug development professionals to make informed decisions in their metabolic modeling projects.
Genome-scale metabolic models aim to encompass all known metabolic reactions within an organism. For E. coli, the iML1515 model represents a state-of-the-art GEM, containing 1,515 genes, 2,712 reactions, and 1,877 metabolites [26] [80]. These models provide a comprehensive representation of metabolism, enabling system-wide investigations and predictions.
In contrast, compact core models focus on central metabolic pathways essential for energy production and biosynthesis of main biomass building blocks. The iCH360 model, a recently developed "Goldilocks-sized" model of E. coli K-12 MG1655, contains 360 genes and provides a manually curated representation of energy and biosynthesis metabolism [26]. Similarly, the classic E. coli Core Model (ECC2) includes 95 reactions and 72 metabolites, covering major pathways like glycolysis, pentose phosphate pathway, TCA cycle, and electron transport chain [8].
Table 1: Key Characteristics of Representative E. coli Metabolic Models
| Model Name | Model Type | Genes | Reactions | Metabolites | Key Features |
|---|---|---|---|---|---|
| iML1515 [26] [80] | Genome-Scale | 1,515 | 2,712 | 1,877 | Most recent comprehensive GEM; includes all known metabolic functions |
| EcoCyc-18.0-GEM [3] | Genome-Scale | 1,445 | 2,286 | 1,453 | Automatically generated from EcoCyc database; frequent updates |
| iCH360 [26] | Compact Core | 360 | - | - | Manually curated core & biosynthesis metabolism; "Goldilocks-sized" |
| ECC2 [8] | Compact Core | - | 95 | 72 | Widely used core model; includes central carbon metabolic pathways |
Quantitative Accuracy Comparison
Evaluating model accuracy is fundamental for establishing their predictive utility. A comprehensive assessment of E. coli GEMs using high-throughput mutant fitness data across 25 carbon sources revealed that the latest models show excellent performance in predicting gene essentiality. The iML1515 model achieved an area under the curve (AUC) of approximately 0.88 when using precision-recall curves to evaluate its ability to predict gene knockout phenotypes, demonstrating high accuracy [6].
Compact models like iCH360 are derived from their genome-scale parents and retain the core predictive capabilities for central metabolism. While specific AUC values for CCMs are not provided in the search results, their reduced scope inherently limits their predictive coverage to central metabolic pathways, whereas GEMs can predict phenotypes across the entire metabolic network [26].
Table 2: Quantitative Accuracy Assessment of E. coli Metabolic Models
| Model Name | Validation Method | Key Performance Metrics | Reported Accuracy |
|---|---|---|---|
| iML1515 [6] | Gene essentiality prediction across 25 carbon sources | Precision-Recall AUC | ~0.88 AUC |
| EcoCyc-18.0-GEM [3] | Gene essentiality prediction | Error rate for gene knockout phenotypes | 95.2% accuracy (4.8% error rate) |
| iCH360 [26] | Not explicitly quantified | Retains core functionality of iML1515 | High accuracy for central metabolism (qualitative) |
| ECC2 [8] | PhPP analysis | Qualitative phenotype prediction | Accurate for central carbon metabolism |
Experimental Protocols for Model Validation
Gene Essentiality Screening Protocol
Protocol Objective: To validate model predictions of gene essentiality against experimental mutant fitness data.
- Experimental Data Source: Utilize published Random Barcode Transposon-Site Sequencing (RB-TnSeq) data, which provides high-throughput fitness measurements for E. coli gene knockout mutants across thousands of genes and multiple environmental conditions [6].
- In Silico Simulation:
- Gene Knockout: For each gene in the model, simulate its deletion by constraining the associated reaction flux(es) to zero.
- Environmental Constraint: Set the model to simulate a specific growth medium (e.g., minimal medium with glucose as the sole carbon source).
- Phenotype Prediction: Use FBA with biomass maximization as the objective to predict growth (a positive growth rate indicates non-essential, while zero growth indicates essential).
- Validation Metric: Calculate the area under the precision-recall curve (AUC) to quantify prediction accuracy, which is robust to the imbalanced nature of essentiality datasets [6].
Phenotype Phase Plane (PhPP) Analysis Protocol
Protocol Objective: To characterize metabolic phenotypes and identify phase shifts in response to varying nutrient uptake rates.
- Model Preparation: Load the metabolic model (e.g., the E. coli core model) and define the objective function (typically biomass synthesis).
- Parameter Variation: Systematically vary the uptake rates of two nutrients of interest (e.g., glucose and oxygen) across a physiologically relevant range [8].
- Shadow Price Analysis: For each nutrient combination, calculate the shadow price of metabolites, which represents the sensitivity of the growth rate to changes in metabolite availability. Phenotypes are classified based on regions where shadow prices remain constant [8].
- SID-Enhanced PhPP (Optional): To overcome limitations of traditional shadow price analysis, employ a System IDentification-enhanced approach. This involves:
- Perturbing the metabolic network through designed input sequences.
- Applying multivariate statistical analysis (e.g., Principal Component Analysis) to the results.
- Visualizing the extracted knowledge against the network map to identify "hidden" phenotypes that share shadow prices but differ in internal flux distributions [8].
Workflow and Pathway Diagrams
Model Validation and Application Workflow
The following diagram illustrates the general workflow for validating metabolic models and applying them to predict phenotypic outcomes, integrating steps from the experimental protocols above.
Diagram 1: Workflow for Metabolic Model Validation and Application. This chart outlines the process for validating both GEMs and CCMs using experimental data and subsequently applying the validated models for phenotype prediction through PhPP analysis.
Central Metabolic Network of E. coli Core Models
The diagram below provides a simplified representation of the key pathways included in compact core models like iCH360 and ECC2, which are the focus of PhPP analysis.
Diagram 2: Key Pathways in E. coli Compact Core Models. This map visualizes the central metabolic pathways included in models like iCH360 and ECC2, highlighting the production of energy, biosynthetic precursors, and common fermentation products.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for Metabolic Model Validation and Analysis
| Tool / Resource | Type | Primary Function | Relevance to Model Comparison |
|---|---|---|---|
| COBRA Toolbox [54] | Software Toolbox | Provides a standardized environment for constraint-based reconstruction and analysis in MATLAB. | Essential for running FBA, pFBA, and implementing methods like ΔFBA for both GEMs and CCMs. |
| EcoCyc Database [3] | Bioinformatics Database | Curated database of E. coli biology; source for automated generation of GEMs via MetaFlux software. | Serves as a reference for model curation and validation. Enables generation of updated GEMs. |
| RB-TnSeq Mutant Fitness Data [6] | Experimental Dataset | High-throughput measurements of gene knockout fitness under different conditions. | Gold-standard dataset for quantitative validation of gene essentiality predictions across models. |
| Precision-Recall AUC [6] | Statistical Metric | Quantifies prediction accuracy for gene essentiality, robust to imbalanced data. | Key metric for objectively comparing the performance of GEMs and CCMs. |
| System Identification (SID) Framework [8] | Analytical Method | Enhances PhPP analysis by using perturbations and PCA to characterize metabolic phenotypes. | Helps uncover subtle differences in model predictions that are not apparent with standard PhPP. |
The choice between genome-scale and compact core models involves a fundamental trade-off between comprehensiveness and curation depth. GEMs like iML1515 excel in providing system-wide predictions, achieving high accuracy (AUC ~0.88) in gene essentiality screening, and are indispensable for discovering non-obvious metabolic interactions or engineering targets outside central metabolism [6]. However, their size can make them prone to predicting biologically unrealistic fluxes through unphysiological bypasses, and their analysis using advanced methods like elementary flux mode analysis can be computationally challenging [26].
CCMs like iCH360 offer superior interpretability and curation quality. Their manageable size facilitates thorough manual refinement, integration with thermodynamic and kinetic data, and detailed visualization, making them ideal for focused studies on central metabolism and for educational purposes [26]. Their primary limitation is their restricted scope, which prevents them from predicting phenotypes involving peripheral pathways.
For Phenotype Phase Plane analysis, which traditionally focuses on central carbon metabolism, both model types are applicable. The E. coli core model (ECC2) has been successfully used in traditional and SID-Enhanced PhPP analysis to characterize phenotypes based on glucose and oxygen uptake [8]. The choice here may depend on whether the research question is confined to core metabolic shifts (favoring a CCM) or requires understanding the system-wide implications of those shifts (favoring a GEM).
In conclusion, the "best" model is contingent on the specific research objective. For projects requiring a system-wide view and the identification of novel gene targets, a genome-scale model is the appropriate tool. For focused studies on central metabolism, algorithm development, or educational applications, a compact core model offers significant advantages in terms of interpretability and computational efficiency. Future directions point toward the development of hybrid approaches that integrate machine learning with mechanistic models to enhance predictive power beyond what either model type can achieve alone [81] [61].
Using PhPP to Interpret Gene Knockout and Mutant Phenotypes
Phenotype Phase Plane (PhPP) analysis is a computational method based on Flux Balance Analysis (FBA) that extends metabolic simulations from single conditions to a two-dimensional space defined by the availability of two key substrates. This technique maps an organism's optimal metabolic phenotype—such as growth rate or product formation—across a continuum of environmental conditions, dividing the plane into discrete phases where distinct metabolic pathways are utilized [82] [7]. For researchers using Escherichia coli models, PhPP provides a powerful theoretical framework to predict the physiological impact of gene knockouts and interpret mutant phenotypes, thereby playing a crucial role in the validation of genome-scale metabolic models (GEMs) [6] [82].
This guide objectively compares the predictive performance of PhPP analysis against other established and emerging computational methods for interpreting gene knockout phenotypes, supported by experimental data from E. coli studies.
## Core Methodology of Phenotype Phase Plane Analysis
### Mathematical and Computational Foundation
PhPP analysis is built upon the framework of constraint-based modeling. A metabolic network is represented by an m × n stoichiometric matrix (S), where m is the number of metabolites and n is the number of reactions. The solution space for possible flux distributions (v) is defined by the mass-balance constraint Sv = 0 and capacity constraints V_i^min ≤ v_i ≤ V_i^max [82].
In PhPP analysis, this solution space is projected onto a two-dimensional plane defined by two specific reaction fluxes, typically the uptake rates of two substrates (e.g., carbon and oxygen sources). For every point (s, t) on this plane, a linear optimization is performed to find the optimal value of a cellular objective function (e.g., biomass growth), effectively creating a landscape of optimal phenotypic behavior [82].
The resulting phase plane is partitioned into distinct regions by isoclines (demarcation lines). Within each region, or "phase," the optimal flux distribution and the partial derivatives of the optimal-value function (known as shadow prices) remain constant, indicating a stable metabolic phenotype [82] [7]. The correct calculation of these shadow prices is critical for accurately identifying the phase boundaries. While traditional methods using linear programming duality theory can be ambiguous, interior point methods provide a rigorous and unambiguous way to compute them, ensuring the correct PhPP structure [82].
### Standard Protocol for PhPP Analysis inE. coli
The following workflow is a standard protocol for conducting PhPP analysis to investigate gene knockout effects in E. coli:
- Model Selection and Preparation: Obtain a curated GEM for E. coli K-12 MG1655, such as iML1515 [6] or a more compact model like iCH360 [26]. Define the gene-protein-reaction (GPR) associations to link genes to metabolic reactions.
- Simulation of Gene Knockout: To simulate the deletion of a target gene, constrain the fluxes of all reactions catalyzed by the corresponding protein to zero, as defined by the GPR rules [25].
- Define the PhPP Axes and Objective: Select two substrate uptake reactions to vary along the axes (e.g., glucose and oxygen). Set the biological objective function, typically the reaction representing biomass synthesis.
- Compute the Phase Plane: Systematically vary the uptake rates of the two substrates across their feasible ranges. For each combination
(s, t), solve the FBA problem to compute the optimal growth rate. Using interior point methods, calculate the shadow prices to accurately determine the phase boundaries [82]. - Visualize and Interpret the Production Envelope: Plot the results as a phase plane. The "production envelope" illustrates the trade-offs between biomass growth and the production of a metabolite of interest under the gene knockout condition [83]. Analyze the shifts in phase boundaries and optimal metabolic strategies compared to the wild-type model.
Figure 1: A standardized workflow for performing Phenotype Phase Plane analysis of gene knockouts in E. coli.
## Performance Comparison with Alternative Methods
The accuracy of predicting gene knockout phenotypes is a key metric for validating any computational method. The table below compares the performance of PhPP analysis—typically implemented within an FBA framework—against other prominent approaches using E. coli K-12 as a benchmark organism.
Table 1: Comparative accuracy of computational methods for predicting gene knockout phenotypes in E. coli.
| Method | Core Principle | Key Inputs | Reported Accuracy (E. coli) | Key Advantages | Key Limitations |
|---|---|---|---|---|---|
| PhPP/FBA [6] [82] [7] | Linear optimization of a cellular objective | GEM, Stoichiometry, Reaction bounds | ~93.5% (gene essentiality on glucose) [25] | Intuitive visualization of trade-offs; Mechanistic basis | Relies on a pre-defined cellular objective function |
| Flux Cone Learning (FCL) [25] | Machine learning on sampled flux distributions | GEM, Monte Carlo flux samples, Experimental fitness data | ~95% (gene essentiality) [25] | Higher accuracy; No optimality assumption required | Computationally intensive; Requires high-quality training data |
| EcoCyc-GEM [3] | FBA with model derived from EcoCyc database | Automatically generated from EcoCyc DB | 95.2% (gene essentiality) [3] | High automation & frequent updates; Excellent readability via website | Database errors can propagate into model |
| Sequence-Based ML (GenePheno) [84] | Deep learning on gene sequences | DNA/protein sequences, Phenotype ontologies | State-of-the-art in AUC/Fmax (multi-label prediction) [84] | Applicable to poorly annotated genes; No GEM required | "Black-box" predictions; Lower mechanistic insight |
The progression of E. coli GEMs themselves also contributes significantly to the accuracy of predictions, regardless of the analysis method. A 2023 evaluation of four successive E. coli GEMs revealed that while the number of genes covered has steadily increased, the accuracy of predicting mutant fitness initially decreased in newer models until key environmental factors were properly accounted for [6].
Table 2: Evolution and performance of E. coli genome-scale metabolic models.
| Model Name | Publication Year | Genes | Key Features/Improvements | Noted Accuracy Issues |
|---|---|---|---|---|
| iJR904 [6] | 2003 | 904 | One of the first comprehensive GEMs | Statistically significant lower prediction accuracy [25] |
| iAF1260 [6] | 2007 | 1,266 | Expanded coverage of cofactor biosynthetic pathways | |
| iJO1366 [6] [3] | 2011 | 1,366 | High-quality standard for over a decade | |
| iML1515 [6] [26] | 2017 | 1,515 | Most recent comprehensive reconstruction | False negatives in vitamin/cofactor genes due to cross-feeding [6] |
| EcoCyc-18.0-GEM [3] | 2014 | 1,445 | Auto-generated from EcoCyc; 3x yearly updates | 70 incorrect essentiality predictions on glucose [3] |
| iCH360 [26] | 2025 | 360 | Manually curated core/biosynthesis; "Goldilocks-sized" | Limited scope; lacks degradation & cofactor pathways [26] |
### Analysis of Model Inaccuracies and Refinement Strategies
A critical application of PhPP and other validation methods is identifying sources of prediction error. A 2023 analysis of the iML1515 model using high-throughput mutant fitness data highlighted specific areas for refinement [6]:
- Vitamin/Cofactor Biosynthesis: Genes in pathways for biotin, folate, NAD+, and pantothenate were frequently false negatives (predicted essential but experimentally non-essential). This was likely due to metabolite cross-feeding between mutants in the library or carry-over from precursor cells, meaning these metabolites were available in the experiment but not in the in silico medium. Manually adding these metabolites to the simulation environment substantially improved model accuracy [6].
- Isoenzyme GPR Mapping: Inaccurate gene-protein-reaction mappings were a key source of error, as isoenzymes can compensate for each other's loss if the mapping is not correct.
- Machine Learning Insights: A machine learning approach identified that metabolic fluxes through hydrogen ion exchange and specific central metabolism branch points were important determinants of the accuracy of iML1515 predictions [6].
Table 3: Key research reagents and computational tools for PhPP and gene knockout phenotype analysis.
| Resource Name | Type | Function in Research | Relevance to PhPP/Knockout Studies |
|---|---|---|---|
| iML1515 GEM [6] | Genome-Scale Model | Most recent comprehensive metabolic network for E. coli K-12. | Primary template for PhPP analysis; represents the wild-type system. |
| iCH360 Model [26] | Medium-Scale Model | Manually curated model of core and biosynthetic metabolism. | A simplified, high-quality network for faster computation and easier interpretation. |
| RB-TnSeq Data [6] | Experimental Dataset | High-throughput mutant fitness data across 1000s of genes and conditions. | Gold-standard data for validating predictions from PhPP and other methods. |
| EcoCyc Database [3] | Bioinformatics Database | Curated database of E. coli biology, including pathways and genes. | Source for automatic GEM generation and functional annotation of results. |
| COBRApy / cameo [83] | Software Toolbox | Python libraries for constraint-based modeling and analysis. | Provides implementations for FBA, FVA, and Phenotypic Phase Plane analysis. |
| Flux Balance Analysis (FBA) [82] [19] | Computational Algorithm | Predicts metabolic fluxes by optimizing a cellular objective. | The core computational engine used to generate data for PhPP construction. |
| Monte Carlo Sampler [25] | Computational Algorithm | Randomly samples the space of possible flux distributions in a GEM. | Used by FCL to generate training features, capturing the shape of the "flux cone" after knockouts. |
PhPP analysis remains a powerful and intuitive method for interpreting gene knockout phenotypes, providing a unique visual representation of metabolic trade-offs and capabilities across environmental conditions. Its strength lies in its mechanistic basis within the framework of GEMs. However, performance comparisons show that emerging data-driven methods like Flux Cone Learning can achieve higher predictive accuracy by leveraging machine learning and extensive sampling of the metabolic solution space [25].
The choice of method depends on the research goal: PhPP is ideal for generating testable hypotheses about metabolic strategies, while FCL and other advanced techniques may be better suited for achieving maximum predictive power. Ultimately, the iterative process of comparing model predictions—from PhPP or any other method—against high-throughput experimental data is the cornerstone of robust FBA model validation and refinement, as vividly demonstrated by ongoing research with E. coli [6].
Flux Balance Analysis (FBA) has established itself as a cornerstone methodology for predicting metabolic behavior in silico, particularly in model organisms like Escherichia coli. However, the predictive power of any computational model hinges on its rigorous validation against empirical data. The integration of multi-omics data provides a powerful framework for this validation, enabling researchers to test, refine, and confirm model predictions against multifaceted biological evidence. This comparative guide examines current methodologies for validating FBA model predictions using multi-omics data, with a specific focus on E. coli Phenotype Phase Plane (PhPP) analysis. We objectively evaluate the performance of various computational and experimental approaches, providing researchers with a clear overview of their strengths, limitations, and optimal use cases.
Performance Comparison of Validation and Integration Methods
The table below summarizes the core methodologies used for model validation and multi-omics integration, comparing their key characteristics and performance metrics.
Table 1: Performance Comparison of Model Validation and Multi-Omics Integration Methods
| Method/Tool | Primary Function | Reported Performance Metric | Key Advantage | Reference Organism/Context |
|---|---|---|---|---|
| Flux Cone Learning (FCL) | Predicts gene deletion phenotypes from metabolic space geometry | 95% accuracy for gene essentiality prediction; outperforms FBA [25] | Does not require an optimality assumption; versatile for various phenotypes | E. coli, S.. cerevisiae, CHO cells [25] |
| iCH360 Metabolic Model | Manually curated medium-scale model of core & biosynthetic metabolism | Enables EFM analysis, enzyme-constrained FBA, & thermodynamic analysis [26] | "Goldilocks" size balances comprehensiveness with ease of curation & analysis | E. coli K-12 MG1655 [26] |
| Flux Balance Analysis (FBA) | Gold standard for predicting metabolic fluxes & gene essentiality | Max 93.5% accuracy for gene essentiality in E. coli on glucose [25] | Well-established, widely used framework with extensive model support | E. coli [25] |
| MOFA+ (Statistical Integration) | Unsupervised multi-omics factor analysis | F1 score of 0.75 for breast cancer subtype classification [85] | Effective feature selection and strong biological interpretability | Human cancer (Breast cancer subtypes) [85] |
| Flexynesis (Deep Learning Toolkit) | Deep learning for bulk multi-omics integration | AUC = 0.981 for MSI status classification [86] | High flexibility for multiple task types (regression, classification, survival) | Human cancer (TCGA datasets) [86] |
| GPGI (Machine Learning) | Genomic and phenotype-based gene identification | Identified key shape-determining genes (pal, mreB) in E. coli [87] | Cross-species predictive power for functional gene discovery | Bacterial Morphology [87] |
| Hybrid dFBA-PLS Framework | Integrates dynamical FBA with statistical learning | NMSE < 0.15 for metabolite prediction in CHO cell culture [88] | Combines mechanistic modeling with data-driven parameterization | CHO cell bioprocessing [88] |
Experimental Protocols for Key Validation Methodologies
Protocol 1: Gene Essentiality Prediction via Flux Cone Learning
Flux Cone Learning (FCL) provides a machine learning framework to predict gene deletion phenotypes, such as essentiality, by learning the shape of the metabolic space [25].
- Model Preparation: Begin with a Genome-Scale Metabolic Model (GEM) like iML1515 for E. coli. The model is defined by its stoichiometric matrix ( \mathbf{S} ) and flux bounds ( Vi^{\text{min}}, Vi^{\text{max}} ), which are adjusted for gene deletions using the Gene-Protein-Reaction (GPR) map [25].
- Monte Carlo Sampling: For each gene deletion strain, use a Monte Carlo sampler to generate a large number of random, feasible flux distributions (samples) within the corresponding "deletion cone." A typical sample size is 100 samples per deletion cone [25].
- Feature and Label Assembly: Construct a feature matrix where each row is a flux sample and each column is a reaction from the GEM. Assign a phenotypic fitness label (e.g., essential or non-essential) from experimental deletion screens to all samples belonging to the same gene deletion [25].
- Model Training and Prediction: Train a supervised machine learning model, such as a Random Forest classifier, on a subset of the gene deletions (e.g., 80%). Use the trained model to predict the phenotype of held-out deletions (e.g., 20%). Sample-wise predictions are aggregated into a final deletion-wise prediction via majority voting [25].
Protocol 2: Multi-Omics Data Integration for Subtype Classification
This protocol describes the use of statistical and deep learning models to integrate multi-omics data for classifying biological subtypes, a process analogous to validating model-predicted phenotypic states [85].
- Data Collection and Preprocessing: Source multi-omics data (e.g., transcriptomics, epigenomics, microbiomics). Perform batch effect correction using tools like ComBat. Filter out features with excessive missing values or zero expression [85].
- Dimensionality Reduction and Feature Selection:
- For MOFA+: Input the processed multi-omics datasets. Train the unsupervised model to derive latent factors that capture shared and specific variations across omics. Select the top features based on their absolute loadings on the most informative latent factors [85].
- For MoGCN: Use an autoencoder to reduce noise and dimensionality. Calculate feature importance scores by multiplying encoder weights by the feature's standard deviation. Select the top features per omics layer [85].
- Model Evaluation: Use the selected features to train supervised classifiers (e.g., Support Vector Classifier with a linear kernel or Logistic Regression). Evaluate performance using metrics like the F1 score with cross-validation to account for class imbalance [85].
- Biological Validation: Perform pathway enrichment analysis on the selected transcriptomic features to interpret results in the context of known biology and generate hypotheses about underlying mechanisms [85].
Workflow Visualization of Multi-Omics Validation
The following diagram illustrates the logical workflow for validating FBA model predictions through multi-omics data integration, synthesizing the methodologies described in the experimental protocols.
Diagram 1: Multi-omics validation workflow for FBA models.
The Scientist's Toolkit: Essential Research Reagents and Solutions
This section details key computational tools and resources essential for conducting robust multi-omics validation of metabolic model predictions.
Table 2: Key Research Reagent Solutions for Multi-Omics Validation
| Item Name | Type | Primary Function in Validation | Example Use Case |
|---|---|---|---|
| Curated Metabolic Model (e.g., iCH360) | Computational Model | Provides a mechanistically structured network of reactions for generating testable predictions. | Serves as the base E. coli model for PhPP analysis and in silico gene deletion studies [26]. |
| Flux Cone Learning (FCL) | Software/Method | Predicts phenotypic outcomes of genetic perturbations by learning from the geometry of the metabolic flux space. | Validates and extends FBA-predicted gene essentiality lists with higher accuracy [25]. |
| Multi-Omics Integration Tool (e.g., MOFA+, Flexynesis) | Software/Method | Integrates disparate omics datasets into a cohesive analysis to identify correlative and driving features. | Discerns if model-predicted metabolic states correlate with measured changes in transcripts, proteins, and/or metabolites [85] [86]. |
| Monte Carlo Sampler | Computational Algorithm | Generates random, thermodynamically feasible flux distributions from a metabolic model under specified constraints. | Creates the training data (flux samples) for FCL from the GEM of both wild-type and mutant strains [25]. |
| Structured Biological Database (e.g., BacDive, TCGA) | Data Resource | Provides curated experimental phenotypic and molecular data for training and validation. | Supplies phenotypic labels (e.g., bacterial shape) for GPGI or clinical outcomes for Flexynesis [87] [86]. |
| Gene Editing System (e.g., CRISPR-Cpf1) | Wet-bench Reagent | Enables targeted genetic modifications in model organisms for experimental validation. | Constructs knockout strains of candidate genes (e.g., pal, mreB) predicted by models or ML tools like GPGI [87]. |
Discussion and Comparative Outlook
The validation landscape is moving beyond simple comparisons of FBA predictions to single data types. The integration of multi-omics data provides a much richer validation framework. Methods like Flux Cone Learning demonstrate that leveraging the mechanistic information in GEMs through machine learning can surpass the predictive performance of traditional FBA, which relies on an often-debated optimality principle [25]. Simultaneously, the emergence of versatile, reusable deep learning toolkits like Flexynesis makes sophisticated multi-omics analysis more accessible, allowing researchers to build custom validation pipelines for complex phenotypes, including drug response and survival outcomes in translational contexts [86].
The choice between a purely data-driven approach (e.g., MOFA+, GPGI) and a model-driven or hybrid approach (e.g., FCL, hybrid dFBA-PLS) depends on the research goal. Data-driven methods excel at discovering novel patterns and biomarkers from large, heterogeneous datasets without prior mechanistic assumptions [87] [85]. In contrast, model-driven methods are inherently grounded in biological knowledge, making their predictions more interpretable within the known metabolic network and ideal for direct hypothesis testing about metabolic function [26] [25] [88]. For the most powerful validation cycle, these approaches are not mutually exclusive but can be used iteratively, where discrepancies between model predictions and multi-omics observations lead to model refinement and new biological insights.
Conclusion
Phenotype Phase Plane analysis provides a powerful, global framework for validating the predictive capabilities of E. coli FBA models, bridging the gap between in silico predictions and observed physiological behavior. By systematically applying the foundational principles, methodological workflows, and troubleshooting techniques outlined, researchers can significantly improve model reliability. The future of FBA validation lies in the continued integration of multi-omics data, the adoption of enhanced constraint-based methods like enzyme-constrained modeling, and the development of automated validation pipelines. These advances will solidify the role of genome-scale models as indispensable tools in rational metabolic engineering and the discovery of novel antimicrobial targets, ultimately accelerating biomedical and clinical research.
References
- 1. Design Principles as a Guide for Constraint Based and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4693187/]
- 2. 11.2 Constraint-based modeling and flux balance analysis [https://fiveable.me/systems-biology/unit-11/constraint-based-modeling-flux-balance-analysis/study-guide/Oo6YYJECiE8REn7w]
- 3. A genome-scale metabolic flux model of Escherichia coli K–12 ... [https://bmcsystbiol.biomedcentral.com/articles/10.1186/1752-0509-8-79]
- 4. Model Validation and Selection in Metabolic Flux Analysis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10055486/]
- 5. Flux Balance Analysis | Virginia - iGEM 2025 [https://2025.igem.wiki/virginia/fba]
- 6. Evaluating E. coli genome‐scale metabolic model accuracy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10698504/]
- 7. a phenotype phase plane analysis - PubMed - NIH [https://pubmed.ncbi.nlm.nih.gov/11745171/]
- 8. A System Identification Enhanced Phenotype Phase Plane ... [https://www.sciencedirect.com/science/article/abs/pii/B9780444642417504122]
- 9. Phenotypic Landscape of a Bacterial Cell [https://www.sciencedirect.com/science/article/pii/S0092867410013747]
- 10. Integrated analysis of metabolic phenotypes in ... [https://bmcgenomics.biomedcentral.com/articles/10.1186/1471-2164-5-63]
- 11. A phenotype phase plane analysis [https://www.semanticscholar.org/paper/Characterizing-the-metabolic-phenotype%3A-A-phenotype-Edwards-Ramakrishna/0608019535b29dc3fd3ebaafa66f7377ad47e287]
- 12. Construction of an enzyme-constrained metabolic network ... [https://microbialcellfactories.biomedcentral.com/articles/10.1186/s12934-024-02415-z]
- 13. Exact Probability Landscapes of Stochastic Phenotype ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8297706/]
- 14. Automatic phenotyping using exhaustive projection pursuit [https://www.nature.com/articles/s42003-025-08581-z]
- 15. Multi-Color Spectral Transcript Analysis (SPECTRA) for ... [https://www.mdpi.com/2218-273X/3/1/180]
- 16. Model | TJUSX - iGEM 2025 [https://2025.igem.wiki/tjusx/model]
- 17. Accurate prediction of gene deletion phenotypes with Flux ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12475001/]
- 18. Model Validation and Selection in Metabolic Flux Analysis and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10922127/]
- 19. Optimization-based framework with flux balance analysis (FBA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12578352/]
- 20. Optimization-based framework with flux balance analysis (FBA ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1013635]
- 21. E. coli filament buckling modulates Min patterning and cell ... [https://www.nature.com/articles/s41467-025-63509-9]
- 22. What is flux balance analysis? - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3108565/]
- 23. The Biomass Objective Function - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC2912156/]
- 24. Metabolic flux balance analysis and the in silico ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC29061/]
- 25. Accurate prediction of gene deletion phenotypes with Flux ... [https://www.nature.com/articles/s41467-025-63436-9]
- 26. A compact model of Escherichia coli core and biosynthetic ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1013564]
- 27. A mathematical framework for yield (vs. rate) optimization ... [https://www.sciencedirect.com/science/article/pii/S1096717617303853]
- 28. Metabolic engineering - Latest research and news [https://www.nature.com/subjects/metabolic-engineering]
- 29. Modeling the metabolic dynamics at the genome-scale by ... [https://www.sciencedirect.com/science/article/pii/S1096717622001501]
- 30. Questions, data and models underpinning metabolic ... [https://www.frontiersin.org/journals/systems-biology/articles/10.3389/fsysb.2022.998048/full]
- 31. A compact model of Escherichia coli core and biosynthetic ... [https://arxiv.org/html/2406.16596v4]
- 32. OptShadow [https://docs.analytica.com/index.php/OptShadow]
- 33. Shadow prices in linear programming [https://math.stackexchange.com/questions/91504/shadow-prices-in-linear-programming]
- 34. pulp shadow price difference with gurobi [https://stackoverflow.com/questions/24480686/pulp-shadow-price-difference-with-gurobi]
- 35. Validated In Silico Population Model of Escherichia coli [https://www.sciencedirect.com/org/science/article/pii/S2161506322001036]
- 36. Measurement and prediction of small molecule retention by ... [https://www.nature.com/articles/s41598-025-10208-6]
- 37. Basic and applied uses of genome-scale metabolic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3658273/]
- 38. An expanded genome-scale model of Escherichia coli K-12 ... [https://genomebiology.biomedcentral.com/articles/10.1186/gb-2003-4-9-r54]
- 39. The evolution of metabolic networks of E. coli - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC3229490/]
- 40. A compact model of Escherichia coli core and biosynthetic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12533975/]
- 41. iCH360: a compact model of Escherichia Coli core and ... [https://github.com/marco-corrao/iCH360]
- 42. Validated In Silico Population Model of Escherichia coli - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9397402/]
- 43. A steady-state model of microbial acclimation to substrate ... [https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1008140]
- 44. A steady-state model of microbial acclimation to substrate ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7478835/]
- 45. NEXT-FBA: A hybrid stoichiometric/data-driven approach to ... [https://www.sciencedirect.com/science/article/pii/S1096717625000461]
- 46. Spatiotemporal development of expanding bacterial ... [https://www.nature.com/articles/s41467-025-60004-z]
- 47. Simulation of Escherichia coli Dynamics in Biofilms and ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2017.02509/full]
- 48. Machine-learning-guided quantitative delineation of cell ... [https://pubs.rsc.org/en/content/articlehtml/2024/nr/d4nr02466d]
- 49. E. coli cells advance into phase-separated (biofilm-simulating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11580401/]
- 50. Glucose and oxygen uptake rates and growth ra [https://bionumbers.hms.harvard.edu/bionumber.aspx?s=n&v=2&id=109689]
- 51. Enhanced uptake of dissolved oxygen and glucose by ... [https://pubmed.ncbi.nlm.nih.gov/18463864/]
- 52. Morphological Phenotypes, Cell Division, and Gene ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8875244/]
- 53. Pressure and Temperature Dependence of Growth and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3736669/]
- 54. ΔFBA—Predicting metabolic flux alterations using genome ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8608322/]
- 55. a phenotype phase plane analysis. [https://systemsbiology.ucsd.edu/publications/2002/characterizing-metabolic-phenotype-phenotype-phase-plane-analysis]
- 56. Comparison of Optimization-Modelling Methods for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7734505/]
- 57. Fast growth phenotype of E. coli K-12 from adaptive laboratory ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5845443/]
- 58. Model - iGEM Toronto [https://2023.igem.wiki/toronto/model]
- 59. Compare FBA Solutions | KBase App [https://kbase.us/applist/apps/fba_tools/compare_fba_solutions/release]
- 60. Model-driven analysis of experimentally determined growth ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2592718/]
- 61. A neural-mechanistic hybrid approach improving the ... [https://www.nature.com/articles/s41467-023-40380-0]
- 62. Simultaneous application of enzyme and thermodynamic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10783817/]
- 63. ECMpy 2.0: A Python package for automated construction ... [https://www.sciencedirect.com/science/article/pii/S2405805X24000565]
- 64. Interrogating the effect of enzyme kinetics on metabolism ... [https://www.sciencedirect.com/science/article/pii/S1096717622001173]
- 65. Model Balancing: A Search for In-Vivo Kinetic Constants and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8621975/]
- 66. Application of dynamic flux balance analysis to an ... [https://www.sciencedirect.com/science/article/abs/pii/S1096717609000627]
- 67. System identification techniques [https://www.sciencedirect.com/science/article/pii/0167473086900068]
- 68. Ch. 18 - System Identification - Underactuated Robotics [http://underactuated.mit.edu/sysid.html]
- 69. Iterative Model Pipeline Refinement and Optimization ... [https://arxiv.org/html/2502.18530v1]
- 70. Power Determination During Drug Development [https://pmc.ncbi.nlm.nih.gov/articles/PMC6430154/]
- 71. Drug Development Process Map • Global Drug Trials [https://globaldrugdevelopment.tghn.org/process-map/]
- 72. A novel linear discriminant analysis approach for in silico ... [https://www.sciencedirect.com/science/article/abs/pii/S3050475925002799]
- 73. UTEX 2973 Bioreactor: Media Optimization - 2025 UBC iGEM ... [https://2025.igem.wiki/ubc-vancouver/bioreactors/utex-2973-bioreactor-media-optimization]
- 74. Statistically sound modelling of the microbial growth rate [https://www.sciencedirect.com/science/article/abs/pii/S0963996917308530]
- 75. Methods to monitor bacterial growth and replicative rates at ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9629498/]
- 76. General quantitative relations linking cell growth and the cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8059327/]
- 77. Quantifying Dynamic Phenotypic Heterogeneity in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10953537/]
- 78. The Top 5 Parameters in Bioprocessing [https://www.scientificbio.com/blog/the-top-5-parameters-in-bioprocessing]
- 79. Bioprocess Development, Optimization & Scale-up [https://evologic.at/fermentation-101-bioprocess-development-optimization-scale-up/]
- 80. Integration of graph neural networks and genome-scale ... [https://www.nature.com/articles/s41540-024-00348-2]
- 81. Modeling host–pathway dynamics at the genome scale ... [https://www.sciencedirect.com/science/article/pii/S1096717625000849]
- 82. Phenotype phase plane analysis using interior point methods [https://www.sciencedirect.com/science/article/pii/S0098135404002388]
- 83. models — cameo 0.8.0+0.g5f365e1.dirty documentation Analyzing [https://cameo.readthedocs.io/en/stable/04-analyze-models.html]
- 84. GenePheno: Interpretable Gene Knockout-Induced ... [https://arxiv.org/html/2511.09512v1]
- 85. Comparative analysis of statistical and deep learning-based ... [https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-025-06662-5]
- 86. Flexynesis: A deep learning toolkit for bulk multi-omics data ... [https://www.nature.com/articles/s41467-025-63688-5]
- 87. Rapid key gene discovery for bacterial shape [https://pmc.ncbi.nlm.nih.gov/articles/PMC12581320/]
- 88. Investigating the effect of media composition on growth and ... [https://www.sciencedirect.com/science/article/pii/S1369703X25003870]